
Abstract
Phosphorylation of N-methyl-D-aspartate (NMDA) receptors is a major regulatory mechanism underlying synaptic plasticity. However, changes in NMDA receptors and phosphorylation after traumatic brain injury (TBI) remain incompletely understood. Using an animal TBI model, we observed that the protein level of NMDA receptor subunit NR2B was downregulated in synaptosomal fractions obtained from the ipsilateral neocortical injury region, whereas the levels of NR2A, NR1, and PSD93 were not significantly altered at 4 and 24 hours after TBI. Further investigation showed that tyrosine phosphorylations of NR2B Y1472 and PSD93 Y340 in synaptosomal fractions were significantly decreased relative to their total protein level after TBI. Correspondingly, phosphorylation of the Src-kinase-inhibitory site Y527 was increased, whereas phosphorylation of the activation site Y416 was decreased, indicating that the activity of Src kinase is significantly inhibited after TBI. In comparison, other Src family kinase substrates of NMDA receptor, NR2A Y1246, NR2A Y1325, and NR2B Y1070 were not obviously affected after TBI. The results suggest that TBI downregulates the Src-kinase-mediated phosphorylation of NR2 and PSD93 to destabilize the synaptic localization of NMDA receptors. Therefore, post-TBI loss of NMDA receptors may contribute to the depression of synaptic activity after TBI.
INTRODUCTION
Traumatic brain injury (TBI) survivors often suffer from depressive, cognitive, and motor disturbances due to disarrangement of glutamatergic neurotransmission and aberrant brain remodeling.1–5 N-methyl-D-aspartate (NMDA) receptors are a subclass of ionotropic glutamate-gated ion channels and has a pivotal role in the regulation of synaptic plasticity in the central nervous system. Most of the central NMDA receptors are heterotetramers formed by two core subunits, NR1, with two regulatory subunits, NR2A or NR2B. The NMDA receptors are not only located in the synaptic membranes, but a significant proportion is also distributed in the intracellular vacuoles and light membranes.6,7 The synaptic membrane NMDA receptors are not static, rather their number and composition are dynamically distributed among the intracellular and synaptic pools. In response to the changes in synaptic activities, NMDA receptors are shuttled among synaptic membranes and the endocytotic degradation pathways.6,7
The Src family kinases (SFKs) are highly expressed in the central nervous system. Src family kinases act as a ‘core or master kinase’ to regulate tyrosine phosphorylation and localization of NR2 subunits of NMDA receptors among different intracellular and synaptic pools. 8 For instance, Src-mediated tyrosine phosphorylation of NR2B subunit at Y1472 residue enhances targeting and binding of NR2B to synaptic scaffolding proteins, PSD95 and PSD93, and promotes the stabilization of NMDA receptors at synaptic membrane surfaces.9,10
To study the regulation of the NMDA receptors after TBI, we developed antibodies to several novel tyrosine phosphorylation sites of NR2. 11 This study shows that both protein level and phosphorylation of NR2 are significantly decreased while NR1 is unchanged after TBI. The decrease in NR2 may be due to the reduction of tyrosine phosphorylation of the NR2B C-terminal Y1472 by SFKs, thus facilitating the endocytotic degradation of NR2B. Downregulation of NMDA receptors may contribute to depression and cognitive dysfunction after TBI.
MATERIALS AND METHODS
Traumatic Brain Injury Model
Experiments were performed with male Sprague-Dawley rats weighing 270 to 320 g (Charles River Laboratories, Raleigh, NC, USA). All experimental procedures were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Maryland Animal Care and Use Committee. All feasible measures were taken to reduce animal suffering and the number of animals used for these experiments. The basic surgical preparation for brain injury was performed according to previously described methods.12,13 Animals were maintained for at least 7 days before the experiment in a temperature-regulated room (23°C to 25°C) on a 12-hour light-dark cycle. The rats were fasted, but allowed free access to water overnight before surgery. Traumatic brain injury was produced with fluid-percussion pressure levels of 2.0±0.2 atmospheres. Rats were anesthetized with 3.0% halothane in a gas mixture of 70% N2O and 30% O2. The femoral artery was cannulated to deliver pancuronium bromide (0.5 mg/kg, intravenously) every 1 hour during the surgical procedure. An endotracheal tube was inserted, and rats were mechanically ventilated with 70% N2O, 30% O2, 0.5% to 1.5% halothane. The animals were then placed in a stereotaxic frame, and a 4.8 mm craniotomy was made over the right parietal cortex (3.8 mm posterior to the bregma, 2.5 mm lateral to the midline). A plastic injury tube (18-gauge modified PrecisionGlide needle hub, Becton Dickinson, Franklin Lakes, NJ, USA), was placed over the exposed dura and fixed with dental acrylic.
Before and after TBI, blood gases and mean arterial blood pressure were monitored and maintained at physiologic levels. Brain temperature was monitored with a thermistor probe placed in the left temporalis muscle, and core temperature was determined with a rectal thermometer. Brain temperature was maintained at 37°C with self-adjusting feedback heating lamps. Blood gases, blood glucose, and hematocrit values were monitored 15 minutes before TBI, 15 minutes after TBI, and then once every hour for up to 4 hours after TBI. All animals were maintained within physiologic ranges for mean arterial pressure (120 to 140 mmHg), pO2 (105 to 170 mmHg), pCO2 (35 to 45 mm Hg), and blood pH (7.38 to 7.41). Sham-operated rats were subjected to identical surgical procedures, but without the fluid-percussion injury pulse. After TBI, anesthesia was discontinued, and the animals were returned to their cages.
For biochemical studies, brains were frozen in situ with liquid nitrogen at 0.5, 4, and 24 hours after TBI according to the methods of Ponten et al. 14 Animals were anesthetized, tracheotomized, and artificially ventilated with ~2.0% halothane in 70% N2O and 30% O2. The femoral artery catheter was recannulated for monitoring the mean arterial pressure. The animals’ respiration was maintained with a ventilator while the brains were frozen in situ with a 50-mL centrifuge tube (open bottom) on the top of the head filled with liquid nitrogen. When the mean arterial pressure dropped below 10 mm Hg, animals were decapitated. 14 The animals experienced few complications during and after the surgeries, thus none was excluded from this study.
Histopathology
Animals were perfusion-fixed with 4% paraformaldehyde and postfixed in the same fixative for 24 hours before sectioning at 40 μm with a vibratome. Brain sections were stained with hematoxylin and eosin and examined with a microscope.
Preparation of Subcellular Fractions
Rat brains were removed from the liquid nitrogen-frozen heads with a saw, a hammer, and a chisel. 14 Frozen brains were sliced into 1 to 2 mm sections in a −15°C glove box freezer. The neocortical tissues in the contusion areas between the bregma 1.6 mm to −4.52 mm from the ipsilateral or the corresponding contralateral side were dissected and chopped into small pieces in the −15°C glove freezer. 15 The contusion area is defined as the cortical area between 1.6 mm lateral from the midline and 5 mm below the horizontal plane of the skull surface at the level between the bregma to lambda. The subcellular fractions were prepared for analyzing protein subcellular redistribution. Rat neocortical tissues from the ipsilateral or contralateral side were homogenized on ice with a Dounce homogenizer (Kontes Glass Co., Vineland, NJ, USA) (100 strokes) in 10 volumes of homogenization buffer containing 15 mmol/L Tris, pH 7.6, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 2.5 mmol/L EDTA, 1 mmol/L EGTA (ethylene glycol-bis (β-amino ethyl ether) tetraacetic acid), 1 mmol/L dithiothreitol, 1. 25 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aprotinin, 0.5 mmol/L phenylmethylsulfonyl fluoride, 0.1 mmol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L Na4P2O7. Homogenates were centrifuged at 800 g at 4°C for 10 minutes to obtain P1 pellets and supernatants (S1). The S1 was centrifuged at 10,000 g at 4°C for 10 minutes to obtain P2 pellets and supernatants (S2). The S2 fractions were centrifuged at 163,000 g at 4°C for 1 hour to obtain pellets (P3) and supernatants (S3). All pellets were suspended in an homogenization buffer containing 0.1% Triton X-100. Protein concentration was measured using the detergent compatible protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA).
Western Blot Analysis
Equal protein amounts (20 μg) from each subcellular fraction were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels and then transferred to Immobilon-P membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 3% bovine serum albumin in Tris buffered saline (TBS) for 1 hour and then incubated with the primary antibodies at 4°C overnight. Primary antibodies against NR1, NR2A, NR2B, PSD93, and β-actin, as well as phospho-Src Y416, phospho-Src Y527 were obtained from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies to phospho-NR2A Y1246, phospho-NR2B Y1325, phospho-NR2B Y1070, phospho-NR2B Y1472, and phospho-PSD93 Y340 were recently developed by Dr Hu's lab and Cell Signaling Technology. 11 The immunoblot membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 60 minutes at room temperature (Bio-Rad, #170-6515). The blots were illuminated using pierce enhanced chemiluminescence (Thermoscientific, Rockford, IL, USA) and developed on Kodak X-omat LS film (Eastman Kodak Company, New Haven, CT, USA). Densitometry was performed with Kodak ID image analyses software (Eastman Kodak Company). β-Actin was used as a sample loading control. All western blot data were first normalized to β-actin and then expressed as the ratio of protein level between experimental animals and mean of sham-operated control animals. One-way analysis of variance followed by Dunnett's tests were used for statistical analysis (mean ± standard deviation, n = 3).
Confocal Microscopy
Double-labeled fluorescence immunocytochemistry was performed on coronal brain sections (50 μm) from sham-operated controls and animals subjected to TBI followed by 24 hours of recovery. The sections were transferred into a 24-well microtiter plate filled half way with 0.01 mol/L citric acid-sodium citrate buffer, pH 6.0, heated for 10 minutes in a 95°C water bath. The sections were washed twice with 0.1% TX100-TBS for 10 minutes. Nonspecific binding sites were blocked with 3% BSA in TBS 0.1% TX100 for 1 hour and incubated with all of the antibodies used in western blot analysis, each at a dilution of 1:300 in TBS containing 0.1% TX100 overnight at 4°C. The sections were washed three times for 10 minutes each in TBS containing 0.1% TX100 at room temperature and incubated in a mixture of fluorescein-labeled anti-rabbit IgG (Thermo Scientific) at a dilution of 1:400 and 0.5 μg/mL propidium iodide for 1 hour at room temperature. The sections were washed three times in TBS 0.1% TX100, mounted on glass slides, and coverslipped using Gelvatol. The stained sections were analyzed on a laser-scanning confocal microscope (Leica, Wetzlar, Germany).
RESULTS
Histology
By means of a fluid-percussion injury model of TBI, we examined under a light microscopy neuronal damage in the neocortical and hippocampal regions during reperfusion periods (Figure 1). No morphologic changes were seen under the light microscopy in the brain sections of sham-operated control rats (Figures 1A–C). There are a few dead neurons in the neocortical areas of the contusion region (Figures 1D and E) and ~30% to 40% dead neurons among the total neurons in the CA3 area (Figures 1D and F) at 24 hours after TBI. Normal neurons are pyramidal like or round in shape (Figure 1B and C, arrowheads), whereas ischemic dead neurons have shrunken and acidophilic cytoplasm, as well as dark shrunken nuclei (Figures 1E and F arrows). These results are consistent with the previous reports.16,17
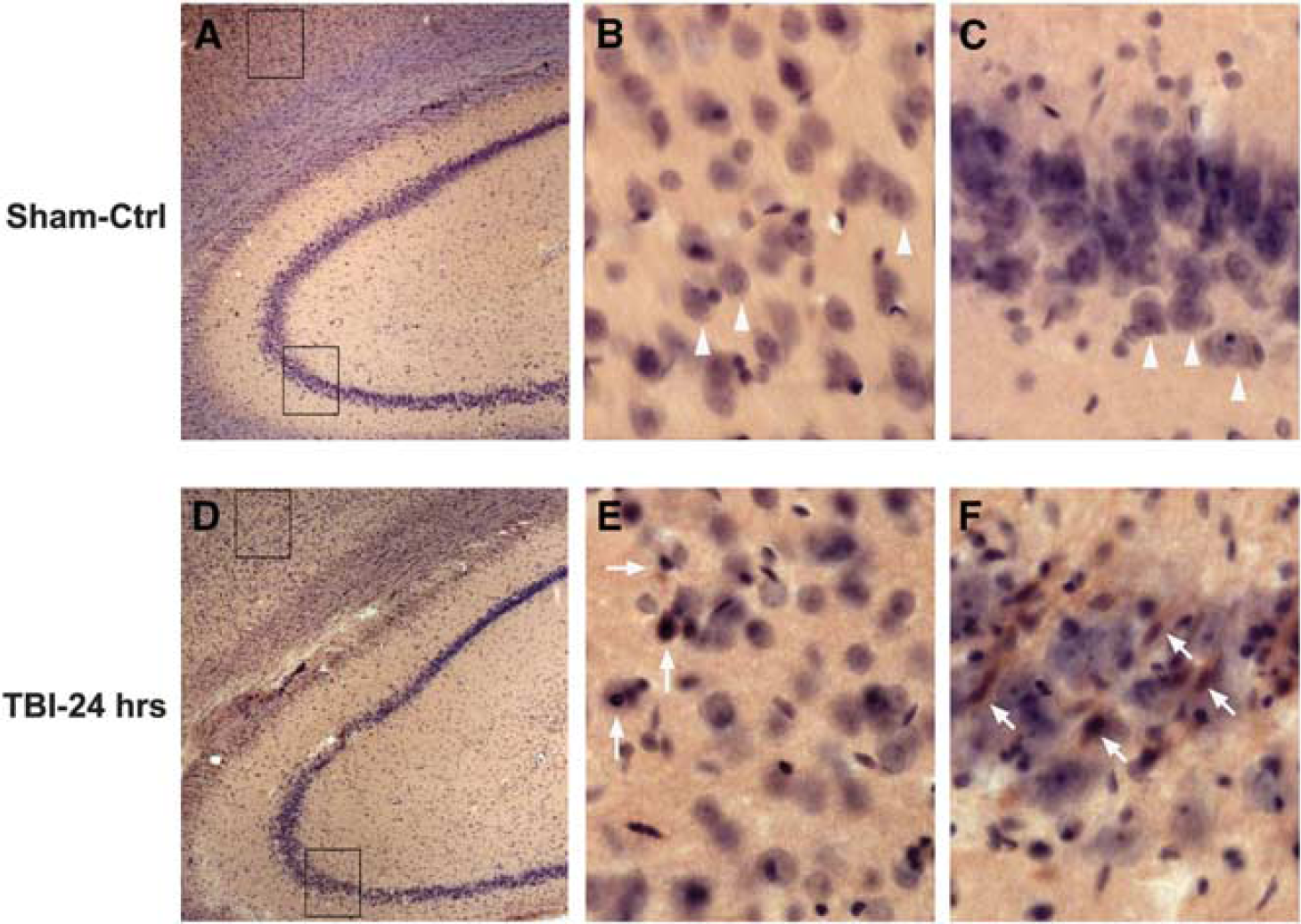
Histopathology after traumatic brain injury (TBI). Brain sections were obtained from a sham-operated control rat (
Decrease in the NR2 Subunit After Traumatic Brain Injury
The protein levels of NR1, NR2A, and NR2B subunits in the homogenate (Homo), crude synaptosomal fraction (P2), and light membrane fraction (P3) from neocortical tissues at 0.5, 4, and 24 hours post-TBI were analyzed and quantified by western blot. These receptors are barely detectable in the S3 cytosolic fractions from both control and post-TBI brain tissues, probably because they are membrane proteins. The protein levels of both NR2A and NR2B were significantly decreased in the brain homogenates and light membrane fractions after TBI (Figures 2A and B). Notably, the protein level of NR2B also was remarkably decreased in the synaptosomal fraction (Figure 2B). In comparison, NR1 was unchanged after TBI (Figure 2C). The protein levels of NR1, NR2A, and NR2B in the contralateral neocortical region were also observed and no change was found after TBI (Supplementary Figure 1A–C).
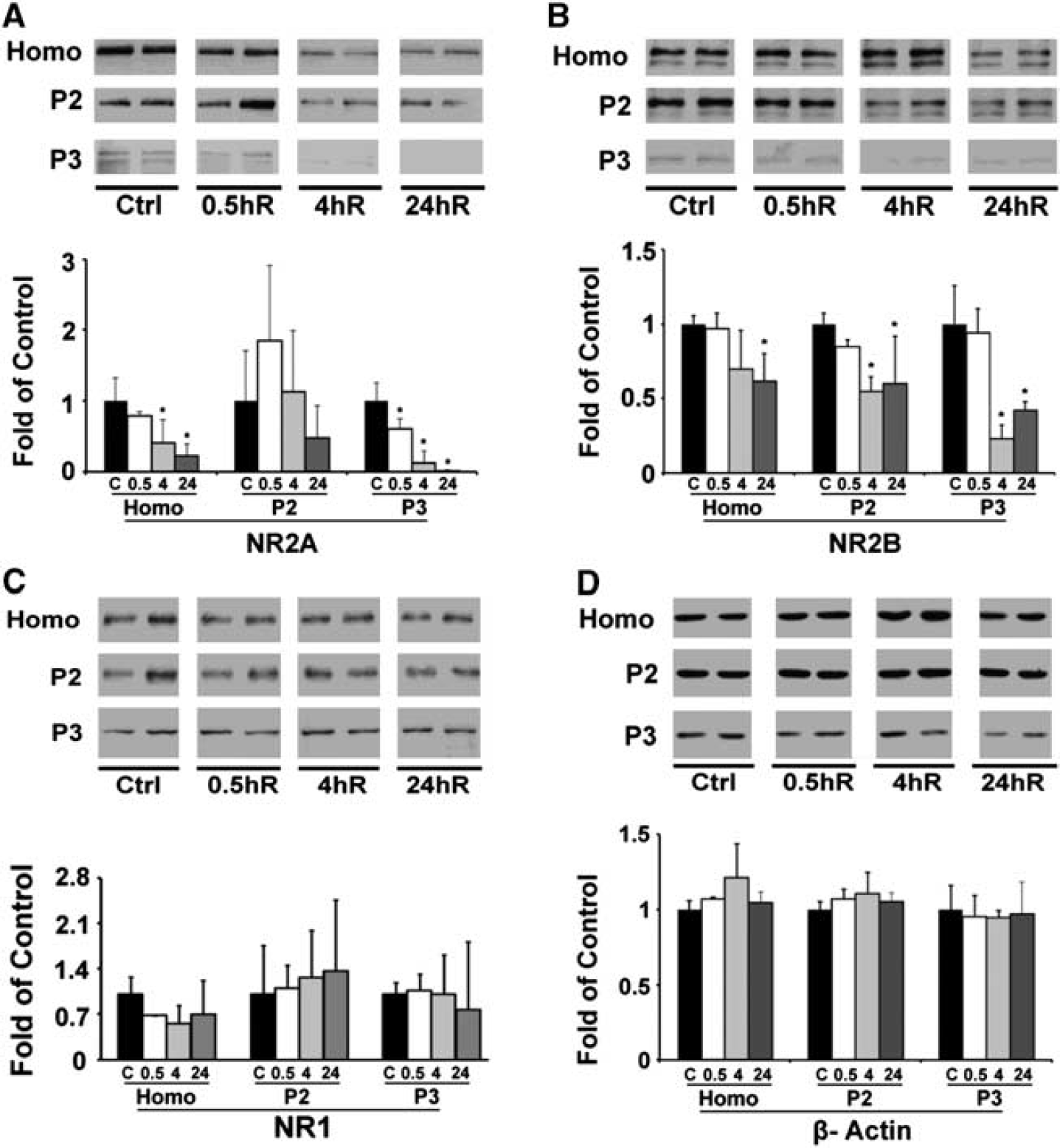
Western blot and quantitative analysis of NR2A, NR2B, and NR1 proteins in the rat cortex after traumatic brain injury (TBI). Neocortical tissues were obtained from the ipsilateral regions of sham-operated control rats and rats subjected to TBI followed by 0.5, 4, and 24 hours recovery (n = 3). An equal amount of protein from each sample in homogenate (Homo) and synaptosomal (P2), and light membrane (P3) fractions was subjected to SDS-PAGE and immunoblotted with antibodies against NR2A, NR2B, NR1, and β-actin respectively. The changes of subunits NR2A, nR2B, and NR1 (
Changes in Tyrosine Phosphorylation of NR2A, NR2B, and PSD93 after Traumatic Brain Injury
NR1 is not a tyrosine-phosphorylated protein, whereas NR2s are phosphorylated by SFKs at multiple tyrosine sites. 11 The decrease in NR2 protein level without a change in NR1 protein level may be associated with changes in their tyrosine phosphorylation status. To study this, we employed phosphorylation site-specific antibodies to analyze NR2A Y1246 and NR2A Y1325 as well as NR2B Y1070 and NR2B Y1472 by western blotting at 0.5, 4, and 24 hours after TBI. Considering the total protein levels decreased after TBI, we calculated the ratios between the phosphoforms of NR2A (Figures 3A and B), NR2B (Figures 3C and D) and their non-phosphoforms (total proteins, Figures 2A and B). The results showed that relative to the sham control, the level of phospho-NR2B Y1472 (Figure 3D) was remarkably reduced in synaptosomal fractions but not in homogenate at 0.5, 4, and 24 hours after TBI, whereas phosphorylation of NR2B Y1070, NR2A Y1325, and NR2A Y1246 was reduced in similar degrees with protein levels because the ratios were not significantly altered after TBI (Figures 3A-C). In comparison, phospho-NR2B Y1472 in the contralateral neocortical region was also observed but no reduction was found after TBI (Supplementary Figure 1D).
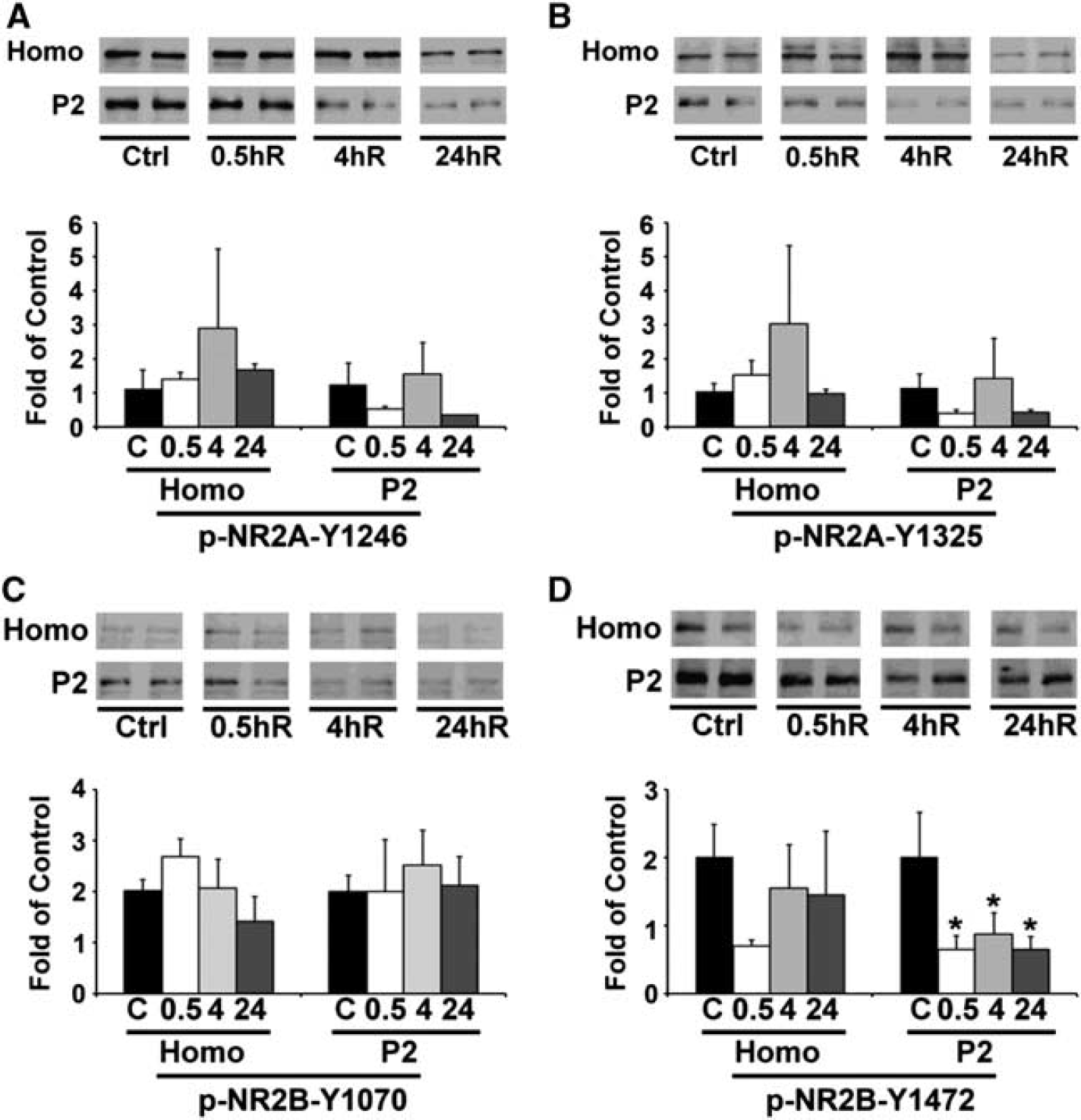
Western blot and quantitative analysis of phosphorylation status of NR2A and NR2B. Neocortical tissues were obtained from the ipsilateral regions of sham-operated control rats and rats subjected to traumatic brain injury (TBI) followed by 0.5, 4, and 24 hours recovery (n = 3). An equal amount of protein from each sample in homogenate (Homo) and synaptosomal (P2) fractions was subjected to SDS-PAGE and immunoblotted with antibodies against phospho-NR2A Y1246, phospho-NR2AY1325, and phospho-NR2B Y1070, phospho-NR2B Y1472. The phosphorylation changes of NR2A -Y1246 and -Y1325 (
Phosphorylation of synaptic scaffold protein PSD93 by SFKs has an important role in stabilizing the NR2s on the synaptic membrane. 18 Therefore, we investigated PSD93 tyrosine phosphorylation of Y340 by western blot after TBI. Figures 4A and B show that the total protein level of PSD93 was unchanged after TBI, but its phosphorylation level of Y340 was significantly decreased at 0.5 and 24 hours after TBI.
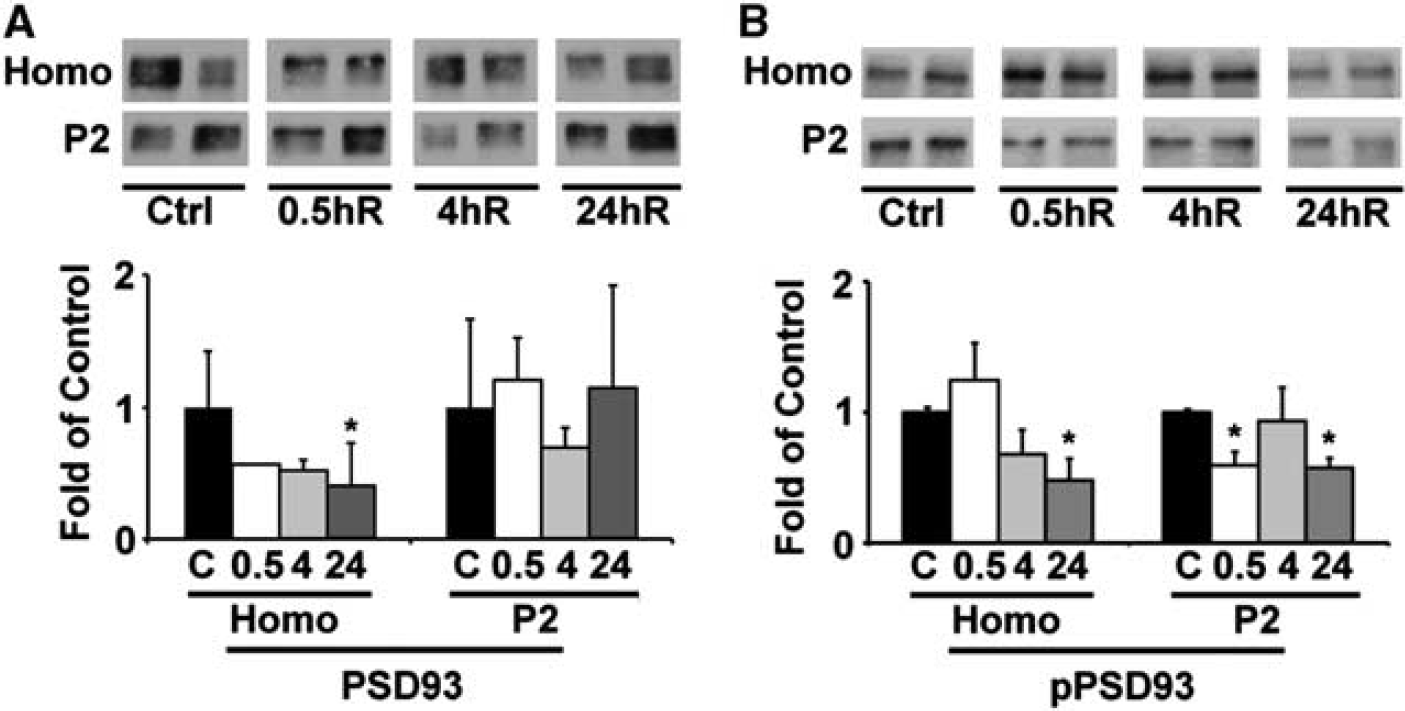
Western blot and quantitative analysis of the protein level of PSD93 and its phosphorylation status. Neocortical tissues were obtained from the ipsilateral regions of sham-operated control rats and rats subjected to traumatic brain injury (TBI) followed by 0.5, 4, and 24 hours recovery (n = 3). Equal amount of protein from each sample in homogenate (Homo) and synaptosomal (P2) fractions was subjected to SDS-PAGE and immunoblotted with antibodies against PSD93 and phospho-PSD93 Y340. The changes of phospho-PSD93 Y340 (
Changes in Src-Kinase Phosphorylation after Traumatic Brain Injury
Stabilization of NR2 subunits at the synaptic surface depends on the Src-kinase-mediated tyrosine phosphorylation of NR2 and PSD93, and dephosphorylation of NR2 triggers their internalization for degradation by the endocytotic lysosomal pathway. 19 Activation of Src kinase requires phosphorylation of the active loop Y416 or dephosphorylation of the inhibitory site Y527. 8 Therefore, we studied Src-kinase phosphorylation status after TBI. As shown in Figure 5, phosphorylation of Src Y416 was significantly downregulated (Figure 5A), whereas phosphorylation of Src Y527 was upregulated in the synaptic P2 fraction at 0.5, 4, and 24 hours after TBI (Figure 5B). In comparison, the total protein of Src was unchanged in the same fractions from the ipsilateral neocortical region, and the phosphorylation of Src Y416 and Y517 was not affected in the contralateral side after TBI. Thus, the data suggest that Src kinase was in its inhibited state in the ipsilateral neocortical area after TBI.
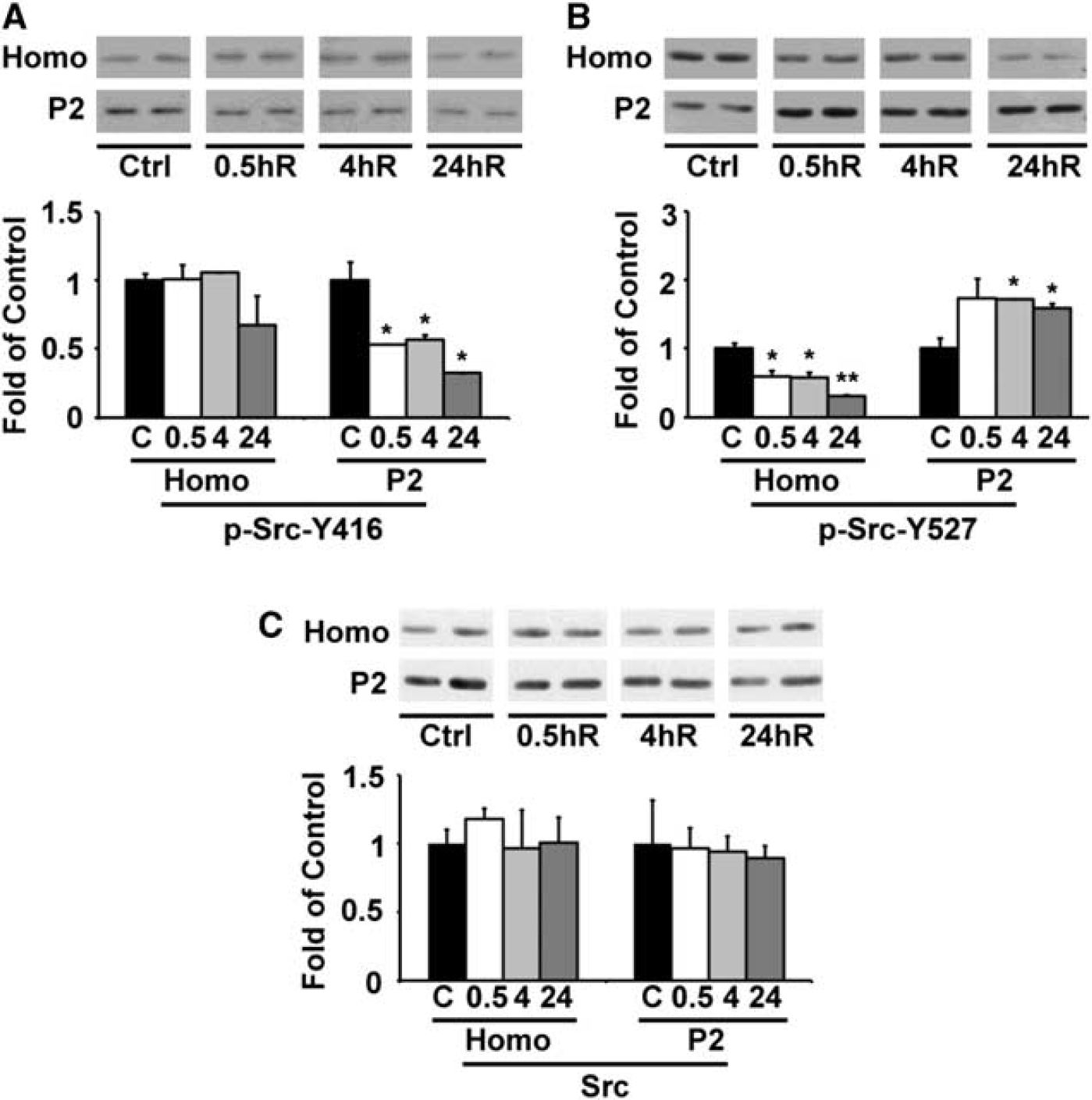
Western blot and quantitative analysis of the phospho-regulation of Src. Neocortical tissues were obtained from the ipsilateral regions of sham-operated control rats and rats subjected to traumatic brain injury TBI followed by 0.5, 4, and 24 hours recovery (n = 3). An equal amount of protein from each sample in homogenate (Homo) and synaptosomal (P2) fractions was subjected to SDS-PAGE and immunoblotted with antibodies against total protein Src, phospho-Src Y416, and phospho-Src Y527. The changes of phosphorylation of Src Y416 (
Confocal Microscopy of NR2B, Src, and PSD93 After Traumatic Brain Injury
To study cellular distribution of the observed proteins in immunoblotting, we conducted confocal microscopy of total PSD93 (green), phospho-PSD93 Y340 (green), total Src, phospho-Src Y527 (green), and total-NR2B (green), and they were co-labeled with propidium iodide (PI, red) in brain sections from sham-operated control rats and rats subjected to TBI followed by 24 hours of recovery. Confocal microscopy of PSD93 immunostaining showed synaptic labeling pattern (dots) in the neocortical region (Figures 6A–D) while Src immunostaining showed cell body and synaptic labeling pattern (Figures 6E–H). Relative to sham controls (Figures 6A and E), the immunostaining levels of total PSD93 and total Src were unchanged (Figures 6B and F), whereas the immunostaining level of phospho-PSD93 Y340 was reduced (Figure 6D) and the staining level of phospho-Src Y527 was increased (Figure 6H) in the ipsilateral neocortical areas at 24 hours after TBI. We were unable to obtain positive immunostaining of phospho-NR2B using brain sections although we have attempted several times. We noticed only a few damaged neurons in the ipsilateral neocortical areas that lost their immunoreactivities of both PSD93 and phospho-PSD93 at 24 hours after TBI (Figures 6B and D), which was consistent with many previous studies. 20 Confocal microscopy also showed that NR2B immunostaining was relatively evenly distributed in sham-operated control samples (Figure 6I), but appeared as dot staining in the ipsilateral neocortical areas at 24 hours after TBI (Figure 6J).
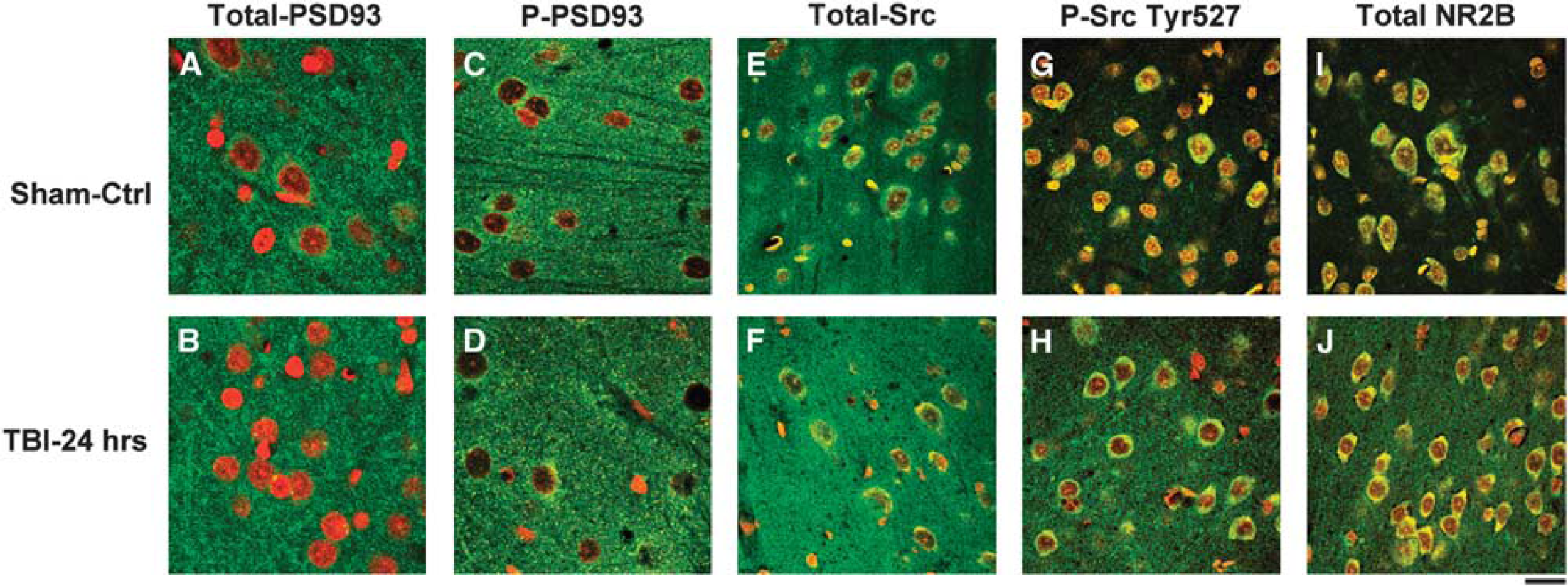
Confocal microscopic imaging of ipsilateral neocortical regions double-labeled with antibody against PSD, Src, NR2B, phospho-PSD93 Y340, or phospho-Src Y527 (right panel), (green), and PI (red) (
DISCUSSION
In this study, we investigated changes in the levels and phosphorylation status of NR2A and NR2B, PSD93, and Src kinase after TBI. We observed that NR2A and NR2B were significantly decreased in the ipsilateral neocortical homogenate and P3 fractions, and to a less degree in the synaptic P2 fractions after TBI. Downregulation of phosphorylation of NR2B Y1472 occurred much earlier and much more significantly than those of its protein levels after TBI. Similarly, TBI also lead to downregulation of phosphorylation of PSD93 Y340 where the total PSD93 protein level was less affected after TBI. Previous reports have identified that PSD93 mediates synaptic targeting of SFKs and tyrosine phosphorylation of NMDA receptors.18,20 Consistently, Src kinase-inhibitory phosphorylation site Y527 was significantly upregulated, whereas its activation site Y416 was downregulated, indicating that SFKs were significantly inhibited after TBI. These data suggest that TBI induces downregulation of Src kinase activity, which may lead to dephosphorylation of NR2B Y1472 and PSD93 Y340. Reduction of these Src-kinase phosphorylation sites leads to destabilization of NR2 localization at synaptic surface, and an increase in the endocytotic degradation of NMDA receptors. These findings may explain a regulatory mechanism that leads to a transient loss of NMDA receptors occurring after TBI.5,21
Changes in Src Kinase Activity After Traumatic Brain Injury
It is interesting to notice that ischemia leads to activation of Src kinase, 11 whereas TBI inhibits Src kinase activity as shown in this study. The underlying mechanism is unknown, but may be related to adenosine triphosphate (ATP) depletion. It is known that ATP is depleted within minutes to below 5% of the control level after cerebral ischemia, 11 whereas ATP level remains at above 60% after mild TBI and 50% after severe TBI in a rat controlled cortical contusion model. 22 Therefore, ATP depletion and depolarization-induced synaptic activation may lead to dramatic activation of Src kinases after ischemia. 11 The evidence suggests that postsynaptic dysfunction may be more severe after brain ischemia, whereas axonal injury is prominent after TBI. Therefore, neuroprotective strategy may be different between ischemic and TBI.
N-methyl-D-aspartate Receptors Synaptic Localization after Traumatic Brain Injury
NR2A, NR2B, Src kinase, and PSD93 are predominantly located in the postsynaptic densities in the central nervous system.23,24 Traumatic brain injury leads to a transient increase in extracellular glutamate level immediately after onset of TBI.21,25 An early study showed that the protein levels of the NR1, NR2A, and NR2B subunits were dramatically and transiently decreased at 6 to 12 hours, and completely recovered to the control level at 24 hours of recovery in a rat controlled cortical impact model. 26 Another study showed that NR1, NR2A, and NR2B were decreased in the cortex at 15 minutes after TBI in a mouse closed head injury model, but this study did not follow up on the receptor changes after 15 minutes. 5 The data of these studies, although they showed a similar trend in the alterations of NR2A and NR2B, are different from the data of the present study. First, the present study employed a rat lateral fluid-percussion injury model that may have resulted in relatively more mild physical damage than the same degree of injury produced by the controlled cortical impact and closed head injury models used in the previous studies. 27 Furthermore, fluid-percussion injury leads to cognitive dysfunction and thus it can be a useful model for posttraumatic dementia. 27 In contrast to the transient decrease shown in the previous studies, the present study shows progressive decreases in the protein levels of NR2A and NR2B, whereas the protein levels of NR1, PSD93, and β-actin, are not significantly affected during the post-TBI phase. The present study further shows that Src kinase activity and its phosphorylation sites of NR2B Y1472 and PSD93 Y340 are reduced more dramatically than their protein levels after TBI. Src-kinase-mediated NMDA receptor and PSD93 phosphorylation have a key role in NMDA receptor synaptic surface expression and stabilization.8,11,28 Therefore, our data strongly suggest that a decrease in Src-kinase-mediated phosphorylation may lead to destabilization of NMDA receptor synaptic surface localization and an increase in its endocytotic degradation after TBI.
Bigford et al, 29 showed that the NR2B protein level was transiently increased at 1, 4, and 8 hours in the ‘cell lysate’ after TBI, and then returned to the baseline thereafter. The results appeared different from the changes in NR2B shown in the present study. The reason for the discrepancy is unknown. A possible reason is that Bigford et al, 29 used 12,000 r.p.m. supernatant as the cell lysate for the immunoblotting of NR2B. It is unclear what centrifuge was used in that study. In comparison, the present study employed different subcellular fractions including the 10,000 g pellet (synaptic) fraction (P2) for the study of the NR2B levels. In addition, the brain sections of the cerebral cortex were used to prepare cell lysates in that study, whereas the present study used the contusion region for preparing subcellular fractions.
Dephosphorylation of NR2B Y1472 and PSD93 Y340 after Traumatic Brain Injury
The synaptic surface expression of NMDA receptors is a dynamic process. This process is regulated by SFKs via tyrosine phosphorylation of NR2 subunits. Src activity is regulated by tyrosine phosphorylation of its two sites Y416 and Y527, but with opposing effects. While phosphorylation at Y416 in the activation loop of Src kinase upregulates its catalytic activity, phosphorylation at Y527 in the carboxy-terminal tail renders the enzyme less active. Worthy of note, the Src Y527 and Y416 peptide antibodies (Cell Signaling Technology) used in this study may detect not only endogenous levels of Src, but also another SFKs member Fyn because SFKs member Src and Fyn share a similar or identical peptide sequences around the inhibitory and activation loop. Therefore, it is possible that both Src and Fyn kinases are inhibited after TBI.
The changes in Src activity after TBI may reflect a balance between Src kinase and the corresponding phosphatases. Changes in the tyrosine phosphatases may also have a role in the changes in the Src activity. For example, recent studies show that protein tyrosine phosphatase a is one of the enzymes that can dephosphorylate C-terminal tyrosine Y527 in chicken cellular Src and thereby increase its activity. 30 Knockout of protein tyrosine phosphatase α in mice leads to inhibition of the Src activity. Therefore, the Src activity may also be regulated by its phosphatases after TBI.
The number and subunit composition of synaptic NMDA receptors are regulated by stimulus-dependent receptor insertion and endocytosis via tyrosine phosphorylation.10,31–33 A well-studied example is NR2B Y1472 phosphorylation by SFKs facilitates NMDA receptor endocytosis via the endocytotic pathway. Downregulation of NR2B Y1472 phosphorylation in the endocytosis YEKL motif by Src or Fyn kinase increases the association of NR2B with AP-2 adaptor protein complex, thus promoting its endocytosis degradation.8,11,34 The present study shows that, relative to the NR2B protein level, NR2B Y1427 phosphorylation are decreased much earlier and much more dramatically after TBI. Concomitantly, Src activation loop Y416 is downregulated, while the inhibitory site Y527 is upregulated after TBI. Together, these data may suggest that NMDA receptor endocytosis followed by degradation may be increased via the Src/Fyn—NR2B Y1472 pathway after TBI.
In addition to NR2B Y1472, tyrosine phosphorylation of PSD93 Y340 also has an important role in the retention of NMDA receptor at synapse after TBI.28,35,36 NMDA receptors, SFKs, and PSD93 interact in the synapses, and PSD93 deletion markedly blocks the SFKs-mediated increase in tyrosine-phosphorylated NR2A and NR2B. 36 Tyrosine phosphorylation of PSD93 Y340 is critical for its interaction with and retention of NMDA receptors at the synapse. 28 The present study shows that PSD93 tyrosine phosphorylation is significantly reduced after TBI. The evidence suggests that reduction of tyrosine phosphorylation of PSD93 may also contribute to the reduced retention of NMDA receptors in synapse after TBI.
Changes in NR2A Y1246, NR2A Y1325, and NR2B Y1070 after Traumatic Brain Injury
In addition to NR2B Y1472, NR2B Y1070 as well as NR2A Y1246 and NR2A Y1325 are phosphorylated by SFKs in the central nervous system. 8 Relative to NR2B Y1472, dephosphorylation of NR2A Y1246, NR2A Y1325, and NR2B Y1070 is less significant after TBI. Recently we found that clustering of NR2A and NR2B to the postsynaptic density membrane from other cellular pools might be associated with upregulation of NR2A Y1246, NR2A Y1325, and NR2B Y1070 phosphorylation after brain ischemia. 11 Dephosphorylation of these tyrosine phosphorylation sites may contribute to the destabilization of NMDA receptors in synapses after TBI.
Potential Role of NR2 Tyrosine Phosphorylation After Traumatic Brain Injury
Evidence of, (i) increased extracellular levels of excitatory neurotransmitters and (ii) NMDA receptor antagonists protection against experimental TBI, have led to the prevalent excitotoxicity hypothesis. However, how neurons adapt to high extracellular excitatory neurotransmitters remains incompletely understood. This study provides evidence that neurons may reduce NMDA receptor density to cope with the increase in extracellular excitatory neurotransmitter after TBI. One such mechanism may be achieved by inhibition of SFKs to dephosphorylate NR2B Y1472 and to increase endocytosis of NMDA receptors after TBI. Therefore, dephosphorylation of NR2 subunits may offer a neuroprotective mechanism.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.