
Abstract
N-arachidonoyl-
Introduction
The endocannabinoid (eCB) Gi/o-protein-coupled receptors CB1 and CB2 were previously identified in the brain (Devane et al, 1988; Munro et al, 1993), followed by identification of the eCB agonists anandamide (arachidonoylethanolamide, AEA) and 2-arachidonoylglycerol (2AG) (Devane et al, 1992; Mechoulam et al, 1995; Sugiura et al, 1995; Van Sickle et al, 2005).
Traumatic brain injury (TBI) leads to an increase in 2AG levels in the brain, which lasts for at least 24 hours after the injury. Treatment with exogenous 2AG following TBI reduces blood–brain barrier permeability, brain water content, lesion volume, and hippocampal cell death. In addition, behavioral function improves significantly in treated mice (Panikashvili et al, 2001). These beneficial effects were abolished when 2AG was coadministrated with the selective CB1-antagonist SR141716A (rimonabant) as well as in CB1−/- knockout mice, suggesting that they are mediated via CB1 receptor signaling (Panikashvili et al, 2001, 2005). Additionally, administration of AEA reduced the volume of cytotoxic edema in a manner that was insensitive to SR141716A (van der Stelt et al, 2001), suggesting that at least part of the beneficial effects of AEA are produced via activation of mechanisms independent of CB1 signaling. Recently, it was proposed that also CB2 receptors mediate neuroprotection after middle cerebral artery occlusion (Zhang et al, 2007). Indeed, injection of CB2 agonist together with CB1 antagonist led to a better outcome in an ischemia–reperfusion model (Zhang et al, 2008).
N-arachidonoyl-
Materials and methods
Reagents
N-arachidonoyl-
Animals
The study was performed according to the Institutional Animal Use and Care Committee guidelines and was approved by the institution's Animal Care and Use Committee. Male Sabra mice (Harlan, Jerusalem, Israel) aged 6 to 7 weeks and weighing 35 to 45 g were used in this study. This is a strain bred at the Hebrew University and provided from the same supplier (Harlan). No variations in dominant/submissive characteristics were observed with this strain. For each experimental group, 6 to 9 mice were used unless otherwise mentioned.
Trauma Model
Mice were subjected to closed head injury (CHI) under isoflurane anesthesia, using a weight-drop device that falls over the left hemisphere, as described elsewhere (Tsenter et al, 2008). In brief, after a longitudinal scalp incision, mice were immobilized under a cylindrical calibrated weight-drop device. A tipped teflon cone was placed (upside down) 2 mm lateral to the midline and 1 mm caudal to the left coronal suture, and a metal rod (94 g) was dropped down on the cone from a height of 11 to 14 cm (adjusted to body weight, to ensure the severity of injury (Tsenter et al, 2008) required to produce CHI). Sham-treated mice were anesthetized with isoflurane, their scalps were incised, but trauma was not induced. It should be noted that < 10% of the injured mice were excluded from the study, mostly because of death by apnea within minutes of injury.
Drug Application
Following preliminary experiments in which dose and time window were determined, AraS 3 mg/kg dissolved in ethanol:cremophor:saline 1:1:18 or vehicle alone was injected intraperitoneally into the mice 1 hour after CHI, as described elsewhere (Panikashvili et al, 2001). To evaluate the involvement of CB1R, CB2R and transient receptor potential vanilloid 1 (TRPV1) in the neuroprotective effects of AraS, CB1-antagonist rimonabant (SR141716A) 1 mg/kg (Barna et al, 2009; Hayakawa et al, 2007), or the CB2-antagonist SR148522, 1 mg/kg (Fride et al, 2005; Hanus et al, 1999), or Capsazepine, a TRPV1 antagonist, 1 mg/kg (E Berry, Laboratory of Human Nutrition Hebrew University Jerusalem Israel; personal communication), together with AraS were also injected into the mice. As AraS was found to be an activator of large conductance Ca+-activated K+ (BK) channels, we also tested the involvement of these channels in the neuroprotective effect of AraS, using the BK channel blocker paxilline (6 mg/kg) (Imlach et al, 2008) alone or with AraS. Paxilline was dissolved in ethanol, sonicated, and diluted in ethanol:-cremophor:saline (1:1:18) to the final concentration.
Evaluation of Functional Outcome
At 1 hour after CHI, the functional status of the mice was evaluated according to a set of 10 neurobehavioral tasks (neurologic severity score, NSS), which examine reflexes, alertness, coordination, motor abilities, and balancing (Beni-Adani et al, 2001). Failure to perform a task scores one point and a success scores 0. Hence, normal animals score 0, reflecting healthy mice, whereas a score of 10 reflects maximal neurologic impairment. Only mice with NSS 6 to 8 at 1 hour after injury (NSS 1hour) were included in the study. Immediately after evaluation of NSS at 1 hour, the mice were randomly assigned to treatment groups (n = 6 to 9 mice/group), which received vehicle or drug and NSS was reevaluated on days 1, 2, 3, 7, and 14 after CHI. The analyses were performed by an investigator that was blinded to treatment.
Lesion Volume
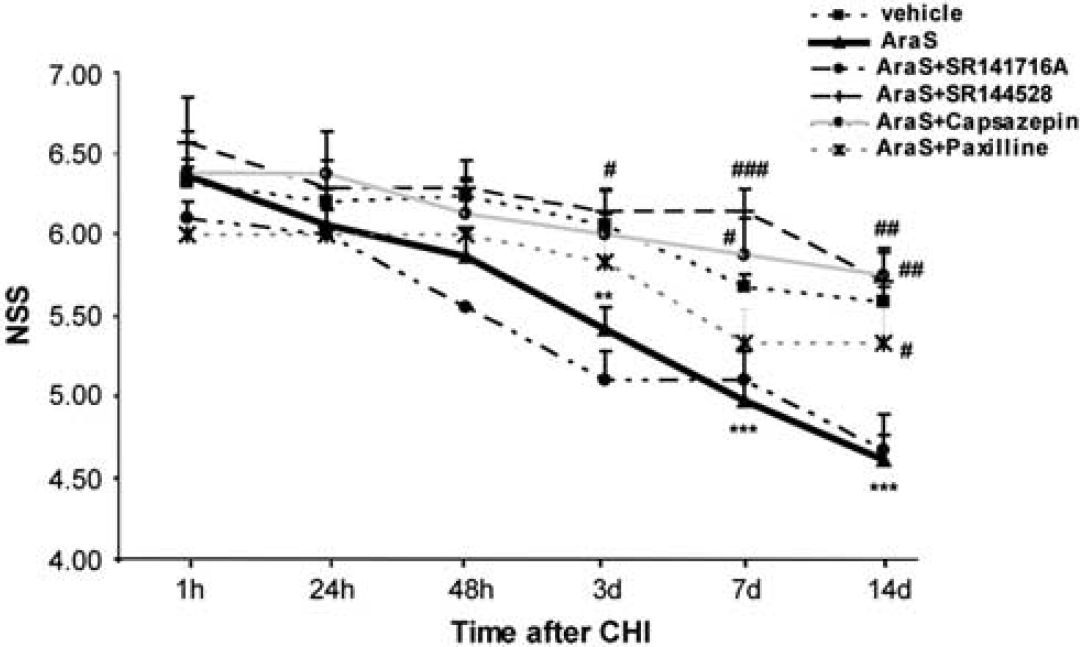
Lesion volume was evaluated with the aid of TTC staining, as described elsewhere (Povlishock and Katz, 2005). The mice were subjected to CHI followed by the different treatments (n = 5 to 16 mice/group). After 24 hours they were killed and their brains were removed and sliced at 2 mm, intervals, using a brain mold. The slices were placed in 2% TTC solution in saline for 30 minutes and preserved in 4% paraformaldehide in PBS. The brains were photographed 24 hours later with the aid of a Stemi SV11 Stereoscope (Zeiss, Goettingen, Germany) and a digital camera (Nikon, Coolpix 4500, Natori, Miyagi, Japan). ImageJ 1.40g software (National Institutes of Health, Bethesda, MD, USA) was used to quantify lesion volume. To avoid inaccuracies due to changes in the ipsilateral hemispheric volume, the lesion volume was calculated as the area of unstained tissue divided by the area of contralateral hemisphere (Swanson et al, 1990).
Cerebral Edema
The edema was determined at 24 hours after CHI, as previously described (Panikashvili et al, 2005). Cortices of both ipsilateral and contralateral hemispheres were removed and weighted immediately and then dried for 24 hours in a 95°C oven (n = 10 to 12 mice/group). The water content of the tissue was calculated as:
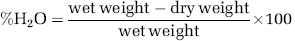
Rectal Temperature Measurements
As cannabinoids have a hypothermic effect, which may contribute to neuroprotection, we assessed the effect of AraS on rectal temperature of mice after trauma (n = 5 to 10 mice/group), which correlates well with brain temperature. The temperature was measured 2, 4, 8, and 24 hours following the insult, using rectal thermistors (Digisense, Thermistor 400 series, Euthech Instruments, Singapore) inserted 3 cm beyond the anal sphincter.
GTPγS Binding (Radio-Labeling) Assay
As AraS was shown to have a very low binding affinity for CB1 and CB2 receptors (Milman et al, 2006), we decided to examine its binding to other candidate receptors from the G-protein-coupled receptor (GPCR) family, using a radio-labeling assay as described elsewhere (Ryberg et al, 2007). A dilution series of AraS was prepared in DMSO in 96-well plates, at 2 μL/well. This gave a final DMSO concentration of 1%. The highest concentration of compound in the assay was 33 μmol/L. [35S]-Guanosine 5'-[γ−35S]-triphosphate binding assays were conducted at 30°C for 45 minutes in membrane buffer (100 mmol/L NaCl, 5, 1 mmol/L ethylenediaminetetraacetic acid, 50 mmol/L HEPES, pH 7.4) containing 0.025 μg/μL of membrane protein with 0.01% bovine serum albumin (fatty-acid free), 10 μmmol/L guanosine 5'-diphosphate, 100 μmmol/L dithiothreitol, and 0.53 nM [35S]-GTPγS in a final volume of 200 μL. Nonspecific binding was determined in the presence of 20 μmmol/L unlabeled GTPγS. The reaction was terminated by the addition of ice-cold wash buffer (50 mmol/L Tris–HCl, 5 mmol/L MgCl2, 50 mmol/L NaCl, pH 7.4), followed by rapid filtration under vacuum through GF/B glass-fiber filters using a cell harvester. The filters were left to dry for 30 minutes at 50°C. Then 15 μL scintillation liquid was added to each well before the bound radioligand was detected with the aid of a microbeta scintillation counter (Wallac 1450 Microbeta TRILUX, Turku, Finland). Antagonist potency was determined versus an EC80 concentration of CP55940 that was determined empirically on the day of the experiment. Data were fitted according to the equation y = A + ((B - A)/1 + ((C/x)^D))) and the EC50 was estimated where A = the nonspecific binding, B = the total binding, C = the EC50, and D = the slope. Antagonist potency was determined versus an EC80 concentration of GPR55 agonist O-1602 that was determined empirically on the day of the experiment. O-1602 (5-methyl-4-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-1,3-benzenediol) is a synthetic atypical cannabinoid analog of cannabidiol, which binds as an agonist to GPR55. It is similar in structure to abnormal cannabidiol but with a methyl group instead of pentyl group substitution of the benzene. The data were fitted as above.
Western Immunoblotting
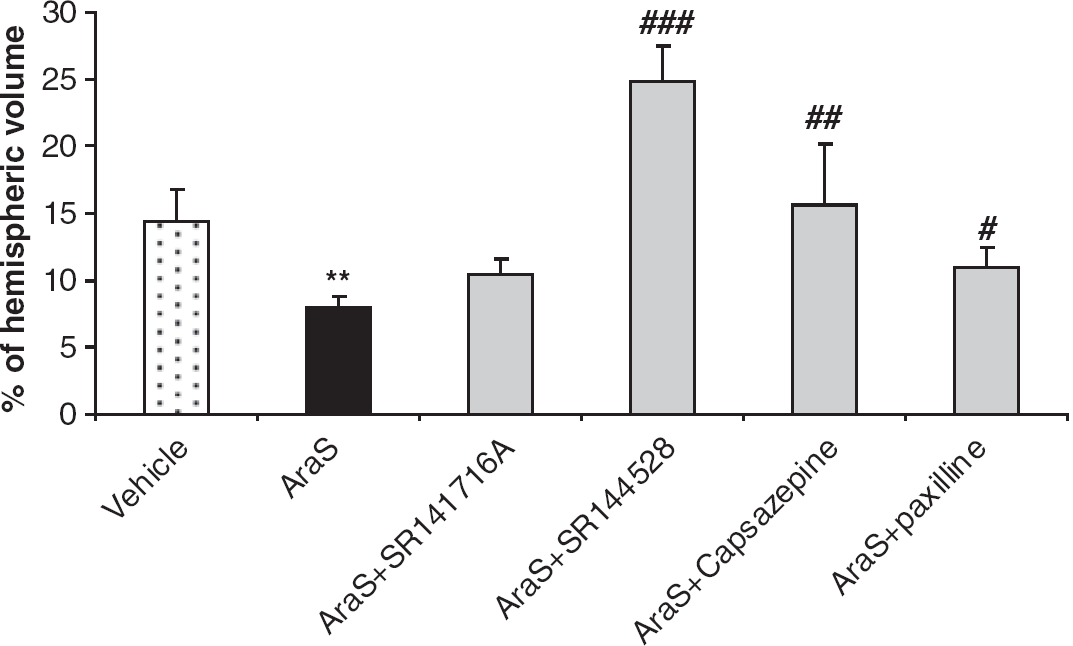
To determine the levels of phosphorylated extracellular-signal-regulated kinase 1/2 (pERK1/2), pAkt, and Bcl-xL, the mice were decapitated under isoflurane anesthesia 2, 4, and 24 hours after injury (n = 5 to 6 mice/group). The cortical tissue adjacent to the site of injury was removed and homogenized using a manual homogenizer, in ice-cold buffer (50 mmol/L Tris–HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid, 1 mmol/L Na3VO4, 50 mmol/L NaF, 10 mmol/L sodium β-glycerophosphate, 5 mmol/L sodium pyrophosphate, 1 mmol/L phenylmethanesulfonyl fluoride, supplemented with 0.1% (v/v) Triton X-100, 0.1% (v/v) 2-β-mercaptoethanol, and protease inhibitor cocktail). For comparison, tissue from the contralateral hemisphere was removed and homogenized separately in the same buffer. After 15 minutes incubation on ice, the extracts were centrifuged at 12,000g for 15 minutes at 4°C and the protein content in the supernatant was determined according to Bradford (1976) and the manufacturer's instructions. A total of 50 μg of the protein extract from each sample was mixed with SDS-sample buffer and boiled at 95°C for 5 minutes before loading. The proteins were separated by 10% SDS–PAGE and transferred onto trans-blot nitrocellulose membranes. The membranes were blocked in 5% nonfat dry milk in Tris buffered saline (TBS), pH 7.4, with 0.1% Tween 20 (TBS–Tween) for 1 h at room temperature. Primary antibodies (phospho-ERK1/2 1:1,000, phospho-AKT Ser473 1:1,000, Bcl-xL 1:1,000, β-actin 1:1,000 (Cell Signaling Technology, Danvers, MA, USA) were diluted in 5% bovine serum albumin solution, and the membranes were incubated overnight at 4°C. The primary antibody was removed, and the blots were washed in TBS–Tween and incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies (1:10,000; Jackson Immunoresearch, Cambridgeshire, UK). Reactive proteins were visualized using Enhanced Chemiluminescence Reagent (Biological Industries, Beit Haemek, Israel) by direct chemiluminescence using FUJIFILM Luminescent Image Analyzer camera LAS-3000 (Tokyo, Japan). The optical density was determined using ImageJ 1.40g software (National Institutes of Health).
Caspase-3 Activity Assay
Caspase-3 activity was measured in the cortical tissue of the injured mice using the commercially available BioVision kit (Mountain View, CA, USA) according to the manufacturer's instructions (n = 5 to 9 mice/group). In brief, the mice were subjected to CHI followed by treatment with AraS (3 mg/kg), AraS + SR141716A, AraS + SR144528, AraS + capsazepine, or vehicle 1 hour after injury. The mice were killed 72 hours later, and the ipsilateral cortices were removed, lysed in 200 μL of the supplied lysis buffer using a manual homogenizer, and frozen immediately at −80°C until assayed. Total protein was calculated and 150 μg of the samples were loaded in each well of a 96-well plate. In all, 50 μL of reaction buffer containing 10 mmol/L dithiothreitol and 5 μL of DEVD-AFC (Asp-Glu-Val-Asp7-amino-4-trifluoromethylcoumarin) substrate were added and the plates were incubated at 37°C for 2 hours. The cleavage product, representing active caspase-3, was detected using a fluorometer (FluoroStar 403, BMG LabTech, Offenburg, Germany) equipped with 390 nm excitation and 520 nm emission filters.
Statistical Analysis
Statistical analysis was performed using SigmaStat 2.03 software (SPSS, Inc., Chicago, IL, USA). The data are presented as the mean ± s.e.m. The statistical significance of differences between means was evaluated by two-way analysis of variance for repeated measures, followed by Tukey post hoc for NSS tests, and with parametric one-way analysis of variance for lesion volume and for fluorometric measurements. The statistical significance of the optical density measurements was calculated using two-way analysis of variance followed by Tukey correction. Probability values (P) < 0.05 were considered statistically significant.
Results
N-Arachidonoyl-l-Serine Improves Motor Function and Reduces Lesion Volume and Water Content After Closed Head Injury
Mice treated with AraS or vehicle had a similar NSS at 1 hour, indicating a similar severity of injury (6.36 ± 0.11 and 6.32 ± 0.09, respectively). The motor function of mice was evaluated for up to 14 days after CHI. Significantly lower NSS values were recorded in AraS-treated mice compared with those in vehicetreated mice, from day 3 up to 14 days of follow-up (Figure 1; P < 0.01 on day 3 and P < 0.001 on days 7 and 14), indicating better recovery rates in the AraS-treated mice. To examine the effect of AraS on lesion volume, brains were stained with the vital dye TTC, which stains live tissue purple and leaves dead tissue unstained. Lesion volumes constituted 8.00% ± 0.87% of the hemispheric volume in the AraS-treated mice, compared with 14.39% ± 2.35% in the vehicle-treated mice (Figure 2). The results indicate a significant reduction of 45% in lesion volume in the AraS-treated mice (P < 0.01). The water content in the brain was determined 24 hours after CHI in both ipsilateral and contralateral hemispheres. A significant reduction in water content was observed in the ipsilateral cortex of AraS-treated mice compared with vehicle (81.82% ± 0.16% and 82.77% ± 0.25%, respectively; P < 0.01, data not shown). No difference in water content was detected in the contralateral hemispheres of both groups.
N-arachidonoyl-
N-arachidonoyl-
N-Arachidonoyl-l-Serine Neuroprotective Effects Involve CB2 and Transient Receptor Potential Vanilloid 1 Receptors
We next sought to identify the receptors involved in mediating the neuroprotective effects of AraS. The CB1 receptors were considered likely candidates as the effects of AraS on motor disability were similar in magnitude to those previously observed for 2AG (Panikashvili et al, 2001). However, coadministration of AraS with the CB1-antagonist rimonabant (SR141716A) did not alter the recovery of the AraS-treated animals (Figure 1) nor reduce the lesion volume (Figure 2; 9.86% ± 1.03%), arguing against a CB1-mediated effect of AraS.
As 2AG also binds to CB2 and TRPV1 receptors, this raised the possibility that the protective effect of AraS may involve these binding sites. Coadministration of AraS with the CB2R-antagonist SR144528 significantly ameliorated the decrease in NSS values compared with those obtained for AraS-treated mice (Figure 1). The values were similar to those observed in vehicle-treated mice, 3, 7, and 14 days after injury (P < 0.05, P < 0.001, and P < 0.01, respectively, versus AraS). Moreover, a threefold increase in lesion volume was observed in mice treated with a combination of AraS and SR144528 (Figure 2; 24.33% ± 2.85%; P < 0.001) compared with those treated with AraS alone (8.00% ± 0.87%). To examine whether AraS acts via ‘nonclassical’ CB receptors, mice were treated with capsazepine, a TRPV1 receptor antagonist. They exhibited significantly higher NSS values than those obtained for the AraS-treated group 7 and 14 days after CHI (Figure 1; P < 0.05 and P < 0.01, respectively) and also showed a twofold increase in lesion volume compared with the volume obtained in AraS-treated mice (Figure 2; 15.61% ± 4.51% versus 8.00% ± 0.87%, respectively; P < 0.01), suggesting that the TRPV1 channels have a role in the AraS neuroprotective effects.
N-arachidonoyl-
Recently, a new G-protein-coupled cannabinoid receptor, GPR55, was identified but it has not been fully characterized (Ryberg et al, 2007). GTPγS binding assays showed no significant agonistic binding of AraS to GPR55 (Table 1A) and no significant antagonistic activity was observed against the GPR55 receptor (Table 1B).
AraS does not bind to GPR55 receptors as an agonist (A) or as an antagonist (B)

AraS, N-arachidonoyl-
Cumulatively, these results suggest that AraS mediates its protective effects in vivo by indirect signaling via CB2R, TRPV1 and partially via BK channels, but not through the CB1R or GPR55 receptors.
N-Arachidonoyl-l-Serine Prevents Hypothermia After Closed Head Injury
Hypothermia, a known neuroprotective mechanism, was found to mediate CB1-induced neuroprotection of the potent synthetic cannabinoid HU-210 (Leker et al, 2003). To evaluate whether AraS induces hypothermia and whether such an effect has a role in AraS-induced neuroprotection, the rectal temperature of mice treated with AraS or vehicle was measured at different time points after injury. Although not a direct measure of brain temperature, rectal and brain temperature are highly correlated with each other. No significant decrease in body temperature was observed in AraS-treated mice 2 hours after injury compared with preinjury temperature (36.77°C ± 0.21°C and 37.64°C ± 0.46°C, respectively), whereas a reduction in body temperature was detected in vehicle-treated mice at the same time point (35.19°C ± 0.37°C and 37.31°C ± 0.09°C, respectively). At all other time points, no significant changes were observed between the different treatment groups (data not shown), arguing against the involvement of a hypothermic mechanism of neuroprotection for AraS.
N-Arachidonoyl-l-Serine Prevents Attenuation in Phosphorylation of Extracellular-Signal-Regulated Kinase 1/2 Levels and Increases pAkt Levels of After Closed Head Injury
To further delineate the downstream signaling involved in the neuroprotective effects of AraS, we studied its effect on ERK, a key antiapoptotic prosurvival mediator. Mice were subjected to trauma, treated with AraS or vehicle 1 hour later and decapitated 2, 4, or 24 hours after injury. The levels of pERK were evaluated in the ipsilateral cortices. Although a significant decline in the levels of pERK was observed at 2 and 4hours after injury in the vehicle-treated mice (P < 0.01 versus sham), no decline was observed in the AraS-treated mice at 2 hours after injury (P < 0.01 versus vehicle). Interestingly, a dramatic decrease in pERK levels was noted 4 hours after CHI (P < 0.001 versus sham) in the AraS-treated mice. In both groups, the pERK levels increased, reverting to normal 24 hours after injury (Figure 3).
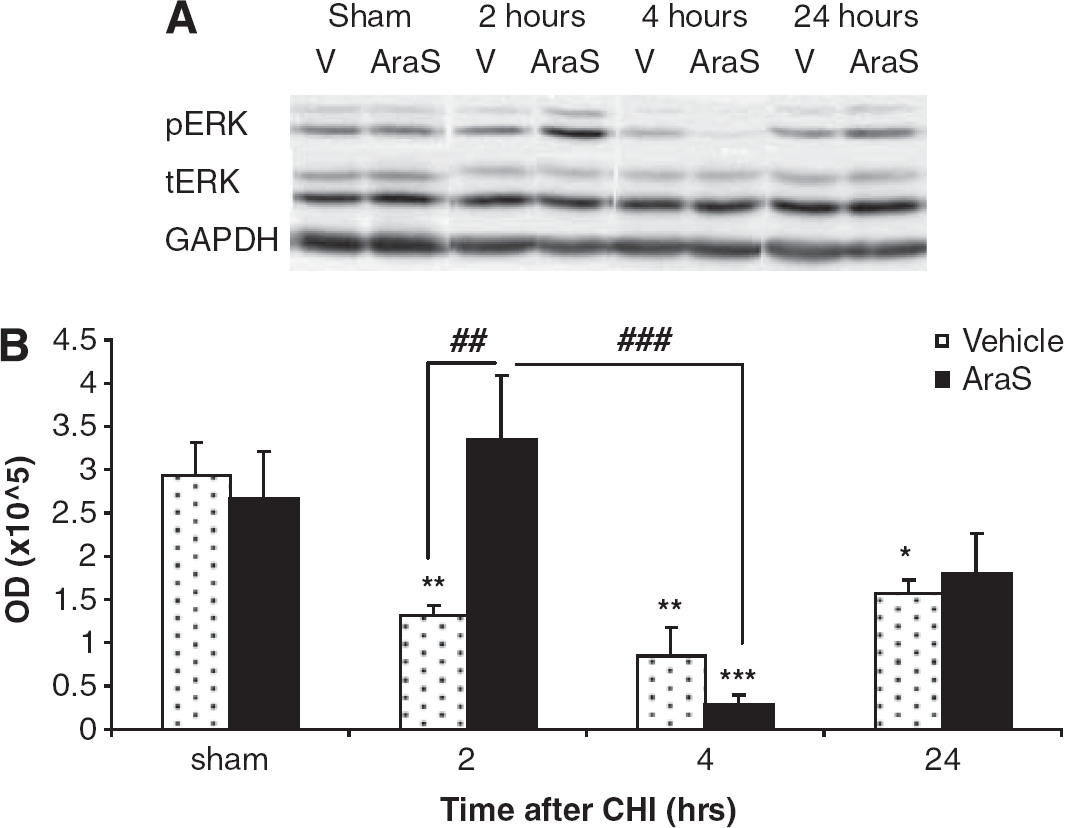
N-arachidonoyl-
The effect of AraS on Akt phosphorylation was next measured by Western blot analysis in the cortex of both hemispheres at 4 and 24 hours after CHI. A significant increase in pAkt levels was observed 4 hours after CHI in the ipsilateral (59.7% ± 19.6%) as well as in the contralateral (54.96% ± 10.72%) cortex of AraS-treated mice, compared with those in the vehicle-treated and sham-operated mice (Figure 4). A drop to basal levels was observed in these regions 24 hours after injury (Figure 4).
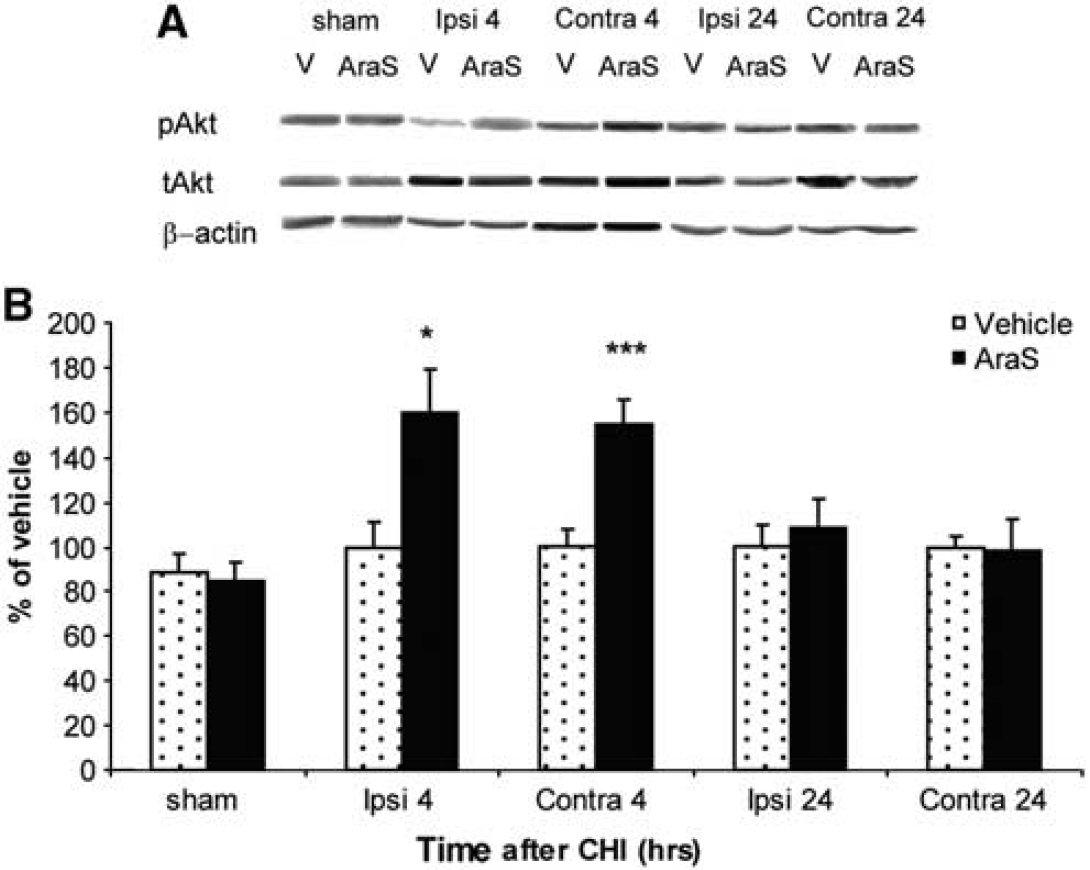
N-arachidonoyl-
N-Arachidonoyl-l-Serine Increases Bcl-xL Levels and Reduces Caspase-3 Activity in a CB2 Involved Mechanism
As AraS reduced lesion volume and since pERK and pAkt are involved in the regulation of apoptosis, we examined the levels of Bcl-xL, an antiapoptotic member of the bcl-2 family, 24 hours after injury. A significant increase of 62.6% ± 12.8% in Bcl-xL level was found in the ipsilateral cortex of mice treated with AraS compared with that in the vehicle (Figure 5). The effect of AraS on the proapoptotic protease caspase-3 activity was then evaluated in the ipsilateral cortex 3 days after CHI. Caspase-3 activity, expressed as fluorescence units, was almost 300% higher in the ipsilateral cortex of vehicle-treated mice compared with that in sham mice. N-arachidonoyl-
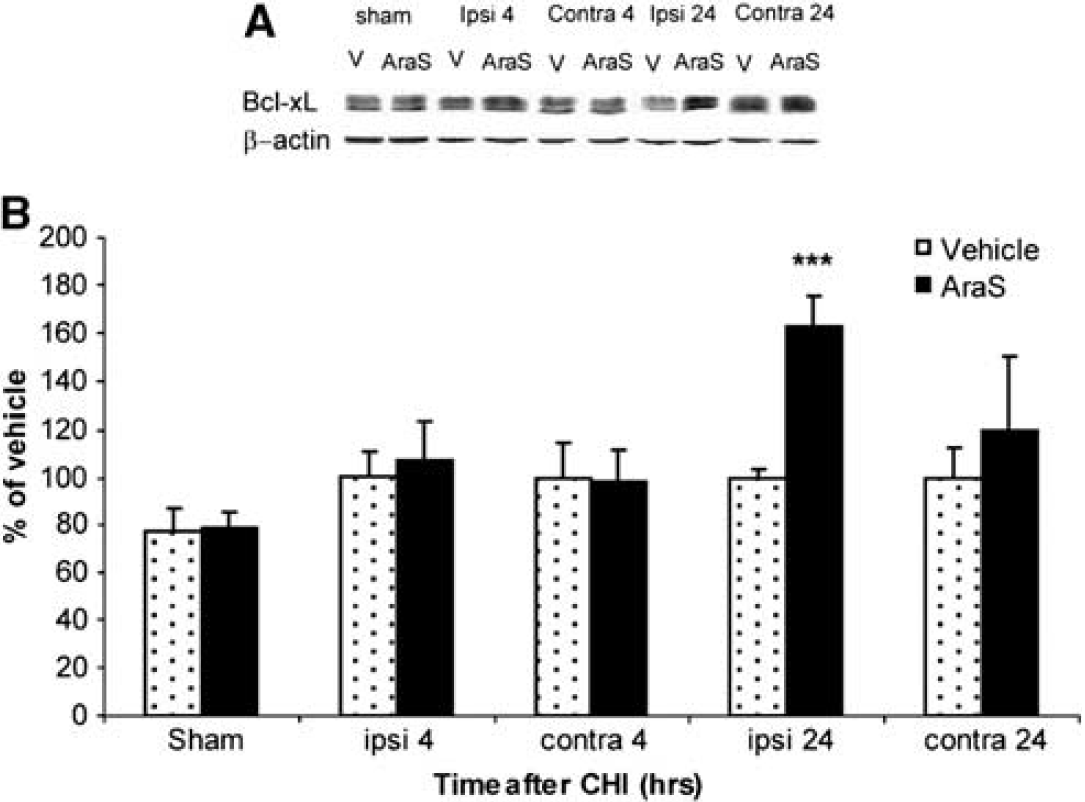
N-arachidonoyl-
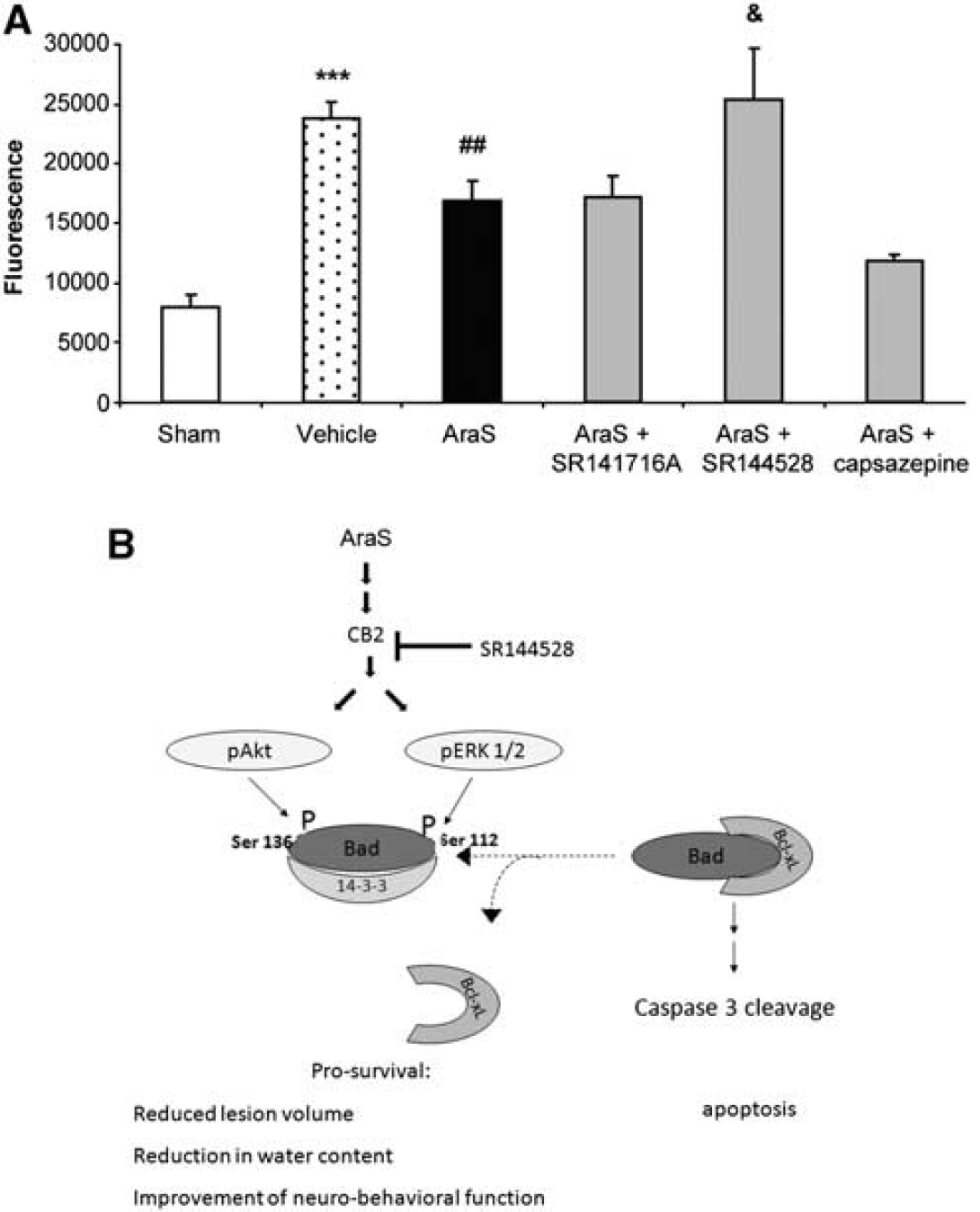
N-arachidonoyl-
Cumulatively, these findings suggest an antiapoptotic mechanism of action for the neuroprotective effects of AraS involving CB2, but not CB1 receptors as is shown in the schematic Figure 6B.
Discussion
In the present study, we show for the first time that the recently described eCB-like compound AraS mediates neuroprotection in vivo after TBI via the induction of a prosurvival and antiapoptotic cascade. This mechanism involves Akt and ERK phosphorylation and its downstream signaling includes increased levels of the antiapoptotic protein Bcl-xL and the reduced activity of the proapoptotic enzyme caspase-3. N-arachidonoyl-
Cannabinoids have been shown to provide neuroprotection via hypothermia, in addition to other mechanisms (Leker et al, 2003). Therefore, we assessed the hypothermic effect of AraS following brain injury. No decrease in body temperature was observed in AraS-treated mice, indicating that the neuroprotective effects of AraS are not mediated via the induction of hypothermia.
Many deleterious and protective processes are induced following TBI, including massive release of glutamate from presynaptic terminals, production of reactive oxygen species, accumulation of inflammatory cytokines in the surrounding tissue, axonal injury, and neuronal cell loss in specific brain regions (Leker and Shohami, 2002; Povlishock and Katz, 2005). In parallel, neuroprotective events are also set in motion and include secretion of antiinflammatory cytokines, growth factors, antioxidative compounds, and enzymes (Leker and Shohami, 2002). The eCB system was proposed to be part of the protective processes (Panikashvili et al, 2001, 2005). The fact that it is activated ‘on-demand’ (Piomelli, 2003) lends support to this supposition.
The neuroprotective effects of 2AG after brain injury were shown to be mediated through CB1 signaling (Panikashvili et al, 2001, 2005). However, since 2AG activates both the CB1 and CB2 receptors, some of the neuroprotective effects may be mediated by signaling via CB2 (Zhang et al, 2007, 2008). These effects may explain our findings on the reduction of cerebral edema in the AraS-treated mice. In addition, CB1 receptor agonists were shown to activate Akt (Gomez del Pulgar et al, 2000) and ERK (Moranta et al 2007), which participate in prosurvival processes, including those related to neuroprotection (Pignataro et al, 2008; Yune et al, 2008). Our present results, in agreement with those of Milman et al (2006), suggest that in the brain, as in endothelial cells, the novel eCB AraS also induces ERK and Akt signaling via mechanisms that probably depend on CB2 and/or TRPV1 and/or slightly on BK channels, but not on CB1. Importantly, Akt kinase participates in many prosurvival and antiapoptotic processes inside and outside the brain (Dasari et al, 2008), including processes that are cannabinoid related (Molina-Holgado et al, 2002; Ozaita et al, 2007), thus corroborating the proposed neuroprotective mechanism of AraS. Here, we show that administration of exogenous AraS also leads to an increase in the antiapoptotic factor Bcl-xL at 24hours and to the decreased activity of the proapoptotic protease caspase-3, 72 hours after injury. BAD is a proapoptotic member of the Bcl-2 family; Bcl-xL intervenes in the apoptotic cascade and attenuates it. When unphosphorylated BAD forms a complex with Bcl-xL, it prevents the antiapoptotic effect of the latter, resulting in apoptosis (Datta et al, 1997; Yang et al, 1995). The phosphorylation of BAD by Akt (Datta et al, 1997) and/or by ERK (Chen et al, 2005), prevents this interaction and allows Bcl-xL to exert its antiapoptotic effect and eventually to inhibit programmed cell death. The attenuation in caspase-3 activity demonstrated by AraS was reversed by CB2R blocker pointing to this receptor as the main conduit of the antiapoptotic effect of AraS.
Importantly, the prosurvival antiapoptotic cellular mechanisms induced in AraS-treated mice led to a 45% reduction in lesion volume, and to a decrease in the neurologic disability, as assessed by the NSS test. A correlation between caspase-3 activity, lesion volume, and neurobehavioral deficits was observed in mice after treatment with the different antagonists. Thus, higher caspase-3 activity found following treatment with AraS in combination with the CB2 blocker led to larger lesions and to a more severe functional outcome. In contrast, in mice treated with AraS in combination with CB1 antagonist, caspase-3 activity, lesion volume, and neurologic outcome were similar to those observed in AraS-treated mice. This indicates that the CB1 receptor is not involved in the neuroportective mechanism. Surprisingly, although lesion volume in mice treated with AraS along with capsazepine was higher and the functional outcome was worse compared with that of the AraS group, caspase-3 activity remained unchanged. These results may imply that the mechanism of AraS-induced neuroprotection mediated by TRPV1 channels does not necessarily involve an antiapoptotic effect or involves an antiapoptotic effect that is not mediated via modulation of caspase-3 activity. As the changes in neurobehavioral function of paxilline-treated mice compared with those of AraS-treated mice were mostly insignificant, lesion volume was hardly affected by the blocker and it appears that the neuroprotective mechanism of AraS does not involve BK channels.
Although AraS does not directly bind to CB2 or to TRPV1, it may exert its neuroprotective effects in two optional mechanisms: (1) It may have an entourage effect. N-arachidonoyl-
Since AraS reduces water content and lesion volume 24 hours after CHI, we postulate that it affects straight on pERK and pAkt directly and rapidly that eventually leads to a reduction in the pathophysiological parameters. Therefore, we believe the effect of AraS on apoptosis is the mechanism by which it provides neuroprotection and that the reduced levels of caspase-3 are not merely the result of smaller lesions.
In conclusion, we show here for the first time that the novel eCB AraS exerts neuroprotective effects in vivo in a CHI model of TBI. These effects are mediated at least in part by the induction of antiapoptotic signaling involving pERK, pAkt, and Bcl-xL, leading to reduced caspase-3 activity. In contrast to the effects seen upon the administration of 2AG, these neuroprotective effects are probably related indirectly to CB2 receptors and TRPV1 and partially to BK channels, but not to signaling via CB1 or GPR55 receptors.
Footnotes
The authors declare no conflict of interest.