
Abstract
Superoxide-dismutases (SOD) catalyze O2− conversion to hydrogen peroxide (H2O2) and with other antioxidant enzymes and low molecular weight antioxidants (LMWA) constitute endogenous defense mechanisms. We first assessed the effects of SOD1 levels on outcome after closed head injury (CHI) and later, based on these results, the effects of SOD−/- deficiency on cellular redox homeostasis. Superoxide-dismutase 1-deficient (SOD1−/-) and -overexpressing (transgenic (Tg)) mice and matched wild-type (WT) controls were subjected to CHI and outcome (neurobehavioral and memory functions) was assessed during 14 days. Brain edema, LMWA, and SOD2 activity were measured along with histopathological analysis. Transactivation of nuclear factor-kappa B (NF-κB) was evaluated by electromobility shift assay. Mortality, motor, and cognitive outcome of Tg and WT mice were comparable. Mortality and edema were similar in SOD1−/- and WT mice, yet, unexpectedly, SOD1−/- displayed better neurobehavioral recovery (P < 0.05) at 14 days after CHI. Basal LMWA were higher in the cortex and liver of SOD1−/- mice (P < 0.05) and similar to WT in the cerebellum. Five minutes after CHI, cortical LMWA decreased only in SOD1−/- mice. One week after CHI, SOD2 activity decreased fourfold in WT cortex (P < 0.001), but was preserved in the SOD1−/- . Constitutive NF-κB transactivation was comparably low in SOD1−/- and WT; however, CHI induced a robust NF-κB activation that was absent in SOD1−/- cortices (P > 0.005 versus WT). At the same time, immunohistochemical analysis of brain sections revealed that astrogliosis and neurodegeneration were of lesser severity in SOD1−/- mice. We suggest that SOD1 deficiency impairs H2O2-mediated activation of NF-κB, decreasing death-promoting signals, and leading to better outcome.
Introduction
Reactive oxygen species (ROS) are widely implicated in the pathogenesis of secondary neuronal damage and apoptotic/necrotic cell death after traumatic or ischemic brain injury (Lewen and Hillered, 1998; Chan, 2001; Fiskum, 2000). Oxidative modifications of cellular macromolecules are often used as indirect markers of tissue oxidative stress that accounts, at least in part, for functional derangement of these essential molecules (Lewen and Hillered, 1998; McDonald et al, 1999; Nishio et al, 1997; Vagnozzi et al, 1999). In addition, ROS apparently regulate signal transduction pathways such as nuclear factor-kappa B (NF-/κB) and activator protein-1 (AP-1) (Vollgraf et al, 1999; Schmidt et al, 1995), which in turn govern both harmful and defensive cellular responses to injurious events (Martindale and Holbrook, 2002; Muller et al, 1997). Nonaka et al (1999) found a persistent activation of NF-κB in the cortex and endothelial cells for at least 1 year after injury and suggested that NF-κB activation plays a role in long-term inflammatory processes after brain trauma. In support of this notion, we recently showed that sustained activation of NF-B κ after closed head injury (CHI) is inhibited by melatonin, the pineal hormone known also for its antioxidant properties, and by the endocannabinoid 2-arachidonoyl glycerol. In both cases, NF-κB inhibition was associated with enhanced functional recovery (Beni et al, 2004; Panikashvili et al, 2005).
In the inactive state, hetero- or homodimerized forms of NF-κB are bound to the inhibitory protein Iκ Bα and sequestered in the cytoplasm. The activation of Iκ kinase complex (IKK) by various cytokines or oxidants (Dopp et al, 2002; Kaltschmidt et al, 1999; Schmidt et al, 1995) results in the phosphorylation of IκB at specific serine residues, thereby promoting its ubiquitination and degradation by the proteosome (Karin and Ben-Neriah, 2000). This in turn allows NF-κB subunits to translocate into the nucleus, activate transcription of cytokines (May and Ghosh, 1998), adhesion molecules (Siebenlist et al, 1994), and redox-regulating enzymes such as inducible nitric oxide synthase 2 (Siebenlist et al, 1994; Xie et al, 1994) and Mn-superoxide dismutase (Maehara et al, 2000).
To combat oxidative stress, aerobic cells are endowed with antioxidant defense systems composed of low molecular weight antioxidants (LMWA) and antioxidant enzymes. Antioxidant capacity differs among tissues, and the central nervous system is particularly vulnerable to oxidative damage (Halliwell and Gutteridge, 1989). The stepwise activity of the enzymatic antioxidant network consists of dismutation of superoxide anions (O2−) by superoxide-dismutases (SOD) to form hydrogen peroxide (H2O2) (Fridovich, 1975), which is then converted to water by mitochondrial and cytosolic glutathione-peroxidase (GSHPx) or paroxisomal catalase. Three SOD isoforms were identified on the basis of their subcellular localization and metal ion requirements: SOD1, a dimeric, cytosolic enzyme, requiring copper and zinc ions as cofactors; SOD2, a tetrameric, mitochondrial enzyme requiring manganese, and SOD3, an extracellular, high molecular weight copper-containing enzyme. Superoxide-dismutase 1 is constitutively present in all cells, while SOD2 is inducible on oxidative or inflammatory stimuli, suggesting distinct roles for the two isoforms (Huang et al, 1999).
The particular role of SOD1 in neuroprotection has been widely investigated. Overexpression of SOD1 was shown to be associated with a dose-related decrease in brain edema, tissue infarction, and blood-brain barrier disruption in models of cold- and contusion brain injury (Chan et al, 1991; Lewen et al, 2001; Mikawa et al, 1995, 1996) and after transient cortical ischemia (Chan et al, 1998; Kinoushi et al, 1991; Murakami et al, 1997; Yang et al, 1994). In contrast, SOD1 deficiency did not affect brain edema, infract volume, or early release of mitochondrial cytochrome c after permanent focal cerebral ischemia (Fujimura et al, 2001). Furthermore, increased SOD1 activity caused pronounced effects on other antioxidant enzymes (Keiner et al, 1995), aggravated oxidative injury (Elroy-Stein and Groner, 1988; Peled-Kamar et al, 1997), and impaired cognitive function in SOD1 transgenic (Tg) mice (Gahtan et al, 1998).
In light of these conflicting data, the present study was performed to assess the effect of SOD1 levels (SOD Tg or SOD1−/-) on neurobehavioral outcome after CHI in mice. Based on the neurobehavioral results, we next hypothesized that deficiency in SOD1 may elicit adaptive mechanisms as an attempt to maintain redox homeostasis at the chronic, pre-injury state or on CHI. To test this hypothesis, levels of LMWA, activity of SOD2, and the pattern of NF-κB transactivation were studied in SOD1-deficient mice before and after CHI, and immuno-staining was performed to investigate the astrogliosis and neuronal cell death after CHI in these mice.
Materials and methods
Animals
Male mice were used in the study. They were maintained and treated according to the regulations of the Animal Care Committee of the Hebrew University, and food and water were provided ad libitum.
Transgenic mice, carrying human SOD1 gene on a CBYB/6 x B6D/2 background, were derived from a founder stock (Epstein et al, 1987). Transgene was confirmed by PCR, followed by immunoblotting and quantitative demonstration of SOD1, using nondenaturating gel electrophoresis and nitroblue tetrazoline staining. Superoxide-dismutase 1 mutant mice (B6/SVl29-Sod1tm1Leb strain, stock number 002972) were purchased from Jackson Laboratories. While male SOD1−/- mice are fertile and have no gross or histologic testicular defects, SOD1−/- female mice exhibit defects in ovarian folliculogenesis, resulting in subfertility and infertility (Matzuk et al, 1998). Therefore, SOD1−/- mice were generated by mating SOD1−/- males with SOD1−/+ females. Genotyping by PCR was performed according to the instructions of Jackson Laboratories, and the absence of SOD1 protein in the brain was confirmed by immunoblotting and activity assays. It should be noted that phenotypically, SOD11/+ mice were indistinguishable from the wild type (WT). Since mice deficient of SOD1 and mice overexpressing SOD1 in our study consist of different backgrounds, each mutant population was compared in each experiment only with its respective background-matched WT controls.
Trauma Model
Closed head injury was induced under ether anesthesia, confirmed by loss of pupillary and corneal reflexes, using a weight-drop device (Chen et al, 1996; Beni-Adani et al, 2001). After a sagittal scalp incision, mice were fixed under the device and a tipped Teflon cone was placed 3 mm lateral to the midline and 1 mm above the left coronal suture. Closed head injury was caused by dropping a 94 g metal rod from a height of 13 cm over the cone. Sham mice were anesthetized, their scalps were incised, but trauma was not induced. Mice were returned to their home cages and maintained under a 12 h light/ dark reversed light cycle to the end of the experiment. Normal body temperature was kept throughout the whole period of anesthesia.
Neurobehavioral Evaluation
Neurologic recovery: Mice were evaluated by a masked examiner, using a set of 10 tasks, collectively termed Neurologic Severity Score (NSS, Table 1), which test reflexes, alertness, coordination, and motor abilities. One point is awarded for failure to perform a particular task; thus, a score of 10 reflects maximal neurobehavioral impairment, whereas a normal mouse scores 0 (Beni-Adani et al, 2001). Preclosed head injury NSS was assessed to detect possible basal functional differences between SOD1-mutant mice and their WT controls. Post-CHI NSS was evaluated at 1 h (NSS1 h) to define severity of injury, and then every other day during 14 days. Mice with preinjury scores of > 2 or with NSS1 h ≤ 4 were excluded from the study.
NSS for CHI mice

NSS = Neurological Severity Score; CHI = closed head injury.
Recognition memory: Object recognition test (ORT) (Ennaceur and Delacour, 1988) which tests memory function (Ennaceur et al, 2005) was performed by a masked evaluator before- and on days 3 and 12 after CHI. Two days before the test, mice were habituated (individually) for 1 h in an empty 40 × 30 cm aquariumlike cage. On the following day, mice were returned to the test cage in which two identical objects were placed, and the time spent exploring each object was recorded for 5 mins, using a computerized program. Twenty-four hours later, one object was replaced with a new one, and the time spent by the mouse exploring the novel object was recorded for 5 mins (test) and compared with the time it had previously spent (pretest) exploring a familiar object placed at the same spot.
Based on the results of the neurobehavioral trials, only SOD1−/- and their respective WT littermates were used for the rest of the study.
Evaluation of Brain Edema
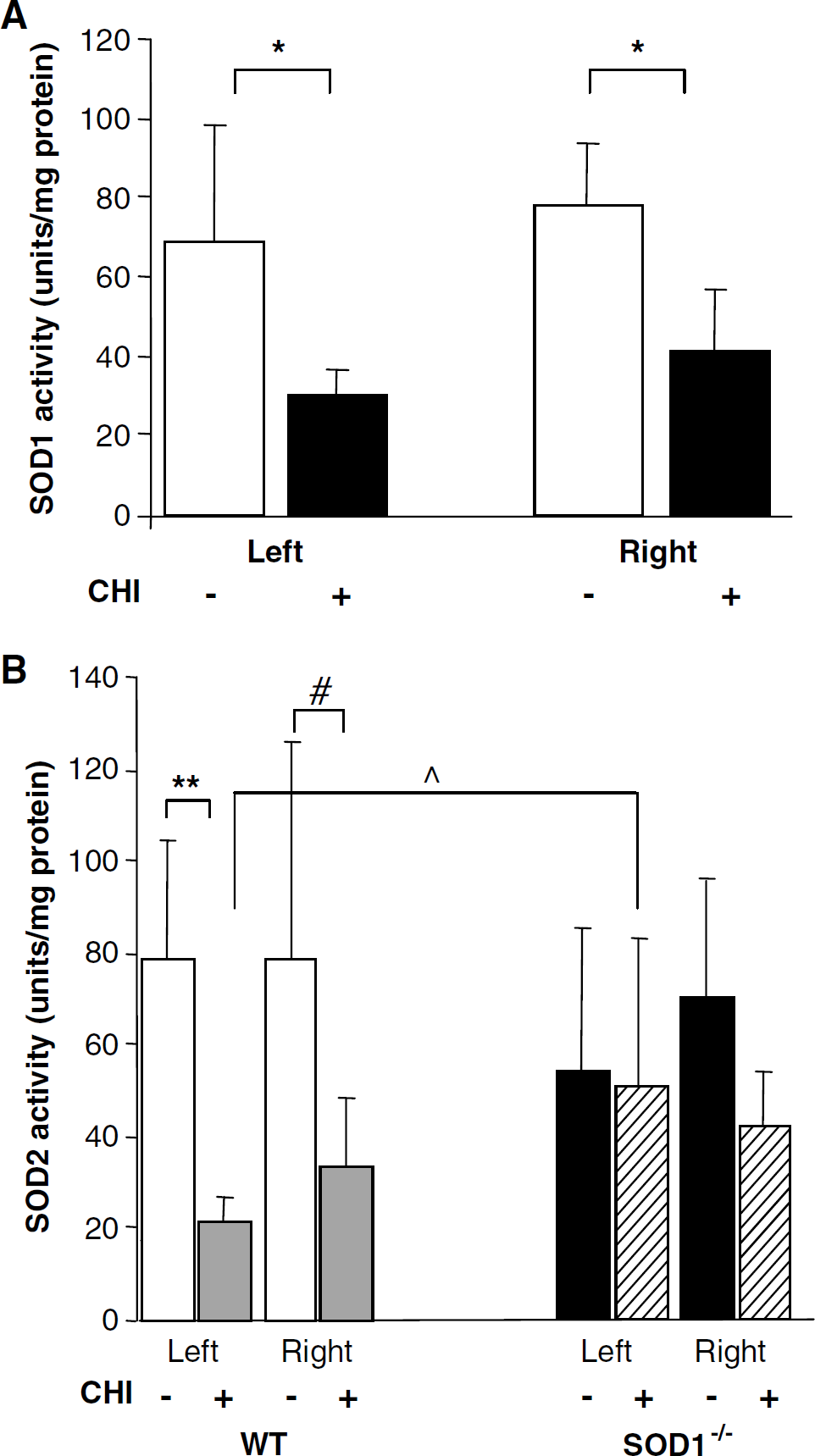
Cerebral edema was evaluated 24 h after CHI [n = 6/group). Cortical segments (30 to 50 mg) from the area bordering the lesion or the contralateral (right) hemisphere were weighted to yield wet weight (WW). Tissues were dried for 24 h in a desiccated oven (100°C), and reweighted to yield dry weight (DW). Water content was calculated as
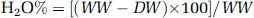
Measurement of Low Molecular Weight Antioxidant
Tissue levels of LMWA were measured before (n = 5 to 6/ group) and 5 mins after CHI (n = 7/group), a time that showed the greatest changes (Beit-Yannai et al, 1997). Cortical, cerebellar, and the liver slices (~120 mg) were each homogenized in PBS (10:1 (w:v); pH 7.4) and centrifuged for 10 mins at 1000g (4°C). Supernatants were analyzed using cyclic voltammeter apparatus (BAS 50 W; West Lafayette, IN, USA), as described previously (Beit-Yannai et al, 1997; Kohen et al, 1999). An electrical linear potential gradient (0 to 1300 mV relative to a reference electrode) was applied at a rate of 100 mV/sec across an electrode-solution interface (working electrode) to oxidize LMWA present in the sample. Computerized analysis (BAS Windows Control Software; Version 2.3 EF-1661) obtained voltammetric waves in a peak-shaped mode, and the potential [Ep), at which the peak current occurs on the voltage axis (x-axis) was determined. The intensity of the anodic current (Ia) was calculated from they-axis, and normalized to protein concentration, as determined by Bradford (Bio-Rad Labs GmbH, Munich, Germany). While Ep correlates with the reducing potency of LMWA, their concentration at a particular Ep is proportional to Ia(Kohen et al, 1999).
Superoxide-Dismutase 2 Activity Assay
Activities of total SOD and SOD2 were measured in sham mice [n = 3/group) and 7 days after CHI [n = 5 to 6/group), as described previously (Asada et al, 1974). Briefly, cortices and cerebellum were dissected on ice, frozen in liquid nitrogen, and stored at —80°C until assayed. Tissue was thawed on ice and homogenized in 800 μl of 0.05 M phosphate buffer (pH 7.2) containing 0.1% Triton X-100 and a cocktail of protease inhibitors (1:100; Roche, Mannheim, Germany). After centrifugation at 50,000g for 1 h, supernatants were collected and used for activity and immunoblotting assays. To measure total SOD activity, xanthine/xanthine-oxidase was used as a source of superoxide, and inhibition of cytochrome creduction was followed spectrophotometrically. The assay mixture contained 50 mmol/L potassium phosphate (pH 7.8), 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.1 mmol/L xanthine, 10/ μmol/L acetylated cytochrome c, and sufficient amount of xanthine-oxidase to reach ΔA550 = 0.05 optical densities (OD)/min. One unit of SOD was defined as the amount that causes 50% inhibition of the initial rate of cytochrome c reduction. Superoxide-dismutase 2 activity was determined by performing the same reaction in the presence of 5 mM potassium cyanide (KCN), a potent inhibitor of SOD1. Superoxide-dismutase 1 activity was calculated by subtracting SOD2 activity from that of total SOD.
Superoxide-Dismutase 1 Immunoblotting
Twenty micrograms of protein were applied to sodium dodecyl sulfate (SDS) 12.5% polyacrilamide gels, electrophoresed and transferred onto immobilon polyvinyl difluoride membrane (Millipore, Bedford, MA, USA). Membranes were incubated overnight with 1:1000 anti-SOD1 antibody (Stressgene Biotechnology, Victoria, BC, Canada) followed by 1 h incubation with 1:7500 goat anti-rabbit secondary antibody and analyzed according to Amersham enhanced chemiluminescence system (ECL). Relative ODs were obtained using Bio-Rad Multi-Analyst/ PC Version 1.1.
Nuclear Transactivation of Nuclear Factor-KappaB
Transactivation of NF- κ B was assessed by electrophoretic mobility shift assay (EMSA) before and 14 days after CHI <(n = 4/group/time point). Ipsilateral cortices were dissected on ice and nuclear extracts were prepared as described previously (Beni et al, 2004). Double-stranded NF-κ B oligonucleotide (5'-AGT TGA GGG GAC TTT CCC AGG C-3'; 3'-TCA ACT CCC CTG AAA GGG TCC G-5'; Promega, E3291) containing 5'-OH blunt ends was labeled with 32P (Perkin-Elmer Life Sciences, MA, USA) using T4 polynucleotide kinase (Promega M4101, WI, USA) according to the supplier's instructions. Ten micrograms of 10 nuclear protein were incubated for 1 h (4°C) with 32P-labeled NF-κB oligomer (20,000 c.p.m.) in a binding reaction mixture (10 mmol/L HEPES at pH 7.9, 60 mmol/ L KCl, 10% glycerol, 2μg bovine serum albumin, 2 μg poly(dI-dC), 0.4 mmol/L DDT). Specificity of protein-DNA complexes and super-shift assay were performed by preincubation (30 mins) of nuclear extracts with 100 × unlabeled NF-κB oligomer, or with 1 μL anti-p65 monoclonal antibody (Santa Cruz Bioctechnology, CA, USA), respectively, before adding 32P-labeled probe. Complexes were resolved at 100 V, on a 5% Polyacrylamide gel made up in tris-glycine EDTA (TGE) (50 mmol/L Tris, 400 mmol/L glycine, 2 mmol/L EDTA). Gels were vacuum-dried and exposed to Kodak X-ray films at —80°C. Optical densities were obtained using Bio-Rad Multi-Analyst/PC Version 1.1.
Histopathology
At 15 days after CHI, mice were anesthetized with a lethal dose of anesthetic and perfused via the ascending aorta with phosphate-buffered saline (PBS) (pH 7.4) followed by cold 4% paraformaldehyde in PBS (pH 7.4). Brain tissue was postfixed by immersion in the same fixative for 24 h at 4°C and embedded in paraffin, thereafter. Brain sections (6 μ thick) were serially taken at 200/μm intervals throughout the neuraxis. Coronal sections were taken anterior and posterior to bregma at various levels like frontal lobe, thalamus and dorsal hippocampus. The frontal and dorsal borders of the trauma were determined by the presence of reactive gliosis and sections within this limited area were further evaluated.
Reactive gliosis was determined by an increase in glial fibrillary acidic protein (GFAP) immunoreactivity after a standard protocol. Briefly, after antigen retrieval with proteinase-K, for 2.5 mins at 37°C, sections were incubated overnight with a 1:1000 dilution of anti-GFAP polyclonal antibody (DAKO), at 4°C. Secondary antibody treatment was performed by incubation with biotinylated anti-rabbit IgG (Novocastra) and finally positive reaction was developed with diaminobenzi-dine (DAB).
The number of damaged neurons in the hippocampus, cortex, and thalamus in both hemispheres were blindly rated by two independent observers under light microscope. Degenerative argyrophylic neurons were recognized according to morphologic criteria. Hippocampal cell counting was performed in both hemispheres, in 12 to 15 randomly chosen sections at the level of dorsal hippocampus. In an attempt to estimate the neuronal loss of the injured side, the percentage of the number of neuronal cells counted in this area was evaluated against the total number of cells, no matter whether they were normal or degenerative.
Cortical and thalamic cell damage as well as GFAP-positive astrogliosis was determined in five standardized microscopic fields, each of 22,500/μm2, defined by an ocular morphometric grid. Seven to eight randomly selected sections were used for measurements. Cortical cell damage per visual field was evaluated by using a grading scale as follows: 0, no degenerative cells; 1, 1 to 50 argyrophylic cells; 2, 51 to 200 argyrophylic cells; 3, 201 to 400 argyrophylic cells; and 4, > 400 argyrophylic cells. Astrogliosis per visual field was also evaluated according to a rating score: 0, no GFAP-positive cells; 1, few astrocytes; 2, mild-to-moderate astrogliosis; and 3, severe astrogliosis.
One series of sections was stained with hematoxylin-eosin for morphologic examination. The following adjacent sections were stained with a modified Bielschowsky's silver impregnation staining for the evaluation of axonal pathology (Litchfield and Nagy, 2001) and Gallyas staining for the evaluation of neurodegeneration. Evaluation of axonal damage was based on the density of axons and the presence of axonal bulbs, spheroids, ovoids, and dilated or dystrophic axons. Axonal pathology was assessed under light microscope in approximately 30 high-power optical fields per section throughout the corpus calosum and the trauma area in both hemispheres according to a rating score as follows: 0, normal morphology and density; 1, scattered patches of axonal damage with mild-to-moderate axonal loss; and 2, extensive morphologic changes with severe axonal loss.
Statistics
Data are expressed as mean ± standard deviation (s.d.) Neurologic Severity Score at various time points after CHI were compared between groups by two-way repeated measures analysis of variance (ANOVA; SigmaStat 2.0), and if differences were statistically significant, NSS at a certain time point were compared using the nonparametric Mann-Whitney U-test. Brain edema and results of ORT were analyzed by two-tailed Student's t-test. Levels of LMWA, enzymatic activity, and ODs were analyzed by one-way ANOVA, followed by post hoc Bonfferoni's multiple comparison test. Histopathological data were evaluated by the Pearson χ2 and Mann-Whitney U-tests using the SPSS 4.0 software.
A P-value < 0.05 was considered significant.
Results
Effect of Superoxide-Dismutase Overexpression on Post-Closed Head Injury Outcome
Neurologic recovery: Based on previous reports on the beneficial effects of SOD overexpression in some models of brain injury, we studied post-CHI neurologic outcome and recognition memory in SOD1-overexpressing mice. Neurologic Severity Score at 1 h were 8.5 ± 1.2 and 8.45 ± 1.0 in Tg (n = 10) and WT groups (n = 11), respectively, indicating quite severe injury, and similar severity of injury in both groups. As depicted in Figure 1A, NSS decreased spontaneously over the studied period reaching 3.4 ± 1.6 in Tg mice (n = 9) and 3.4± 2.0 in the WT (n = 8) at 14 days after injury (P < 0.05 comparing time points within a group). However, neither the rate of recovery nor the extent of residual neurologic impairment differed between groups at any time point during this period. Mortality rate at this period was 10% and 27% in Tg and WT groups, respectively (χ2 = 1.46, P=0.23).
Effect of superoxide-dismutase (SOD) levels on post-closed head injury (CHI) outcome. Neurologic Severity Score (NSS) (see text) was followed during 14 days after CHI. (
Recognition memory: Noninjured Tg and WT mice spent significantly more time exploring a novel object as compared with the time at the pretest (70% ± 12.9% versus 49.9% ± 5.3% and 65% ± 13% versus 50.7% ± 3.5%, respectively; P < 0.05 within group), indicating that recognition memory was not affected by excess of SOD1 in the intact mouse. However, 3 days after CHI, neither Tg nor WT mice were able to differentiate between novel and familiar objects (52.9% ± 24.8% versus 61.5% ± 12.4% and 52.1% ± 15.3% versus 55.1% ± 5.1%, respectively), indicating impairment of recognition memory. This transient effects was resolved at 12 days to a similar extent in both groups (66.4% ± 9.8% versus 49.5% ± 2.4% and 63.8% ± 3.4% versus 51.2% ± 4.1%, respectively; P < 0.05 within group). Hence, overexpression of SOD1 in mice neither relieved the extent nor shortened the duration of memory deficits caused by CHI.
To ascertain that these unexpected results reflected a true lack of effect of SOD1 overexpression on neurobehavioral outcome of mice after CHI, experiments were performed to evaluate recovery of SOD1−/- mice of a different genetic background. Based on the literature, we hypothesized that SOD1 deficiency might be deleterious.
Effect of Superoxide-Dismutase Deficiency on Post-Closed Head Injury Outcome
Neurologic Severity Score at 1 h was 7.3 ± 1.0 in SOD1−/- mice (n = 27) and 7.2 ± 1.0 in matched WT littermates (n = 28), indicating similarly moderately severe injury (Beni-Adani et al, 2001). As depicted in Figure 1B, neurologic impairment was moderately, yet significantly attenuated over a period of 14 days in both SOD1−/- mice and their WT littermates. However, temporal changes in NSS differed between these groups (P < 0.05, ANOVA), and, unexpectedly, NSS was significantly lower in SOD1−/- mice (5 ± 0.8, n = 13) as compared with WT (5.9 ± 0.8, n = 14) at 14 days after injury (P < 0.05, Mann-Whitney U-test). Rates of mortality during the study period were similar in both groups (~13%). It should be noted that the initial NSS (1 h) was lower in these two groups, as compared with those described above (8.5) and this difference may account for the higher mortality seen in the TG and respective WT mice. However, the relevant comparison is within each set, and these were not different.
Effect of Superoxide-Dismutase 1 Deficiency on Brain Edema
Water content examined at 24 h after CHI (time for maximal edema; Chen et al, 1996) in the ipsilateral cortex increased significantly as compared with the contralateral cortex in both SOD1−/- and WT mice (80.54%± 1.32% versus 78.05%+0.25% and 80.11% ± 1.19% versus 78.02%± 0.73%, respectively; P< 0.005 within group). Hence, surprisingly, SOD1 deficiency was not associated with aggravation of brain edema.
Effect of Superoxide-Dismutase 1 Deficiency on Low Molecular Weight Antioxidant
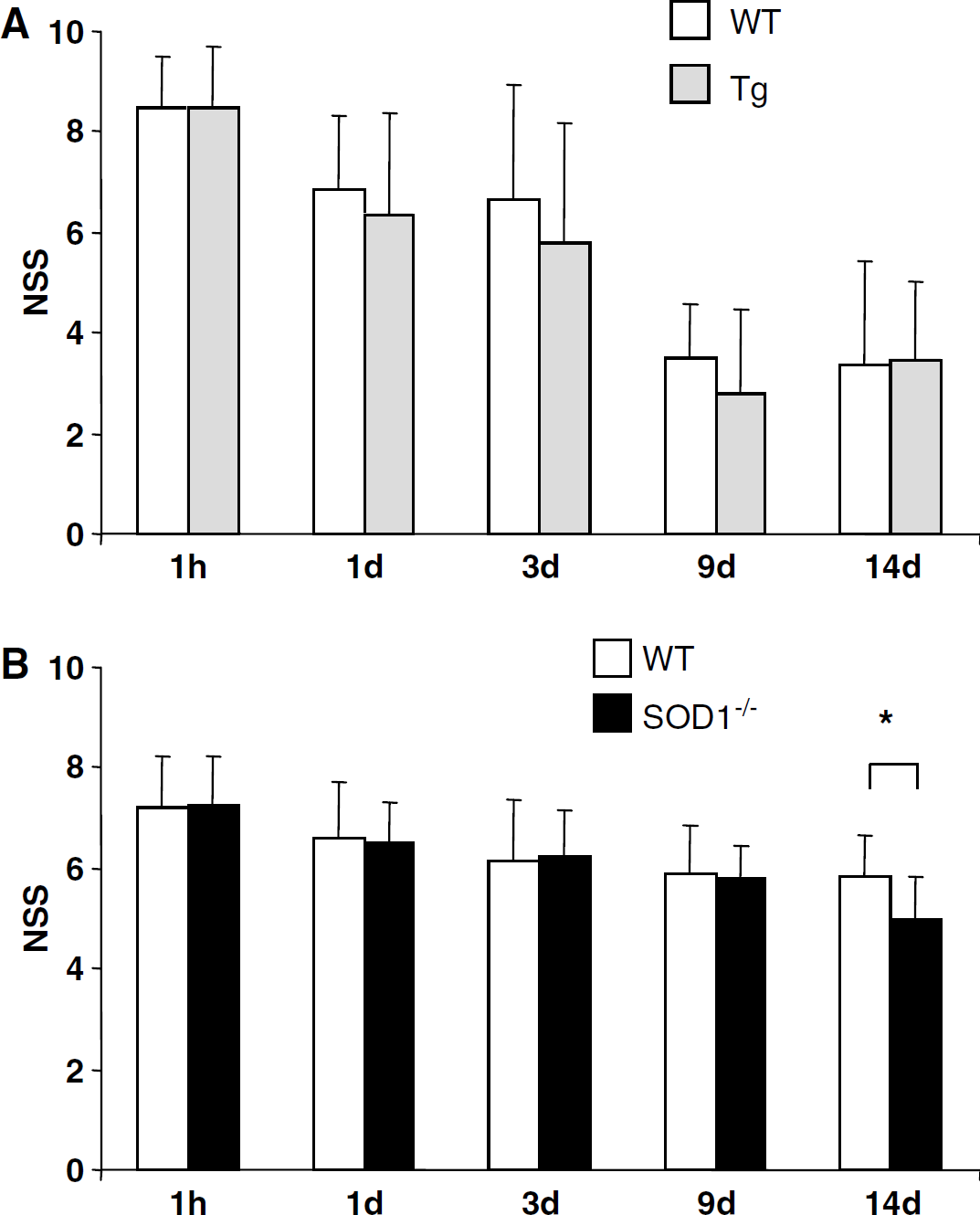
Closed head injury induces oxidative stress in the brain as well as in peripheral organs such as the liver (Shohami et al, 1999). Since the most pronounced effect was shown to occur at 5 mins after CHI, we assessed whether SOD1 deficiency modifies central or peripheral responses to CHI at this time point. Oxidation potentials (Ep) were detected in cortical and cerebellar homogenates at 350 to 400, 750 to 850, and 1110 to 1400 mV. Basal levels of cortical LMWA (expressed as anodic currents, Ia) composing the first Ep (Figure 2A) were higher in SOD1−/- as compared with WT mice (0.81 ± 0.08 versus 0.66 ±0.08 μA/mg protein; P < 0.05). Five minutes after CHI, these LMWA decreased significantly in SOD1-−/- (0.66 ± 0.13 μA/mg protein; P < 0.05) but not in WT (0.59 ±0.12 μA/mg protein). Neither CHI nor SOD1 deficiency affected levels of cortical LMWA at the other two Ep. Basal and post-CHI levels of LMWA were similar in SOD1−/- and WT cerebella (data not shown).
Low molecular weight antioxidants in thebrain and liver. Peak potentials (Ep) and anodic current intensities (***la) were determined by cyclic voltammeter (CV) in tissue homogenates derived from the ipsilateral cortex and the liver. Anodic current intensities (la), which are proportional to LMWA levels at a particular Ep, were measured before and 5 mins after closed head injury (CHI), normalized to protein concentration and expressed as the mean ± s.d. (
The electrochemical properties of the liver (Figure 2B) differed from those of the brain: The wave at 350 to 400 mV was undetectable in some sham mice (3/5 WT and 1/7 SOD1−/-) and the wave at 1110 to 1400 mV was absent in all mice. Five minutes after CHI, LMWA at 350 to 400 mV increased significantly in WT (0.13 ± 0.04 versus 0.04 ± 0.05 μA/mg protein in sham; P <0.05) but not in SOD1−/- (0.12 ± 0.04 versus 0.08 ± 0.04 μA/mg protein). The most prominent voltammetric wave appeared in the liver at 750 to 850 mV. Low molecular weight antioxidants at this potential were significantly higher in SOD1−/- as compared with WT sham mice (0.56 ± 0.02 versus 0.35 ± 0.06 μA/mg protein; P <0.05), but were not affected by CHI (0.56 ± 0.19 and 0.48 ± 0.06 ±A/mg protein, respectively) in either group. Thus, SOD1 deficiency exerted tissue-specific effects on basal and post-CHI levels of LMWA.
Effect of Superoxide-Dismutase 1 Deficiency on Superoxide-Dismutase 2 Activity
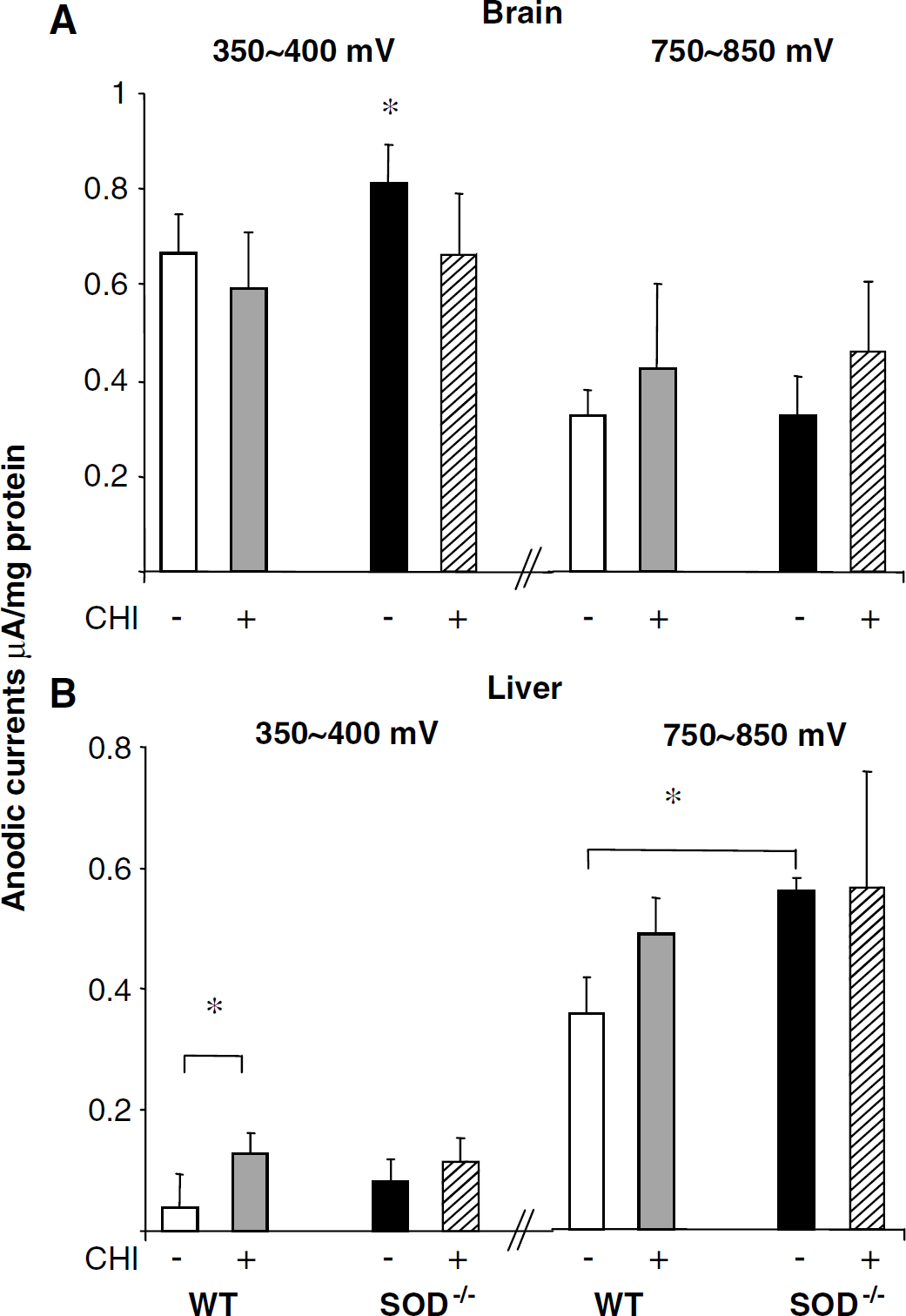
As expected, basal total SOD activity was significantly lower in SOD1−/- mice as compared with WT (39.74 ± 5.52 versus 74.96 ± 11.7 U/mg protein; P <0.01) and was unaffected by KCN, a selective SOD1 inhibitor, indicating lack of any SOD1 activity. Seven days after CHI, ipsilateral protein levels of SOD1 were fourfold higher than in the contralateral cortex in WT mice (data not shown). Yet, as depicted in Figure 3A, cortical activity of SOD1 in injured WT mice was significantly lower as compared with sham WT (30.8 ± 5.7 versus 69.9 ± 30.3 and 41.4 ± 15.7 versus 78.2 ± 15.5 U/mg protein, in the ipsi- and contralateral cortices, respectively; P <0.05), while it was preserved in the cerebella (data not shown).
Enzymatic activity of superoxide-dismutase (SOD) 1 and SOD2 before and 7 days after closed head injury (CHI). (
Figure 3B depicts the effect of CHI on the activity of SOD2. Basal activity of SOD2 was similar in the cortex and cerebellum of WT and SOD1−/- mice. Seven days after CHI, SOD2 activity in WT mice markedly decreased in the ipsilateral cortex (21.6 ± 5.4 versus 78.4 ± 26.3 U/mg protein; P <0.001), and less so in the contralateral cortex (33.5 ± 14.7 versus 79 ± 46.9 U/mg protein; P = 0.055) but not in the cerebellum (data not shown). In contrast, injured SOD1−/- mice maintained ipsi- and contralateral SOD2 activity at near-basal levels (50.1 ± 32.8 versus 53.9 ± 31.2 and 42.3 ± 11.9 versus 69.8 ± 25.8 U/mg protein, respectively), and manifested a trend towards higher level of activity at the ipsilateral cortex as compared with injured WT mice (P = 0.03 by ANOVA; P = 0.06 between CHI groups). As a result of these differential responses to CHI, ipsilateral total activity of SOD after injury was surprisingly similar between injured WT and SOD1−/- mice, despite the lack of SOD1 in the latter.
Effect of Superoxide-Dismutase 1 Deficiency on Nuclear Factor-KappaB Translocation
Two bands, consisted of distinct NF-κB DNA–protein complexes, were identified by EMSA (Figure 4). Their disappearance in the presence of excessive unlabeled (cold) NF-κB oligomer confirmed specificity, while a supershift assay confirmed that the upper complex consisted of p65-subunit. Constitutive NF-κB was comparably low in cortical extracts of intact WT (1.08 ± 0.27) and SOD1−/- mice (0.79 ± 0.24). However, 14 days after CHI, NF-κB was dramatically activated at the ipsilateral cortex of WT but not of SOD1−/- mice (8.95 ± 1.74 versus 1.82 ± 1.7; P <0.005).
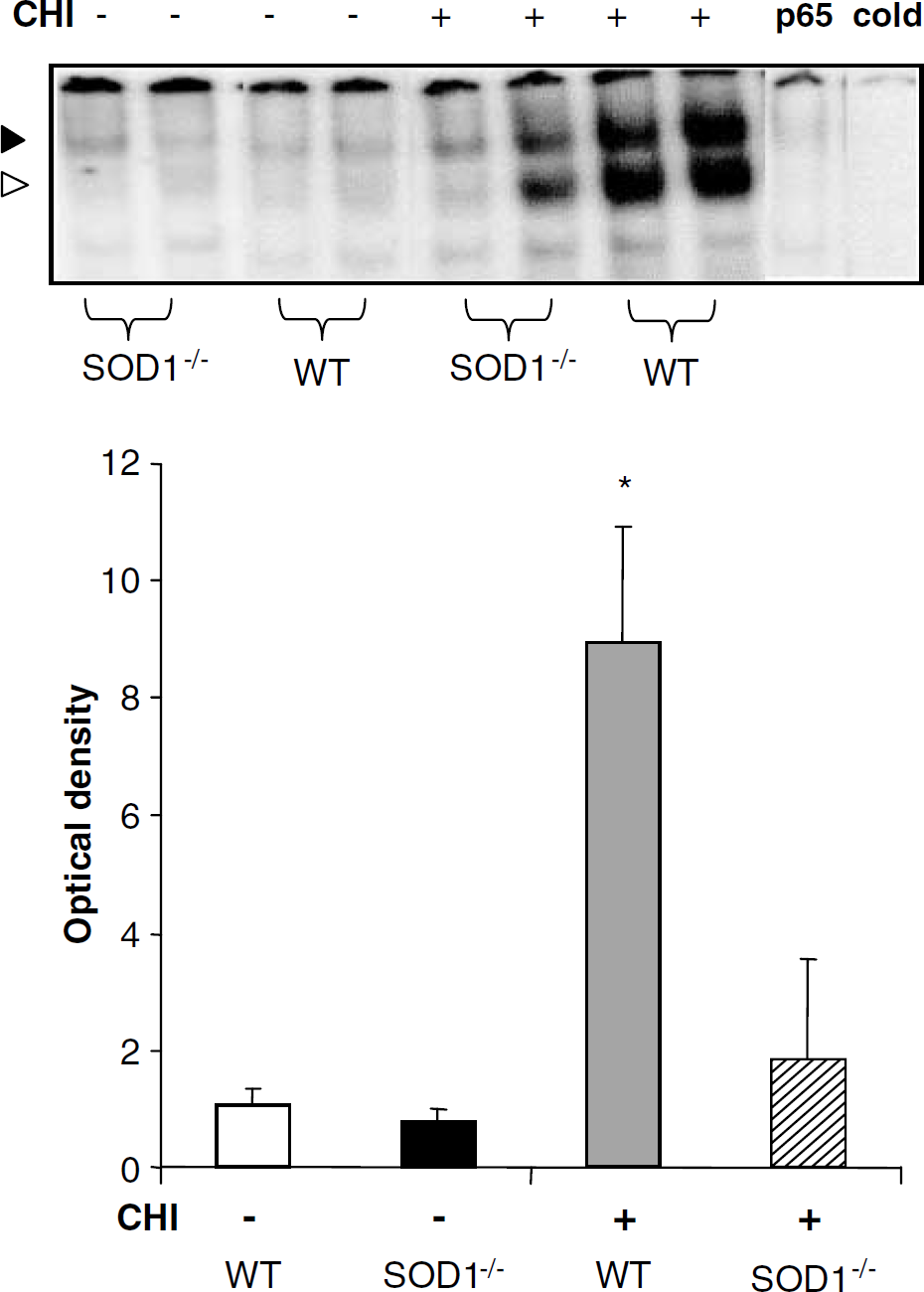
Nuclear factor-kappaB (NF-κB) activation is inhibited in SOD1−/- mice. Two nuclear NF-κB complexes were resolved by electromobility shift assay (EMSA). Their disappearance in the presence of excessive unlabeled (cold) NF-κB oligomer confirmed specificity, and a supershift assay confirmed that the upper complex consisted of p65-subunit (upper inset). Optical densities are presented as the mean ± s.d. Constitutive nuclear NF-κB was comparably low in wild-type (WT) and SOD1−/- cortices. Fourteen days after closed head injury (CHI), NF-κB was dramatically (> 8-fold) transactivated in the ipsilateral cortex of WT but not SOD1−/- mice (P <0.00005 by ANOVA; *P <0.005 comparing injured WT with all other groups).
Effect of Superoxide-Dismutase 1 Deficiency on Histopathology
Glial fibrillary acidic protein immunestaining: Activated astrocytes were identified by GFAP staining that revealed different distribution and severity of astrogliosis between the SOD−/- and WT (Figures 5A to D). Astrogliosis-free areas (grade 0) were detected in 14% of the evaluated visual fields in the injured hemisphere of SOD−/- mice and in only 1.7% of the WT. In contrast, astrogliosis was robust in WT, with 45% and 43% of the evaluated visual fields displaying mild-moderate and severe astrogliosis (grades 2 and 3, respectively), as compared with only 29% and 15% in SOD−/- mice (Figure 5E; P < 0.0001 for all pairs). In the contralateral hemisphere, no grade 3 (severe) astrogliosis was found, 63% and 27% of the studied fields were normal in SOD−/- and WT, respectively, (grade 0) and grades 1 and 2 were twice as high in WT as compared with SOD−/- mice (Figure 5F; P < 0.0001 for all pairs).
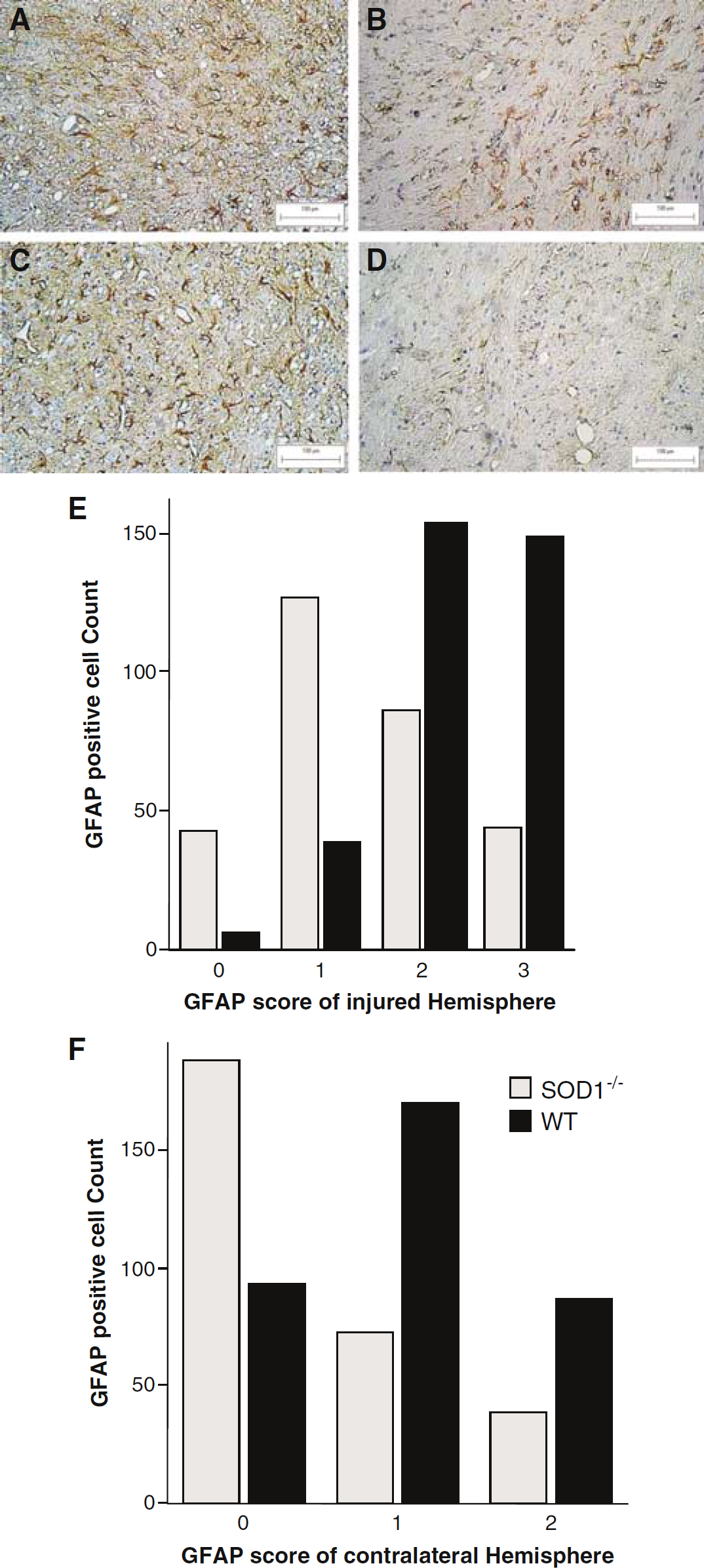
Glial fibrillary acidic protein (GFAP) immunohistochemistry. Glial fibrillary acidic protein immunohistochemistry in the injured (
Gallyas silver staining: Figure 6 shows argyrophylic degenerative neurons stained with Gallyas in injured hippocampi, taken from severely injured mice (indicated by NSS = 9 at 1 h after CHI). Areas of severe damage are presented in WT (A, B, and C) and SOD−/- (D, E, and F) mice. The massive degeneration throughout several hippocampal areas is evident in WT compared with SOD−/- mice. CA4 and CA3 areas of the hippocampus were vulnerable to degeneration in both groups, yet not to the same extent. Figure 6G shows the quantification of the Gallyas-positive cells according to the 0 to 4 scale described, and it is evident that WT mice displayed significantly more positive cells (P < 0.0001), indicating more degenerative neurons, as compared with SOD−/- mice. Similarly, in the contralateral hippocampus (data not shown) absolute amount of Gallyas staining was lower than in the ipsilateral, yet the difference between WT and SOD−/- was also evident, such that the amount of total positive, degenerating neurons was about fourfold higher than that in the SOD−/- (P <0.001).
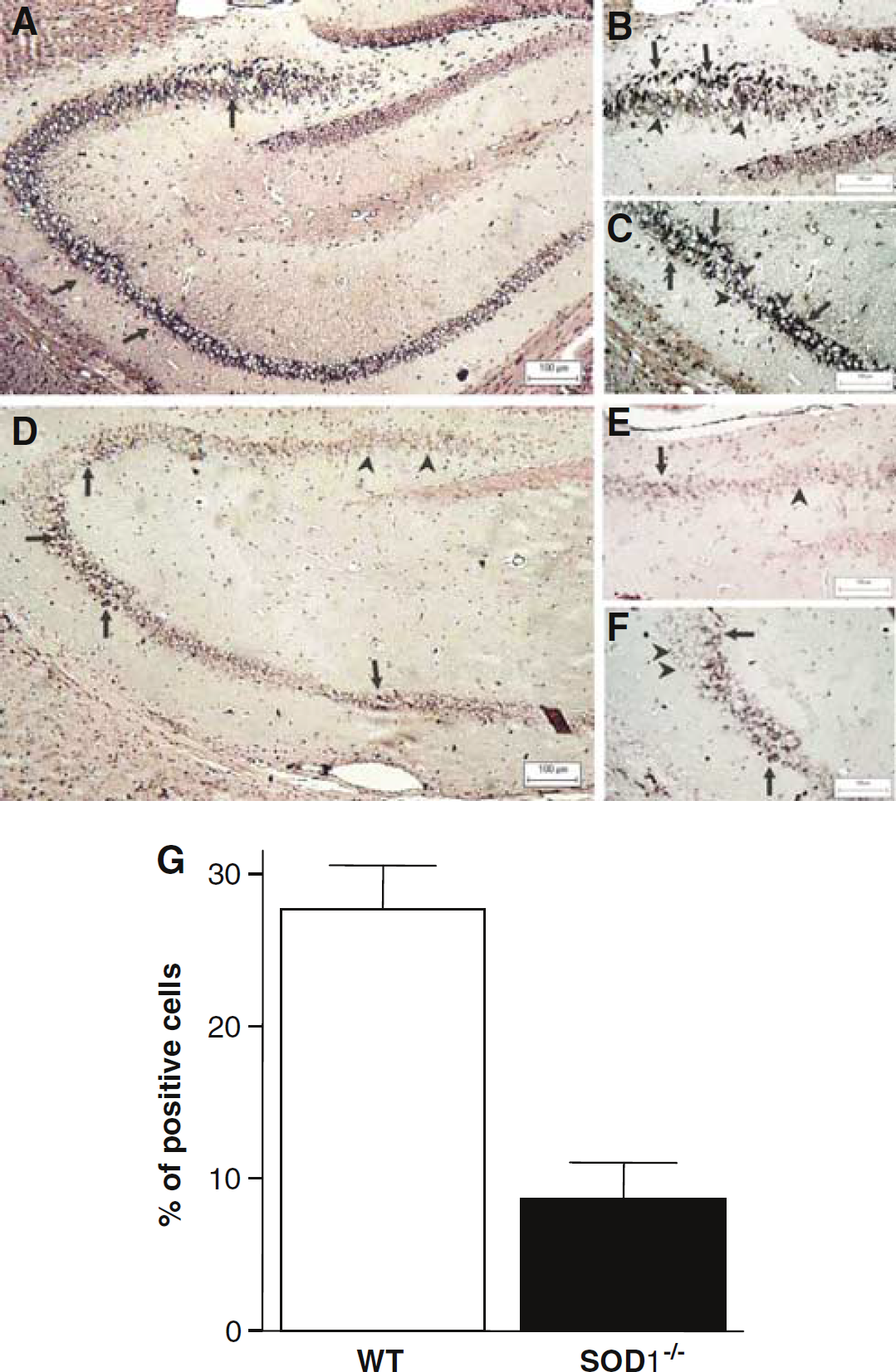
Analysis of Gallyas-positive staining in the hippocampus. (
Figure 7 depicts the analysis of the Gallyas-positive staining in cortical tissue adjacent to the site of injury. While 40% and 35% of the total studied fields were either normal (grade 0) or grade 1 in SOD −/- mice, only ~5% and 20%, respectively, were in these grades in the WT (Figure 7A). The reverse pattern was found for grades 2 and 3. No difference was found between the groups in the contralateral hemisphere (Figure 7B).
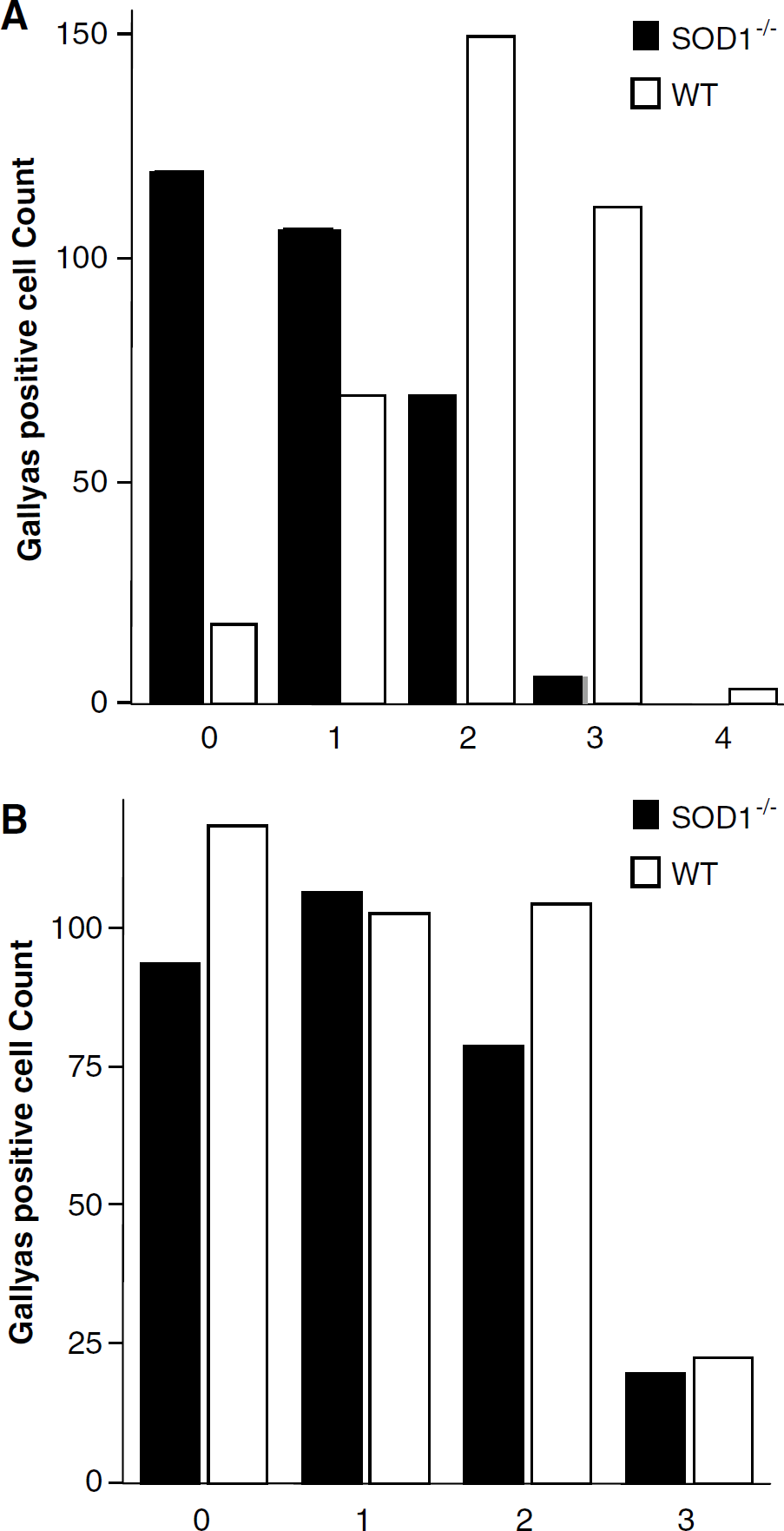
Semiquantitative analysis of Gallyas-positive staining in the cortex. Degenerative argyrophylic neurons were recognized according to morphologic criteria. The number of neuronal cells counted in the seven to eight randomly selected sections is presented for cortical tissue adjacent to the site of injury (
Discussion
Transgenic and knockout animal models have contributed to our understanding of endogenous mechanisms in normal and pathologic conditions (Longhi et al, 2001). However, data from studies using SOD1-mutant mice have been at times contradictory about whether excessive SOD1 is pathogenic or protects from disease. The present study demonstrates that SOD1 overexpression failed to enhance recovery of neurobehavioral and cognitive functions after CHI. Rather, SOD1 deficiency was associated, unexpectedly, with better long-term (2 weeks) functional outcome, accompanied by robustly less severe astrogliosis and neuronal degeneration, and less activation of NF-κB. Antioxidant capacity was also enhanced in these mice. Consistent with these data, brain edema was not exacerbated by SOD1 deficiency and mortality after CHI was unaffected by SOD1 levels.
In contrast to our findings, earlier studies in models of contusion and cold brain injury showed milder motor deficits, smaller tissue necrosis, reduced blood–brain barrier permeability, and less brain edema in SOD1 Tg mice (Chan et al, 1991; Mikawa et al, 1995, 1996). Lewen et al (2001) found that brain trauma resulted in greater cortical lesion in SOD1−/- mice, but functional outcome was not reported in that study. Brain injury models in these studies differed form our model of CHI, and this may, at least in part, account for the different results. Background variations may also account for differential sensitivity of mutant SOD1−/- and Tg mice to traumatic brain injury.
Tissue-targeted disruption of SOD1 resulted in apoptosis in transient cerebral ischemia (Kawasa et al, 1999; Kondo et al, 1997), while neither mitochondrial signaling for apoptosis nor neural damage were affected in permanent cerebral ischemia (Fujimura et al, 2001). Hence, SOD1 may specifically confer protection in ischemic–reperfusion injuries and in paradigms of O2−-mediated neural damage (Fujimura et al, 2000; Lewen et al, 2001; Yoshida et al, 2000) while aggravating damage in H2O2/OH•--mediated pathologies (Busciglio and Yanker, 1995; de Haan et al, 1996; Peled-Kamar et al, 1997). While overexpression of SOD1 facilitates O2− removal, it also enhances H2O2 production, which in turn favors generation of OH•- via Fenton reaction. Neurons from SOD1 Tg mice manifested high levels of H2O2, decreased cellular glutathione, (Bar-Peled et al, 1996), and increased lipid peroxidation that correlated with altered SOD1/glutathione peroxidase-1 (GpX1) ratio (Ceballos-Picot et al, 1992). In addition, Gahtan et al (1998) found impaired spatial learning in SOD1 Tg mice and their hippocampi failed to express long-term potentiation. They suggested that these effects were mediated via excessive H2O2. These findings emphasize that a concerted activity of SOD together with GpX or catalase (de Haan et al, 2003: Rodriguez et al, 2000) is required for redox homeostasis, and provides a mechanism to explain the current lack of neuroprotection in Tg mice.
The neurobehavioral data prompted us to assess whether the improved outcome after CHI in SOD1−/- mice is because of adaptive mechanisms that preserved cellular redox homeostasis. We previously demonstrated that LMWA are mobilized and/or consumed soon after CHI to counteract oxidative burst (Beit-Yannai et al, 1997; Shohami et al, 1997, 1999). The present study shows that basal and post-CHI levels of LMWA are modified in SOD1−/- mice in a tissue-specific manner, suggesting differential adaptive responses to prolonged imbalance between O2− and H2O2. Yet, it appears that the beneficial effect of SOD1 deficiency on late-phase recovery was not mediated by LMWA.
In the present study, basal brain SOD2 activity was similar in SOD1−/- and WT mice, yet 7 days after CHI, a robust decrease was found only in the WT but not in SOD1−/- mice. From that time point on, the neurobehavioral score of the SOD1−/- indicated better recovery, suggesting that SOD2 plays a pivotal role in neuronal resistance to oxidative stress (Khan and Black, 2003). Similar compensatory/functional interactions of SOD's have been reported for fibroblasts transfected with SOD1 gene that showed decreased SOD2 activity (Keiner et al, 1995), while higher brain SOD2 activity was found in SOD1 Tg mice (Ceballos-Picot et al, 1992). One may thus speculate that a chronic, yet subtle, oxidative stress in the SOD1−/- mice affords tolerance to additional injury. Possibly, increased oxidative stress in SOD1−/- mice at the immediate post-CHI phase led to the sparing of SOD2 activity at a later phase, thereby promoting neuroprotection. This was evidenced by less astrogliosis and neuronal cell death, as well as better functional outcome. Along the same line, Sullivan et al (1999) reported that mice overexpressing SOD2 showed smaller infarct after trauma.
Reactive oxygen species not only determine cellular redox-state but they also act as second messengers in signal transduction pathways such as NF-κB and AP-1 (Kaltschmidt et al, 1999; Kamata et al, 2002a; Schmidt et al, 1995; Vollgraf et al, 1999), thereby affecting cellular choice between death and survival (Martindale and Holbrook, 2002). Recently, we reported a remarkable induction of NF-κB in mouse cortex 24 h after CHI, reaching > 12-fold activation 8 days later (Beni et al, 2004). Nuclear factor-kappaB is a primary regulator of stress response in the central nervous system, and is induced after spinal cord injury (Bethea et al, 1998), cerebral ischemia (Clemens et al, 1998; Huang et al, 2001), excitotoxicity (Lerner-Natoli et al, 2000), and neurodegeneration (Combs et al, 2001).
In the present study, SOD1 deficiency abolished the robust late-phase (14 days) activation of NF-κB in response to CHI, while it had no effect on its constitutive activity. Redox regulation of NF-κB is affected by dynamic changes in cellular superoxide/H2O2 ratio (Bernard et al, 2002). We propose that H2O2-mediated activation of NF-κB is largely prevented in SOD1−/- mice as a result of less H2O2 formation. Indeed, ample evidence suggest that H2O2 is the key ROS responsible for NF-κB activation, while O2− is apparently involved indirectly as a source of H2O2 (Vollgraf et al, 1999; Schmidt et al, 1995; Li et al, 2001). Several studies suggest that H2O2 modulate crucial phosphorylation/dephosphorylation events in NF-κB signaling pathway, including MAPK, phospholipase Cκ, and IκB kinases (IKK) (Anderson et al, 1994; Kamata et al, 2002a; Li et al, 2001). In particular, phosphorylation of serine residues in activation loops of IKK, a complex targeting the ubiquitination and proteasomal degradation of IκB, was essential for IKK activation via H2O2 (Kamata et al, 2002b). However, H2O2 seems to modify the secondary structure and activity of certain tyrosine and serine/threonine phosphatases by oxidizing specific cysteine residues on these proteins (Carballo et al, 1999; Denu and Dixon, 1998). Therefore, H2O2 could possibly prevent cessation of upstream signaling cascade of MAPK or IKK.
In the present study, blockage of NF-κB activation in SOD1−/- mice occurred concomitantly with less astrogliosis and neuronal degeneration as well as with neurobehavioral improvement, suggesting that prolonged activation of NF-κB after brain injury is harmful. These findings agree with our recent reports that showed association between the neuroprotective effects of melatonin (Beni et al, 2004) and the endocannabinoid 2-arachidonoyl glycerol (Panikashvili et al, 2005) and inhibition of late-phase induction of NF-κB after CHI. Similarly, Clemens et al (1998) reported that the antioxidant LY231617 prevented persistent but not transient activation of NF-κB and induced neuroprotection against global ischemia. Indeed, the long-term (1 year) activation of this factor after brain injury may point to its role in chronic inflammatory response (Nonaka et al, 1999). Nuclear factor-kappaB regulates a wide array of target genes, some of which play a dual role in mechanisms of cell survival (e.g., cytokines), others that are neuroprotective (e.g., SOD2 and BCL-2), and yet others that mediate cell death (e.g., inducible nitric oxide synthase (iNOS) and cytosolic phospholipase A2) (Martindale and Holbrook, 2002). Such a diversity of downstream cellular responses implies that transcription is regulated by specificity of NF-κB-subunits (Perkins et al, 1992) and/or by duration of their activation. Taken together, prolonged activation of NF-κB apparently shifts the transcriptional balance in favor of cell death.
In conclusion, SOD1 deficiency, rather than overexpression, afforded neuroprotection in the late post-CHI phase. Superoxide-dismutase 1−/- and SOD1 Tg mice in the present study carried distinct genetic backgrounds, suggesting that the current data reflect the effect of SOD1 levels rather than that of strain variation. The higher basal levels of LMWA in SOD1−/- mice and the spared activity of SOD2 after CHI point to the development of compensatory mechanisms and better tolerance to CHI, which are expressed by less activation of NF-κB and less astrogliosis and neuronal death. Since H2O2 is a major mediator of cell injury/death via NF-κB activation, less H2O2 formation in the SOD1−/- mice may account for the observed neuroprotection. Taken together, these findings corroborate the role of redox homeostasis in cellular events and point to the complexity of tissue responses to genetic manipulations.