
Abstract
Traumatic brain injury activates N-methyl-D-aspartate receptors (NMDAR) inducing activation of the Ras protein (a key regulator of cell growth, survival, and death) and its effectors. Thus, trauma-induced increase in active Ras-GTP might contribute to traumatic brain injury pathology. Based on this hypothesis, a new concept of neuroprotection is proposed, examined here by investigating the effect of the Ras inhibitor S-trans, trans-farnesylthiosalicylic acid (FTS) in a mouse model of closed head injury (CHI). Mice subjected to CHI were treated systemically 1 h later with FTS (5 mg/kg) or vehicle. After 1 h, Ras-GTP in the contused hemisphere showed a significant (3.8-fold) increase, which was strongly inhibited by FTS (82% inhibition) or by the NMDA-receptor antagonist MK-801 (53%). Both drugs also decreased active (phosphorylated) extracellular signal-regulated kinase. FTS prevented the CHI-induced reduction in NMDAR binding in cortical, striatal, and hippocampal regions, measured by [3H]-MK-801 autoradiography, and decreased lesion size by 50%. It also reduced CHI-induced neurologic deficits, indicated by the highly significant (P < 0.0001) 60% increase in extent of recovery. Thus, FTS provided long-term neuroprotection after CHI, rescuing NMDAR binding in the contused hemisphere and profoundly reducing neurologic deficits. These findings suggest that nontoxic Ras inhibitors such as FTS may qualify as neuroprotective drugs.
Traumatic brain injury is one of the leading causes of mortality and disability in younger individuals and accounts for an estimated 2 million new cases per year in the Unites States (Sosin et al., 1995). The primary mechanical impact activates cellular and molecular responses that lead to active processes mediated by many biochemical pathways, resulting in secondary damage (for review see Kochanek et al., 2000). Studies of traumatic brain injury in experimental animal models have contributed to our understanding of the pathophysiology of this condition (Laurer and McIntosh, 1999). Excessive glutamate release, which occurs within minutes of trauma, activates various subtypes of glutamate receptors and contributes to injury processes (Faden et al., 1989). A post-traumatic decrease in binding of ligands to NMDAR has also been reported (Miller et al., 1990). Neuroinflammation (including cytokine production, reactive astrocytosis, microglial activation, and macrophage infiltration) is another of the early responses to traumatic brain injury (Feuerstein et al., 1998; Shohami et al., 1999; Morganti-Kossmann et al. 2002), and is therefore considered a potential target for therapy. Recently, we showed that neuroinflammation, like excessive glutamate release, is also associated with a significant decrease in N-methyl-D-aspartate receptors (NMDAR) (Biegon et al., 2002). NMDAR antagonists and antiinflammatory agents have indeed been shown to modify the outcome in several animal models of traumatic brain injury. An alternative approach to singlemechanism drugs is to identify and attempt to target a component that is common to both neuroinflammation and NMDAR-mediated damage. We have identified a possible target, namely, the small GTP-binding protein Ras.
In its active (GTP-bound) state, Ras activates a multitude of effector molecules associated with regulation of cell growth and differentiation, cell death and survival, and cell adhesion and migration (Shields et al., 2000). Ras-GTP is formed by receptor-mediated activation of guanine nucleotide exchange factors (GEFs) that induce an exchange of GDP for GTP, whereupon the signal is turned off by GTPase-activating proteins (Scheffzek et al., 1997). The classic Ras pathway is the one in which growth factors induce activation of Ras that activates the Raf/MEK/extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) cascade (Shields et al., 2000).
Calcium influx through the NMDAR also activates the Ras/ERK pathway (Chen et al., 1998). ERK is associated with NMDAR functions (Atkins et al., 1998; Brambilla et al., 1997; English and Sweatt, 1996; Fukunaga and Miyamoto, 1998; Kaminska et al., 1999; Rosenblum et al., 1997). Active Ras and MAPKs also participate in neuroinflammatory responses (Dalakas, 1995) and in excitotoxicity (Ferrer et al., 2002). Recent studies have demonstrated that trauma induces Ras-dependent MAPK activation (Mandell et al., 2001; Mori et al., 2002; Otani et al., 2002). We therefore postulated that Ras would be an appropriate target for neuroprotection. To substantiate our hypothesis, we used a mouse model of closed head injury (CHI) (Chen et al., 1996) and examined whether inhibition of active Ras by the Ras inhibitor S-trans, trans-farnesylthiosalicylic acid (FTS) (Kloog et al., 1999) has protective effects in this trauma model.
The mechanism of FTS action is known. In earlier studies, we demonstrated that FTS inhibits Ras-dependent cell growth in vitro and inhibits both receptor-mediated and constitutively active Ras-mediated ERK activation (Kloog et al., 1999). FTS affects Rasmembrane interactions, dislodging Ras from its anchorage domains and facilitating its degradation (Haklai et al., 1998). It thus seems that Ras must be anchored to the inner leaflet of the cell membrane to receive and transmit signals (Shields et al., 2000), and that FTS, acting directly on saturable Ras-anchorage sites in the cell membrane, prevents Ras from associating with these sites (Niv et al., 2002). It is important to note that Ras, when in its GTP-bound active state, interacts with sites distinct from those with which inactive GDP-bound Ras interacts (Niv et al., 2002; Prior et al., 2001), and that FTS affects primarily the interactions of Ras-GTP with the cell membrane (Haklai et al., 1998; Kloog et al., 1999). This shows that FTS acts as an activity-dependent drug and may explain why FTS is not toxic and has no adverse side effects in animals (Kloog et al., 1999). FTS thus appears to be a reasonable candidate for inhibiting trauma-induced Ras/MAPK activation (Mandell et al., 2001; Mori et al., 2002; Otani et al., 2002).
MATERIALS AND METHODS
Drug treatments
FTS was a gift from Thyreos (Newark, NJ, U.S.A.). Its purity, assessed by thin-layer chromatography, [1H]-NMR, and mass spectral analysis, was greater than 95%. FTS powder was diluted in chloroform (35.8 mg/mL FTS = 0.1 M) and kept in aliquots. Aliquots were evaporated under nitrogen and their contents (per aliquot) were dissolved in 4 μL of absolute ethanol and 7 μL of NaOH, to which 890 μL of phosphate-buffered saline (PBS) was then added. Each mouse received 0.1 mL of this solution at a dosage of 5 mg/kg. This dose was selected for the present study based on our earlier experiments in other models in which dose response for inhibition of Ras was studied. This dose was found to be both effective and safe. MK-801 was purchased from Sigma (St. Louis, MO, U.S.A.), dissolved in saline, and injected at a dose of 1 mg/kg. The drugs were injected intraperitoneally 1 h after CHI.
Pharmacokinetics of FTS in the brain
Farnesyl 1-3H-thiosalicylic acid ([3H]-FTS), 12.5 Ci/mmol, 1 mCi/mL, was purchased from American Radiolabeled Chemicals (ARC; St. Louis, MO, U.S.A.). The labeled drug was isotopically diluted with unlabeled FTS. Mice (ICR strain) were injected intraperitoneally with 0.1 mL of this FTS solution (14.8 μCi, 351 nmol, 3 mg/kg). At each time point (2.5, 5, 10, 20, 60, and 120 minutes) after the injection, two mice were killed and their brains were removed. The forebrains were washed in PBS, weighed and homogenized, and samples were counted in a scintillation fluid using an LKB β-counter with automatic correction for quenching. Data are expressed as [3H]-FTS in disintegrations per minute per gram of tissue as a function of time.
Animals and trauma
The study was approved by the Institutional Animal Care Committee of Hadassah Medical Center and the Hebrew University. Sixty male C57bl mice weighing 25 to 35 g were used. Closed head injury was induced with the animal under ether anesthesia, as previously described (Chen et al., 1996) and modified (Yatsiv et al., 2002). Briefly, after induction of ether anesthesia, the skull was exposed by a midline longitudinal incision. A tipped nonstick-coated (Teflon) cone was placed in the midcoronal plane above the left anterior frontal area, 1 mm lateral to the midline. A weight (74 g) was dropped onto the cone (from a height of 15 cm), resulting in a focal injury. After trauma, animals received supporting oxygenation with 100% O2 for no longer than 2 minutes and were then returned to their cages. Sham-injured mice were anesthetized and their skin was incised, but they were not subjected to CHI.
Ras-GTP and phospho-ERK assays
In the first experiment, mice were killed 10, 30, or 120 minutes after sham injury or CHI to assess levels of Ras-GTP and phospho-ERK. In the second set of experiments, the animals received FTS (5 mg/kg intraperitoneally) or vehicle, 1 h after CHI and sham-injured mice received the vehicle (n = 4 per group). The mice were killed 2 or 24 h after CHI, their brains were removed, and cortical tissue samples adjacent or contralateral to the site of injury were homogenized in homogenization buffer containing protease inhibitors and 0.5% Triton X100 as described (Haklai et al., 1998). Debris was removed by centrifugation and protein in the extract was determined with the aid of the Bio-Rad protein assay (Bio-Rad Laboratories, GmbH, Munchen, Germany). Total Ras protein was determined by Western immunoblotting of 30 μg of protein with 1:2,500 pan-Ras Ab (Oncogene Research Products, Darmstadt, Germany), followed by 1:7,500 peroxidase goat anti-mouse immunoglobulin G (Haklai et al., 1998). For determination of GTP-bound Ras in protein samples (1 mg), Ras-GTP was pulled down by glutathione-S-transferase fused to the Ras-binding domain of Raf (GST-RBD), which binds Ras-GTP only. The GST-RBD–Ras-GTP was pulled down with glutathioneagarose beads, and Ras was then determined by immunoblotting with pan-Ras Ab as described above, followed by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) (Paz et al., 2000). The bands were quantified by densitometry with Image Master VDS–CL (Amersham Biotech) using TINA 2.0 software (Ray Tests, Germany).
ERK and phospho-ERK were determined in 30 μg of brain extract proteins by immunoblotting, enhanced chemiluminescence, and densitometry (Paz et al., 2000). ERK immunoblots were incubated with 1:2,000 rabbit anti-ERK1/2 Ab (Santa-Cruz) and then with 1:1,000 peroxidase-goat anti-rabbit immunoglobulin G. Phospho-ERK immunoblots were incubated with 1:10,000 mouse anti-phospho-ERK Ab (Sigma) and then with 1:10,000 peroxidase-goat anti-mouse immunoglobulin G.
Neurobehavioral evaluation
The neurologic severity score (NSS) is a tool for assessing an animal's functional status. It is based on the ability of the animal to perform different motor and behavioral tasks representing motor ability, balancing, and alertness (Beni-Adani et al., 2001). Scores range from zero, achieved by healthy uninjured animals, to a maximum of 10, indicating severe neurologic dysfunction, with failure of all tasks. The NSS obtained 1 h after trauma reflects the initial severity of injury and is inversely correlated with neurologic outcome. Animals were evaluated 1 h after CHI, and again at 24 and 48 h and at 5 and 7 d. To assess the effect of FTS on neurologic recovery, mice were randomly assigned to treatment with either FTS or vehicle (n = 10 per group), administered immediately after the initial NSS evaluation. Each animal was assessed by an observer who was blinded to the treatment it had received. The extent of recovery (ΔNSS) was calculated as the difference between the NSS at 1 h and at any subsequent time point. Thus, a higher ΔNSS reflects better recovery. This parameter therefore serves as a tool for evaluation of drug effects.
Quantitative autoradiography and assessment of infarct volume
Seven days after CHI, vehicle-treated (n = 5) and FTS-treated (n = 4) mice were decapitated and their brains were removed (within 1 minute), frozen in powdered dry ice, and kept at −70°C until used. Consecutive 20-μm cryostat sections of the whole forebrain were cut in the coronal plane, and 1 in every 10 sections (i.e., at intervals of 200 μm) was thawmounted onto coated microscope glass slides.
NMDAR autoradiography was performed as described (Bowery et al., 1988), with some modifications. After being prewashed for 30 minutes in 50 mmol/L Tris–acetate buffer at pH 7.4, the sections were incubated for 2.5 h at room temperature in the same buffer containing 10 nmol/L [3H]MK-801, 30 μmol/L glutamate, and 10 μmol/L glycine (200 μL per section). Nonspecific binding was determined in the presence of excess (100 μmol/L) unlabeled MK-801. At the end of incubation, the sections were dipped for 5 seconds in ice-cold buffer, washed for 90 minutes in cold fresh buffer, and then dipped in ice-cold distilled water. The dried tissue sections were exposed to tritium-sensitive film accompanied by commercial calibrated tritium standard scales (Amersham). After exposure for 36 days, the films were developed in Kodak D-19, fixed, and dried. The sections were then stained with cresyl violet for anatomic region placement according to a mouse brain atlas (Paxinos and Franklin, 1997) and for identification of lesions.
Quantitative image analysis
The films were scanned and digitized using PhotoShop (Adobe Systems, Inc., U.S.A.) and a large-bed Umax scanner, and saved in tiff format for accessibility to NIH image software. Using NIH Image routines, the standard curve was measured and used to calibrate regional brain measurements. Morphometry routines were used to measure the lesion area on each section where it was visible. The volume was calculated by multiplying the summed lesion areas by the distance between sections (0.2 mm).
Statistical analysis
Values of NSS are expressed as means ± SD, and analyzed using the Kruskall-Wallis nonparametric test. NMDAR binding densities in various brain regions ipsilateral and contralateral to the trauma were compared by a side by region analysis of variance (ANOVA) followed by regional post hoc analyses. Ras, ERK, and phospho-ERK were quantified as described above, expressed as means ± SD, and analyzed using Student's t-test. A P value of <0.05 is considered significant.
RESULTS
Systemically injected FTS accumulates in the brain
First it was necessary to determine whether FTS can enter the brain, and if so, whether it can exist there in the micromolar concentration range previously found to be relevant to its efficacy as a Ras inhibitor in vitro (Kloog et al., 1999). To this end, we performed pharmacokinetic experiments in which mice received [3H]-FTS (3 mg/kg, intraperitoneally), and the amounts of labeled inhibitor in the brain were then determined by counting radioactivity in samples of brain homogenates. Results of a typical experiment demonstrate that FTS enters the mouse brain, reaching peak amounts within 20 to 30 minutes and remaining relatively high for at least 2 h (Fig. 1). Based on the specific activity of [3H]-FTS and assuming a homogeneous distribution in the brain, we estimated that the peak concentration of FTS was 4.5 μmol/L. Thus, under the conditions employed here, it appears that a pharmacologically relevant concentration of FTS in the brain was achieved.
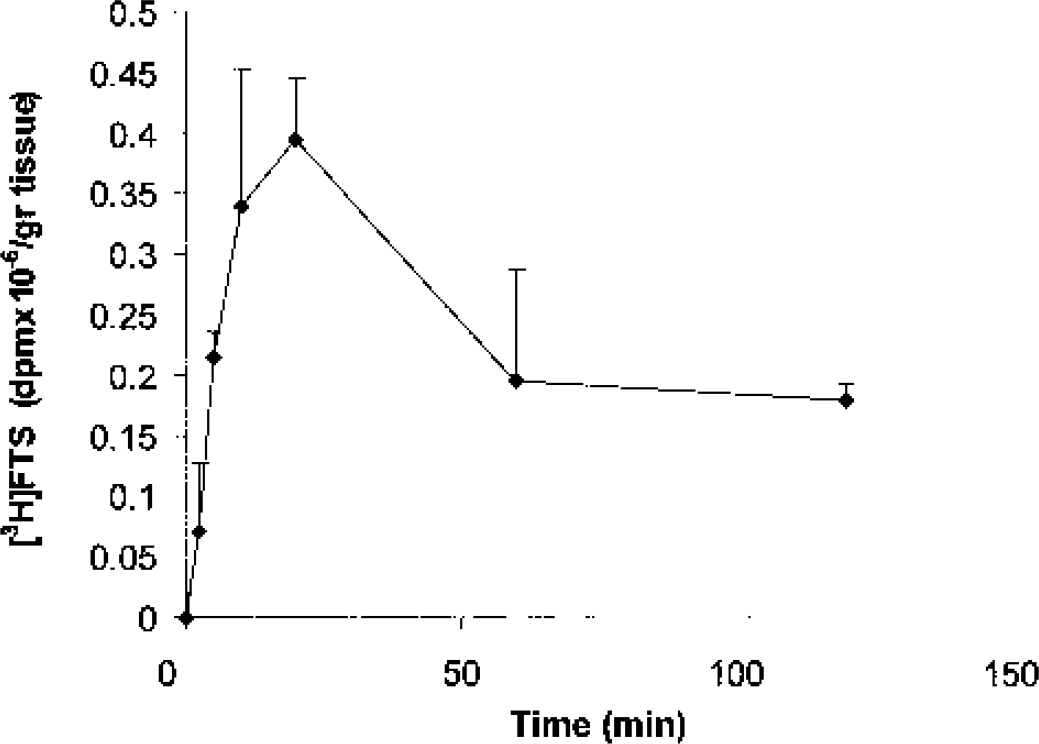
Time course of [3H]-S-trans, trans-farnesylthiosalicylic acid (FTS) accumulation in the mouse brain. Mice received [3H]-FTS (3 mg/kg, intraperitoneally) and the amounts of labeled drug in the forebrain were then determined at the indicated times, as described in Materials and Methods. Mean values (disintegrations per minute per gram tissue) of two separate determinations at each time point are shown.
Closed head injury–induced increase in brain Ras-GTP is inhibited by FTS
Next, we examined whether CHI induces activation of the Ras pathway, as it does in other trauma models (Ferrer et al., 2002). Since previous studies indicated changes in basal levels of active phospho-ERK after traumatic brain injury, we first determined the temporal changes in the levels of active Ras-GTP and of phospho-ERK after CHI. Mice were killed at 10 minutes, 30 minutes, and 2 h after CHI or sham surgery (n = 3/time point), and the left (contused) hemisphere was analyzed by Western blot. It can be seen that under these conditions, the basal levels of Ras-GTP and of phospho-ERK are relatively low in the sham animals (Fig. 2). Closed head injury induced an increase in Ras-GTP and phospho-ERK, observed already at 10 minutes after injury and remaining relatively high even 2 h later (Fig. 2). Here too, only small variations in the levels of Ras-GTP and phospho-ERK were observed between the triplicates (Fig. 2). Notably, there were no changes in the amounts of total Ras or total ERK proteins. In a second set of experiments, the effect of CHI on the total amounts of Ras and of active Ras-GTP in the brains of injured mice was examined, 2 and 24 h after the injury, in the contused (left) and contralateral hemispheres. Figure 3A shows that although CHI did not alter the total amount of Ras in either hemisphere, there was a marked increase in Ras-GTP 2 h after CHI compared to the amounts of Ras-GTP observed in the sham-injured mice. The increase was significantly more pronounced in the contused (left) hemisphere (3.8-fold increase, P = 0.016) than in the contralateral hemisphere (1.6-fold increase) (Fig. 3B). In both hemispheres, however, the increase in Ras-GTP was observed 2 h but not 24 h after the injury, indicating the transient nature of this response.
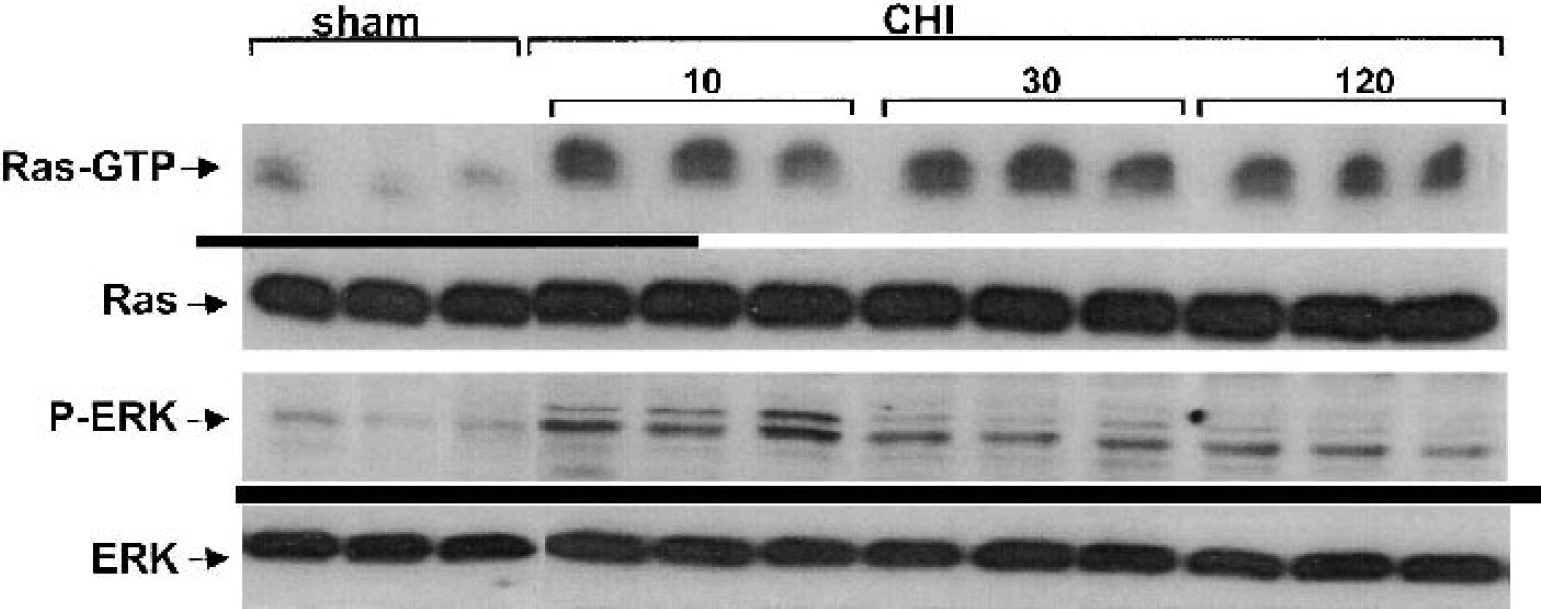
Closed head injury (CHI) induces an increase in Ras-GTP and in phospho-extracellular signal-regulated kinase (ERK) in the contused hemisphere. The brains of either sham mice (n = 3) or of CHI mice at the indicated times after the injury (n = 3) were removed and the left (contused) hemispheres were used for immunoblotting assays. The amounts of total Ras and Ras-GTP were then determined by immunoblotting with pan-Ras antibody, and the amounts of total ERK and phospho-ERK were determined by immunoblotting with anti-ERK and anti-phospho-ERK antibody as described in Materials and Methods.
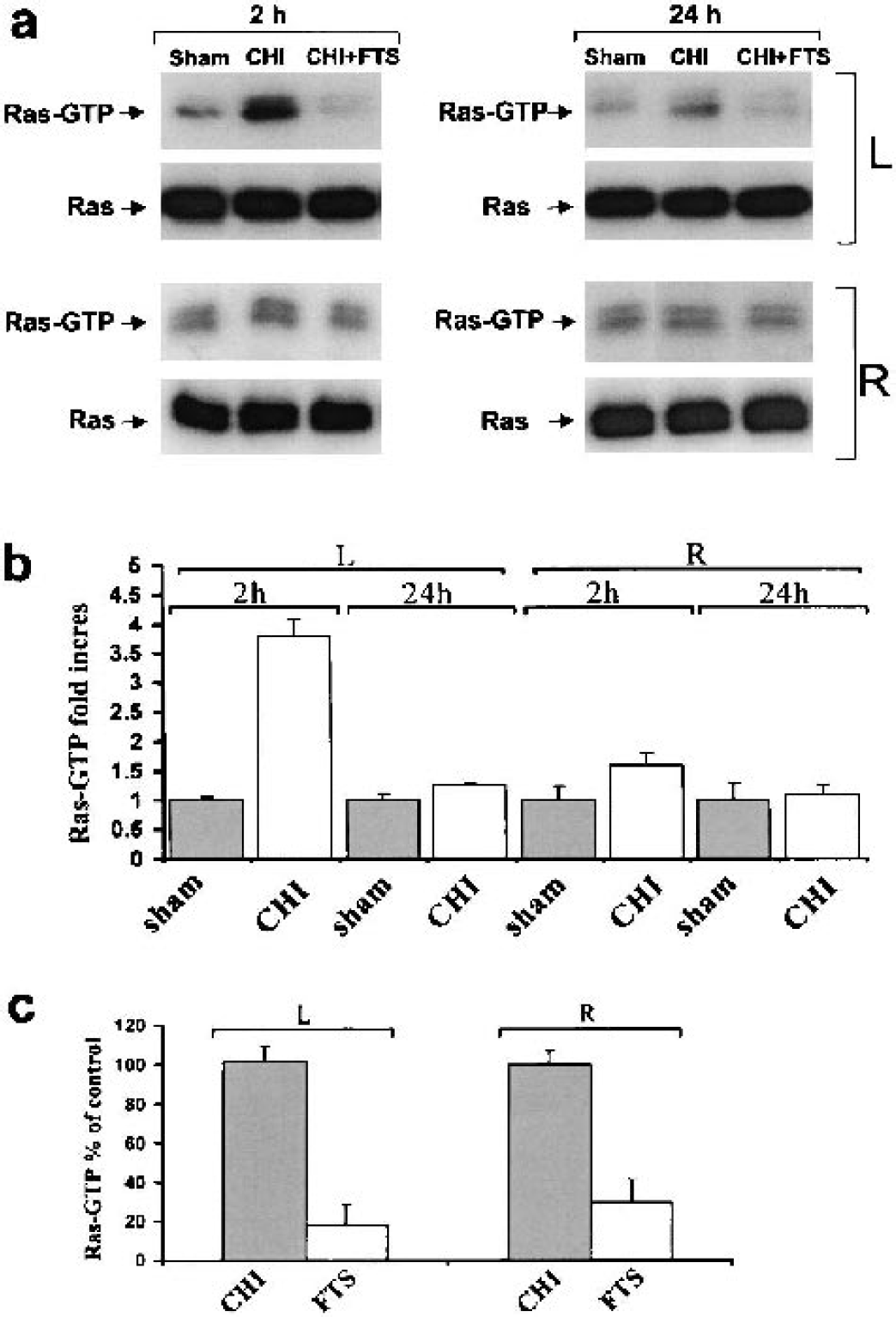
A transient increase in Ras-GTP induced in the brain by closed head injury (CHI) is inhibited by treatment with the Ras inhibitor S-trans, trans-farnesylthiosalicylic acid (FTS). One hour after CHI, mice were treated with the vehicle (CHI) or with 5 mg/kg FTS (CHI + FTS). The mice were killed 2 h or 24 h after the injury, as described in Materials and Methods (n = 4 per group). The amounts of total Ras and Ras-GTP were then determined in both the left (contused, L) and the right (R) hemispheres by immunoblotting with pan-Ras antibody, as described in Materials and Methods. Sham-injured mice received vehicle but were not injured, and their representative immunoblots are shown
We next examined in a second set of experiments whether the Ras inhibitor FTS can alter the CHI-induced increase in Ras-GTP levels in the brain. One hour after CHI, mice were injected intraperitoneally either with vehicle or with 5 mg/kg FTS. In previous studies, this dose of FTS was found to suppress neoplasticity in a number of animal models without adverse side effects (Jansen et al., 1999; Kloog et al., 1999). The amounts of Ras and Ras-GTP were determined 2 and 24 h after injury. The results of a typical experiment show that the CHI-induced increase in Ras-GTP observed 2 h after the injury was strongly inhibited by FTS (Fig. 3A, left panels). This was seen in both the contused and the contralateral hemispheres. As shown in Fig. 3C, the extents of inhibition recorded in the left and right hemispheres were 82 ± 10% and 70 ± 12%, respectively (n = 3). Notably, FTS had almost no effect on Ras-GTP levels when assessed 24 h after CHI (Figs. 3A and 3B), when the amounts of active Ras were already as low as in the sham-injured mice. Also, FTS had no effect on the total amount of Ras in the brain (Fig. 3). Taken together, these results support earlier studies on other brain-insult models (Ferrer et al., 2002; Mandell et al., 2001; Mori et al., 2002; Otani et al., 2002) in which activation of the Ras/MAPK pathway was demonstrated, and are consistent with an FTS action mechanism that acts preferentially on active GTP-bound Ras (Kloog et al., 1999).
MK-801, like FTS, reduces the amounts of Ras-GTP and phospho-ERK in the brains of CHI mice.
To determine whether the CHI-induced increase in Ras-GTP was associated with NMDAR functions, we assayed Ras-GTP in CHI mice injected intraperitoneally with FTS (5 mg/kg) or the NMDAR antagonist MK-801 (1 mg/kg) 1 h after injury. Amounts of Ras-GTP were determined 2 h after CHI, a time point at which we observed a marked increase in Ras activation (Fig. 4). MK-801 partially inhibited the transient CHI-induced increase in Ras-GTP in both the contused (left) and the contralateral hemispheres (Fig. 4A). The calculated extent of inhibition (Fig 4A) was 53 ± 8% in the left hemisphere and 31 ± 5% in the right (n = 4). These results show that the increase in Ras-GTP in the brains of the CHI mice was associated, at least in part, with activation of NMDAR.
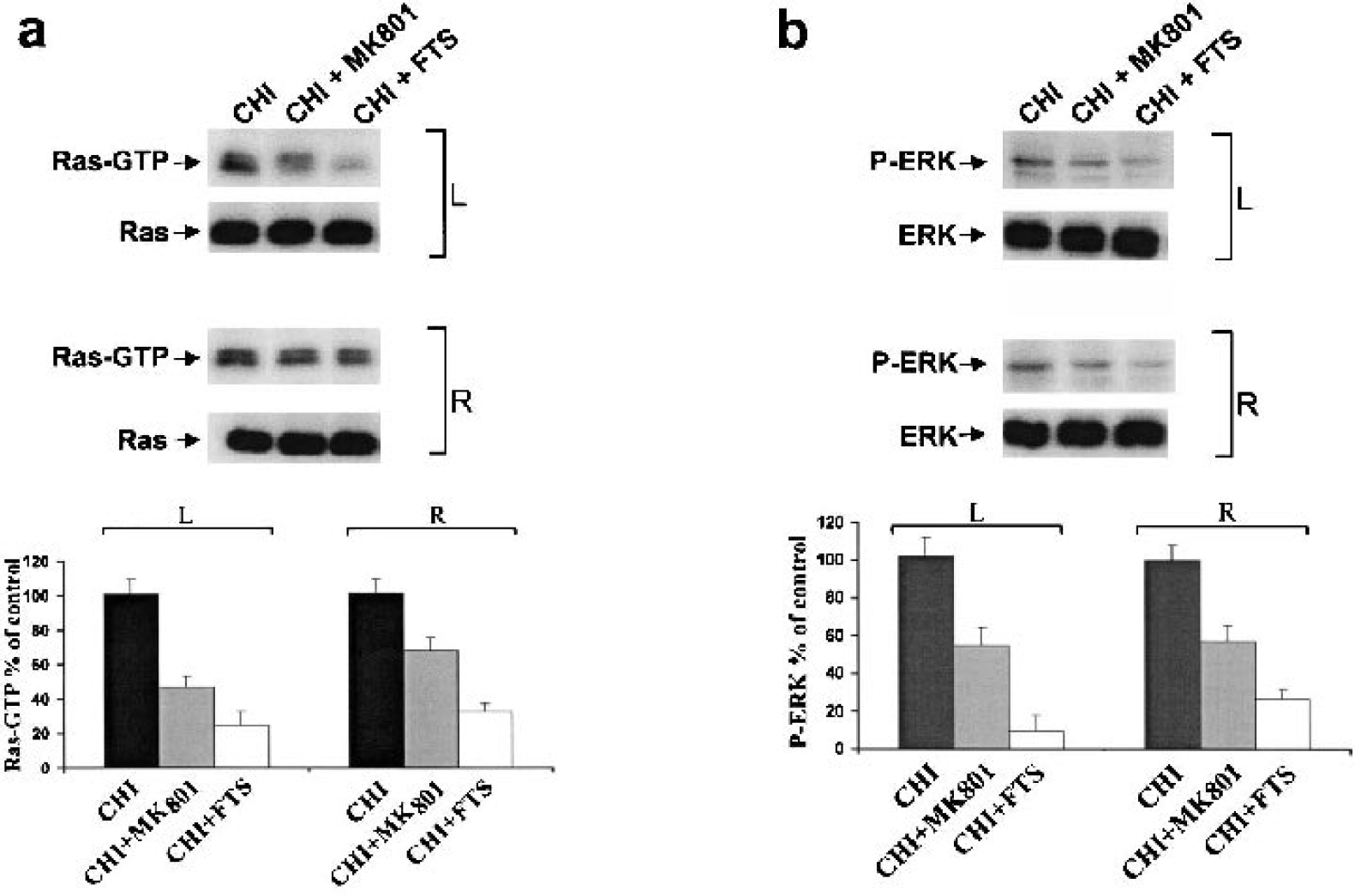
MK-801, like S-trans, trans-farnesylthiosalicylic acid (FTS), reduces the amounts of Ras-GTP and phospho-extracellular signal-regulated kinase (ERK) in the brains of closed head injury (CHI) mice. One hour after CHI, mice were treated with the vehicle (CHI) or with 0.5 mg/kg MK-801 (CHI + MK-801) or with 5 mg/kg FTS (CHI + FTS). The mice were killed 2 h after the injury, as described in Materials and Methods (n = 4 per group). The amounts of total Ras and Ras-GTP were then determined in both left (contused, L) and right (R) hemispheres, by immunoblotting with pan-Ras antibody (
In the set of experiments described above, we also determined the effects of MK-801 and FTS on the amounts of activated (phosphorylated) ERK in the brains of the injured mice. Active ERK and total ERK protein were assayed 2 h after CHI. Neither inhibitor altered the amounts of total ERK (Fig. 4B). However, treatment with each of the inhibitors resulted in a decrease in phospho-ERK in both the contused (left) and the contralateral hemispheres (Fig. 4B). FTS treatment reduced phospho-ERK by 90 ± 9% in the left hemisphere and by 73 ± 8% in the right hemispheres (n = 4), and treatment with MK-801 reduced it by 46 ± 8% and 43 ± 5%, respectively (n = 4). These results show that the inhibitory effects of both the NMDAR antagonist MK-801 and the Ras inhibitor FTS on Ras-GTP were also manifested in the Ras-dependent Raf/MEK/ERK cascade.
Effect of FTS on NMDAR binding
The results described above showed that in CHI, as in other types of trauma, the Ras/MAPK pathway is activated, the activation is at least partially dependent on NMDAR, and the Ras inhibitor FTS strongly inhibits the CHI-induced activation of the Ras/MAPK pathway in the brain. In light of these results and the reported participation of the Ras/MAPK cascade in excitotoxicity (Ferrer et al., 2002), we examined whether treatment with FTS exerts neuroprotective effects. Binding of [3H]-MK-801 to NMDAR was taken to be a measure of NMDA glutamate-receptive neurons. As expected, there were no differences in regional MK-801 binding between the left and right side of the brain in sham-treated animals (Table 1). In agreement with previous reports (Biegon et al., 2002), we found that the binding of [3H]-MK-801 to NMDA glutamate receptors in the brain was substantially decreased after CHI, although the extents of decrease were not uniform: ANOVA with repeated measures revealed a significant effect of side (P < 0.0001) and a significant side by region interaction (P < 0.001) (Table 1B). NMDAR levels in the contralateral (right) hemisphere followed the known regional distribution pattern (Bowery et al., 1988) also seen in the shaminjured animals, with the largest amounts in the hippocampus and cortex. The contralateral hemisphere showed a trend towards lower binding in the frontal motor cortex, but this was not statistically significant. The largest reductions (>20%, or significant at P < 0.05 by post hoc analysis, or both) were observed in regions close to the lesion, including the perilesion area (>40% decrease), parietal cortex, perirhinal cortex, piriform cortex, frontal motor cortex, and dorsal striatum (Table 2, Fig. 5). More moderate reductions, which were not statistically significant on post hoc analysis (13% to 20%), were seen in the ventral striatum and hippocampal CA3 and CA1 fields.
NMDA-receptor density in various brain regions in sham-treated mice
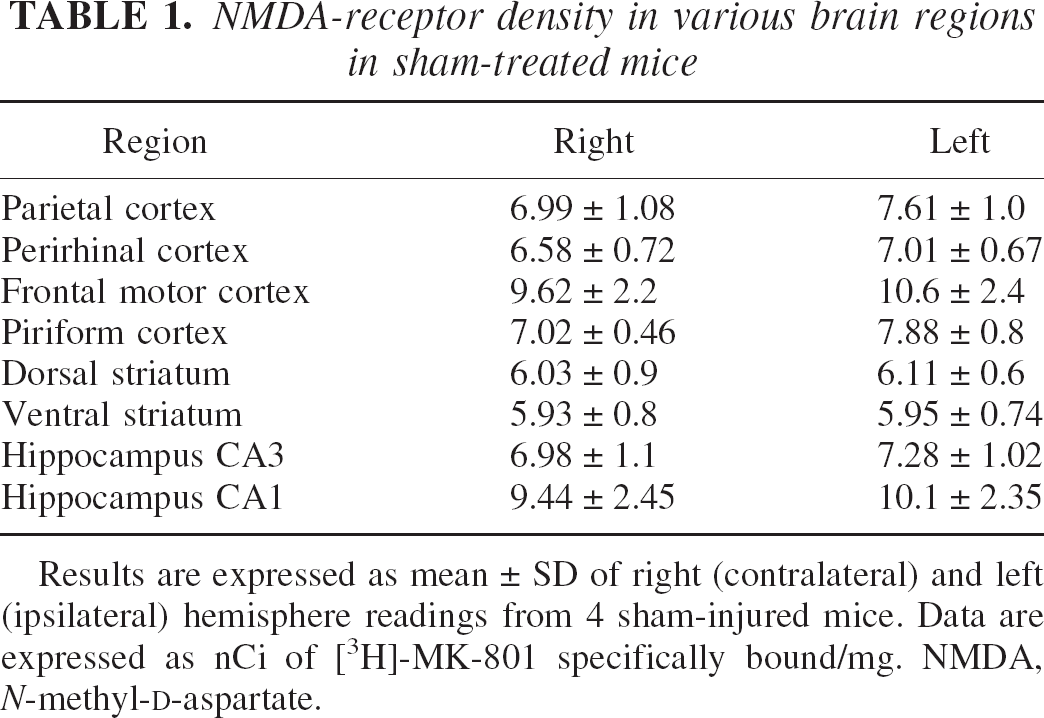
Results are expressed as mean ± SD of right (contralateral) and left (ipsilateral) hemisphere readings from 4 sham-injured mice. Data are expressed as nCi of [3H]-MK-801 specifically bound/mg. NMDA, N-methyl-D-aspartate.
NMDA-receptor density in various brain regions 7 days after CHI without or with FTS treatment
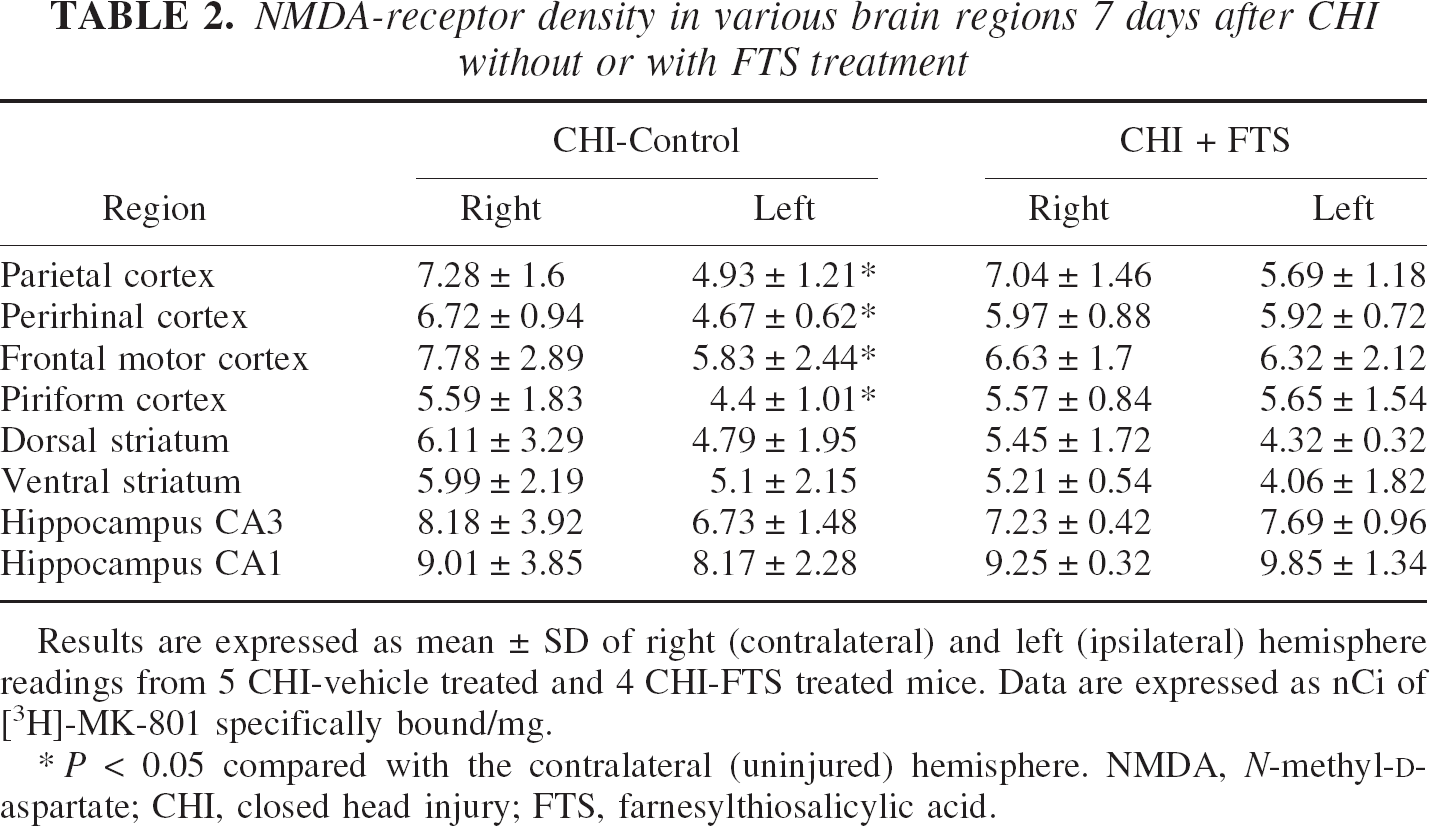
Results are expressed as mean ± SD of right (contralateral) and left (ipsilateral) hemisphere readings from 5 CHI-vehicle treated and 4 CHI-FTS treated mice. Data are expressed as nCi of [3H]-MK-801 specifically bound/mg.
P < 0.05 compared with the contralateral (uninjured) hemisphere. NMDA, N-methyl-D-aspartate; CHI, closed head injury; FTS, farnesylthiosalicylic acid.
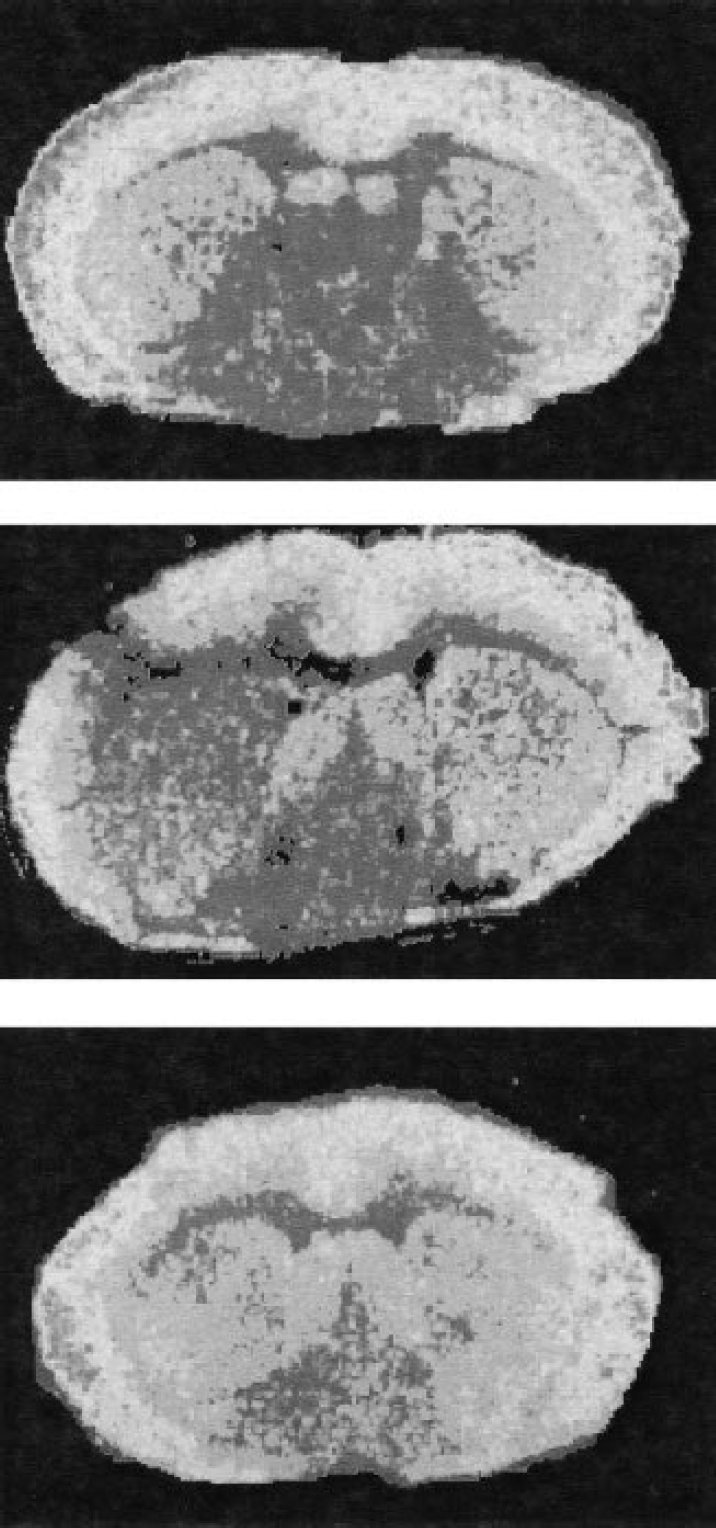
Long-term loss of N-methyl-D-aspartate receptors (NMDAR) binding is prevented by S-trans, trans-farnesylthiosalicylic acid (FTS). Mice were injected with FTS or vehicle 1 h after CHI, evaluated for neurologic deficits for 7 days, and then decapitated. Their brains were sectioned for quantitative autoradiography and histology, and pseudocolored (rainbow spectrum, purple = low, red = high) images are shown. The top image is from a sham animal, showing symmetrical MK-801 binding. The middle image is from a traumatized mouse at the same anatomic level, just posterior to the lesion, showing profound loss of NMDAR binding relative to the contralateral hemisphere in the cortex and striatum. The bottom image shows a section at the same anatomic level from an FTS-treated mouse, with symmetrical binding of MK-801 indicating preservation of the NMDAR.
Treatment with FTS completely reversed the effect of trauma on the binding of [3H]-MK-801 to the NMDA receptors, as indicated by the finding that ANOVA with repeated measures showed no significant difference between sides and no significant interaction (P = 0.5, Table 2, Fig. 5). Complete reversal of the trauma effect was seen in the piriform cortex, perirhinal cortex, posterior cingulate cortex amygdala, frontal motor cortex, and all dorsal hippocampal fields. A nonsignificant trend towards lower binding in the ipsilateral hemisphere of the FTS-treated animals was seen only in the parietal cortex and striatum (Table 2).
Effect of FTS on lesion volume
We next examined the effect of FTS on the size of the CHI-induced lesion. This model results in progressive tissue loss and cavitation of the cortex in the injured side that reaches a stable size within 3 to 7 days. A distinct lesion was indeed observed in all of the mice that were subjected to CHI in this study. As expected, the lesion was located in the left frontoparietal cortex. The mean lesion volume calculated in the control mice was 155 ± 41 μL (mean ± SD of 5 animals, range 113 to 211 μL). FTS treatment resulted in a trend towards a reduction in lesion volume by almost 50%, to 87 ± 53 μL (mean ± SD of 4 animals, range 37 to 161, P < 0.07, Student's t-test, two tailed). The range of lesions is illustrated in Fig. 6, with the largest cross-sectional representation, from a vehicle-treated animal shown in Fig. 6A and the section with the smallest lesion, from an FTS treated animal, shown in Fig. 6B.
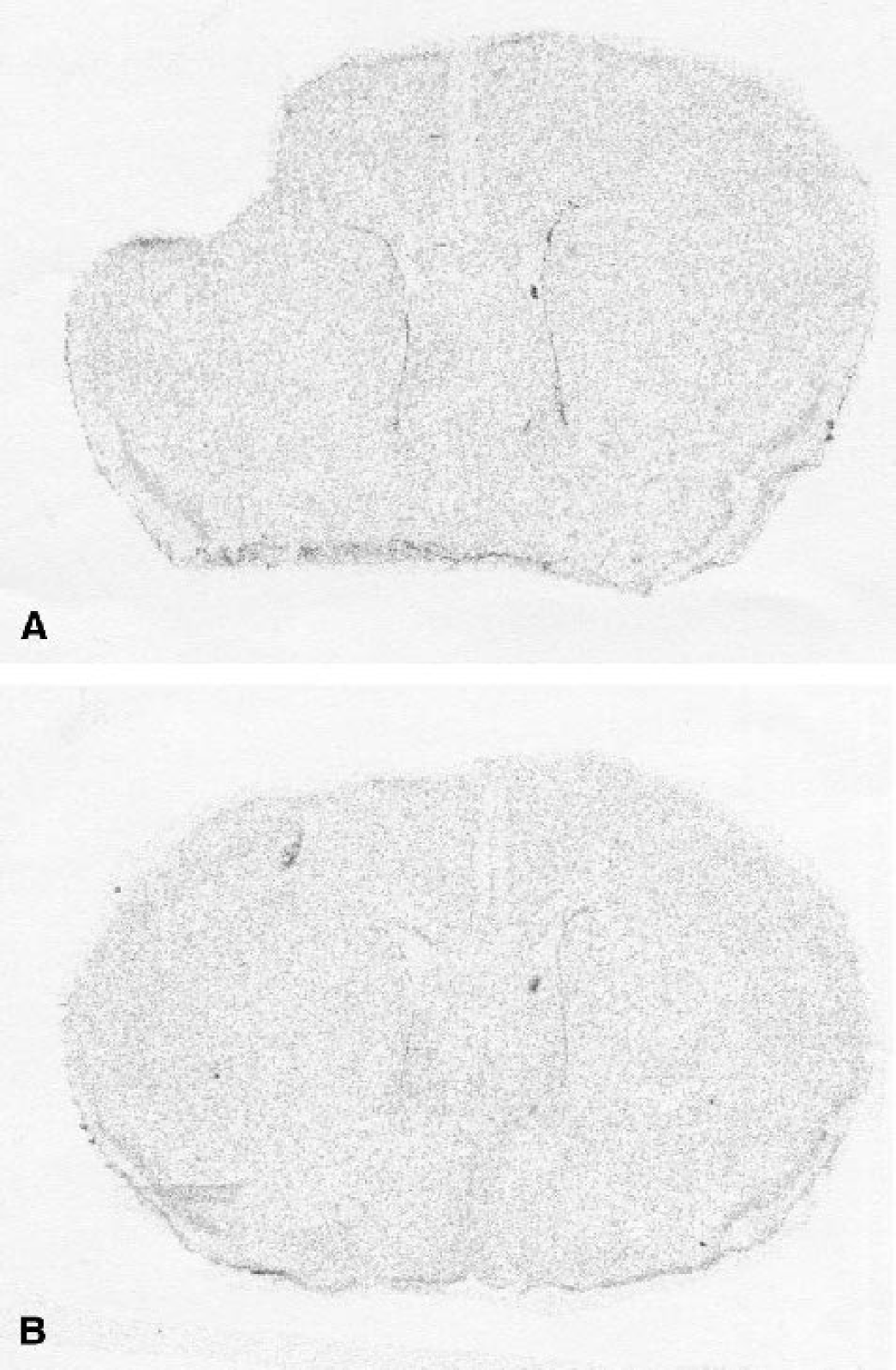
Effect of S-trans, trans-farnesylthiosalicylic acid (FTS) on lesion size after closed head injury (CHI). Brain sections used for autoradiography on day 7 post-CHI (see Fig. 5) were stained with cresyl violet and magnified at low power (4×). The illustration shows a section through the area of maximal lesion in one vehicle-treated CHI mouse
Effect of FTS on functional recovery
The results described above indicated that FTS exerts a profound neuroprotective effect after CHI in mice. It was therefore of interest to examine whether these FTS-induced effects are also manifested in a decrease in the neurologic impairment induced by CHI. The extent of neurologic damage was determined, as described (Beni-Adani et al., 2001) (also see Materials and Methods), in terms of the NSS, which was first evaluated 1 h after the injury. The mice were then divided into vehicletreatment (control) and FTS-treatment groups (n = 10 per group), ensuring that the severity of injury in the two groups was similar (mean NSS ± SD = 6.9 ± 0.38 and 6.7 ± 0.3, respectively). Immediately thereafter the mice received either FTS (5 mg/kg, intraperitoneally) or vehicle, and the NSS was then evaluated at different time points, and both the spontaneous and the drug-related recovery (in terms of ΔNSS) in the two groups were compared. As shown in Fig. 7, a significantly better recovery was observed as early as 24 h after injury in the FTS-treated mice. This effect was maintained for up to 7 days and became even more pronounced over time (P < 0.0001 Mann Whitney). The mean ΔNSS values recorded on day 7 after injury were 4.2 in the FTS-treated mice and 1.7 in the controls (Fig. 7). Thus, single-dose treatment with FTS provided a robust, long-lasting beneficial effect that reduced the CHI-induced neurologic deficits by 60% (P < 0.0001).
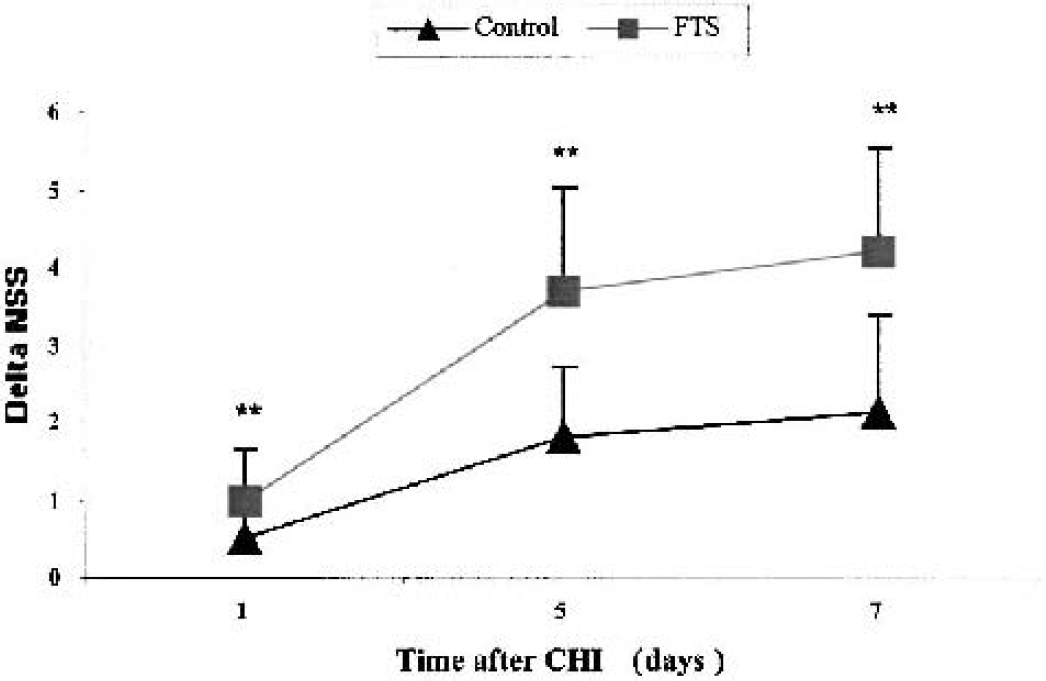
Time course of neurologic recovery of mice after CHI. Mice were subjected to closed head injury (CHI) and their neurologic severity scores (NSSs) were assessed 1 h after the injury. Immediately thereafter they were treated with 5 mg/kg S-trans, trans-farnesylthiosalicylic acid or vehicle (n = 10 per group). The NSS was reevaluated between 24 h and 7 days after CHI. ΔNSS was calculated as described in Materials and Methods, and is shown for both groups.
DISCUSSION
In its active GTP-bound state, Ras activates a multitude of effectors associated with regulation of a variety of cell functions. Ras-GTP activates its effectors (Shields et al., 2000) and is then turned off by Ras-GTPase-activating proteins (Scheffzek et al., 1997). The consequences of Ras activation are evident long after it is turned off. Recent studies have demonstrated that trauma induces Ras-dependent MAPK activation (Mandell et al., 2001; Mori et al., 2002; Otani et al., 2002). In addition, active Ras and MAPK are associated with excitotoxicity (Ferrer et al., 2002). On the basis of these observations, we postulated that inhibition of early posttraumatic responses, such as Ras-dependent signaling, may provide long-term neuroprotection. Because Ras acts at the apex of many signaling pathways, it seemed reasonable to utilize a Ras inhibitor to test this hypothesis.
Our results show that after CHI in mice, the Ras inhibitor FTS exerts robust neuroprotective effects. This was evident from the better neurologic recovery observed in the FTS-treated mice than in vehicle-treated controls, with a highly significant, long-lasting improvement of 60% of neurologic status seen even at 7 days after trauma, as well as from the observed rescue of the binding of [3H]-MK-801 to NMDAR and the smaller size of lesions recorded in the brains of FTS-treated injured mice. To the best of our knowledge, this is the first demonstration that inhibition of active Ras by a nontoxic Ras inhibitor can confer neuroprotection and lead to a better neurologic recovery after traumatic brain injury. Our results thus support a new concept of neuroprotection, achieved through the inhibition of active Ras in the brain. The present results also support the possible development of FTS as a brain-active Ras inhibitor. They imply that FTS crosses the blood–brain barrier, that a pharmacologically relevant concentration of the inhibitor (4.5 μmol/L) is rapidly achieved (Fig. 1), and that FTS inhibits the CHI-induced transient increase in active Ras-GTP and in active phospho-ERK in the brain (Figs. 3 and 4).
The well-documented release of glutamate after CNS injury is a critical event, which is followed by activation of NMDAR and accumulation of intracellular calcium (Faden et al., 1989). A subsequent decrease in NMDAR binding as early as 2 h after traumatic, ischemic, inflammatory, or NMDA-provoked injury has been reported. This decrease, which appears to precede neuronal death, can be prevented by the NMDA antagonist MK-801 and is believed to contribute to the neurologic deficits caused by these injuries (Biegon et al., 2002; Miller et al., 1990; Goebel and Poosch, 2001). Calcium influx through the NMDAR activates the Ras/ERK pathway (Chen et al., 1998), and ERK in the brain is activated in response to various NMDAR-related stimuli, including long-term potentiation (English and Sweatt, 1996), long-term memory (Brambilla et al., 1997), visual stimulation (Kaminska et al., 1999), associative learning (Atkins et al., 1998), and ischemia (Farnsworth et al., 1995). Knockout mice for the brain-specific Ras exchange factor Ras-GEF, which activates Ras through binding to calcium/calmodulin (Farnsworth, 1995), indeed exhibit impaired long-term potentiation and memory consolidation (Brambilla et al., 1997). It still is not clear, however, how NMDAR activation causes an increase in Ras-GTP. A possible link between the NMDAR-mediated calcium influx and the Ras/ERK cascade is provided by the Ras-GTPase-activating protein SynGAP, which is highly enriched in the brain and is inhibited by calcium/calmodulin kinase II (Chen et al., 1998; Kim et al., 1998). Such a decrease in GAP activity would cause an increase in Ras-GTP. Nonetheless, the present finding that the CHI-induced transient increases in Ras-GTP and phospho-ERK are inhibited by MK-801 (Fig. 4) is consistent with previous reports that traumatic brain injury and NMDAR activation trigger activation of the Ras/MAPK pathway in the brain. Moreover, the early increase in Ras-GTP observed after CHI (Fig. 2) correlates well with the previously described transient increase in Ras-GTP after an excitotoxic insult (Ferrer et al., 2002) and with the time course of the increase in glutamate release under similar conditions (Faden et al., 1989). The greater increase in Ras-GTP in the injured hemisphere (3.8-fold) than in the contralateral hemisphere (1.6-fold) would indicate that the release of glutamate occurs mainly at the site of injury, as reported earlier (Faden et al., 1989).
The above considerations lead us to believe that the most likely sequence of events after CHI in our experiments is the release of glutamate followed by NMDAR activation, subsequently resulting in increased calcium influx and Ras activation. It is worth noting that although both inhibitors, MK-801 and FTS, inhibited the CHI-induced increase in Ras-GTP and in phospho-ERK, they acted through entirely different mechanisms. As discussed above, MK-801 (which blocks NMDAR and calcium influx) would either prevent a receptor-mediated exchange of GDP for GTP on Ras or decrease a receptor-mediated inhibition of Ras-GAP activity. FTS, however, is known to act mainly on membrane Ras once it has become active (GTP-bound) through the action of Ras exchange factors (reviewed in Kloog et al., 1999). Thus, the decrease in Ras-GTP observed with MK-801 treatment represents inhibition of exchange, or decrease in GTP hydrolysis by Ras, or both, whereas the decrease observed with FTS treatment represents a direct effect of the inhibitor on membrane association of the active Ras-GTP formed as a consequence of CHI and activation of NMDAR. The preferential effect of FTS on Ras-GTP without affecting the total amount of Ras (Figs. 3 and 4) is consistent with the above mentioned mechanism of drug action. Additional recent and significant evidence supports the mechanism whereby FTS affects mostly the active (GTP-bound) forms of Ras. FTS was shown to compete with binding of Ras to Ras escort proteins (e.g., galectin-1) that bind only the active forms of Ras (Elad-Sfadia et al., 2002; Paz et al., 2000). These recent findings, together with earlier observations in nonneuronal cells in which active Ras-GTP and ERK activation were preferentially inhibited by FTS, and the similar results presented here for mice with CHI, can explain the lack of toxicity of FTS, since it affects only a small fraction of the total amount of cellular Ras (reviewed in Kloog et al., 1999).
It is important to note that in addition to regulating the Raf/MEK/ERK pathway, Ras directly and indirectly regulates many other signaling cascades, including phosphoinositide 3-kinase pathways, the Ral-GTPase pathways, the Rac and Rho GTPases, and the p38 and Jun kinase pathways (Shields et al., 2000). The latter two cascades were recently studied in the context of traumatic brain injury in vitro and in vivo (Mori et al., 2002; Otani et al., 2002), but results obtained were conflicting. However, the results of those two studies with respect to trauma-induced ERK activation are mutually consistent, and are in agreement with the results presented here. Our results obviously do not rule out the possibility that an increase in Ras-GTP after CHI might lead to the activation of Ras signaling pathways other than the Raf/MEK/ERK, which might also be involved in CHI-induced neuronal deficits.
Whether or not the CHI-induced increase in Ras-GTP results in activation of a multitude of Ras effectors, our results show that both the increase in Ras-GTP (Figs. 2 and 3) and the loss of NMDAR binding (Fig. 5 and Table 2) are inhibited by treatment with FTS. This strongly suggests that the inhibition of NMDAR-dependent Ras activation has a neuroprotective effect. The post-CHI loss of NMDAR, which is believed to contribute to neurologic deficits (Biegon et al., 2002; Miller et al., 1990; Goebel and Poosch, 2001), is strongly inhibited by FTS and is indeed manifested in a decrease in the CHI-induced neurologic deficits (Fig. 7). A significant, long-lasting improvement in neurologic status was recorded between 24 h and 7 days in the FTS-treated mice. During the entire period of follow-up, the ΔNSS was greater in the FTS-treated mice than in the vehicle-treated mice, and this effect, although already significant 24 h after CHI, became more pronounced with time. A pattern similar to that found in mice was also observed in rats (not shown). It is interesting to note that MK-801 was shown to reduce edema in the same rat model. Neurologic evaluation could not be performed in that study, however, because of the strong sedative effect of the drug (Shapira et al., 1990). FTS had no sedative effects at any of the times tested.
Many of the clinical signs of CHI, including memory impairment (Chen et al., 1996), are probably manifestations of functional loss of NMDAR and NMDA-receptive neurons. NMDAR are indeed vulnerable to traumatic, ischemic, and inflammatory brain damage, and this effect is reversed by early administration of MK-801 (Biegon et al., 2002; Miller et al., 1990; Goebel and Poosch, 2001). In this study, we showed that FTS is capable of complete reversal of NMDAR loss in the traumatized hemisphere. Taken together with the findings that both FTS and MK-801 treatments reduced the relatively large amounts of Ras-GTP observed in the brains of CHI mice, as well as the improvement in neurologic status after FTS treatment, these observations strongly suggest a direct protective effect of FTS on NMDA-receptive neurons. Finally, our study also showed that FTS significantly reduces the mean lesion area.
The significantly stronger effect of FTS than of MK-801 on Ras-GTP (inhibition of 70% to 82% compared to 31% to 53%) suggests that some of the positive effects of the Ras inhibitor might be mediated by inhibition of NMDAR-independent processes. One such process might be neuroinflammation, which is among the early posttraumatic responses sustained over a long period (7 days and more) (Feuerstein et al., 1998). Active Ras participates in neuroinflammatory responses, and mechanical trauma induces Ras-dependent astroglial MAPK activation (Dalakas, 1995). Indeed, many forms of brain injuries, including trauma and inflammation, induce astrogliosis and activation of astroglia (Mandell et al., 2001). The possibility that FTS may suppress a neuroinflammatory response after CHI is supported by previous studies demonstrating that FTS can suppress inflammatory processes (Kloog et al. 1999).
In conclusion, the neuroprotective effect of FTS, expressed in the results of behavioral testing and in the rescue of NMDAR-receptive neurons, supports the role of Ras-GTP activation as an early upstream signal in the late consequence of traumatic brain injury, and suggests that early inhibition of this pathway or intracellular events further downstream could provide new strategies for the management of head injury.
Footnotes
Acknowledgments:
ES is affiliated with the David R. Bloom Center for Pharmacy, The Hebrew University School of Pharmacy.