
Abstract
We investigated the occurrence of endotoxin (lipopolysaccharide, LPS) preconditioning in traumatic brain injury (TBI), evaluating the time window of LPS-induced protection, its persistence, and the associated molecular mechanisms. Mice received 0.1 mg/kg LPS or saline intraperitoneally and subsequently TBI (by controlled cortical impact brain injury) at various time intervals. Mice receiving LPS 3, 5, or 7 days before TBI showed attenuated motor deficits at 1 week after injury compared with mice receiving saline. Those receiving LPS 5 days before injury had also a reduced contusion volume (7.9 ± 1.3 versus 12 ± 2.3 mm3) and decreased cell death. One month after injury, the protective effect of LPS on contusion volume (14.5 ± 1.2 versus 18.2 ± 1.2 mm3) and neurologic function was still present. Traumatic brain injury increased glial fibrillary acidic protein, CD11b, CD68, tumor necrosis factor-α, interleukin (IL)-10, and IL-6 mRNA expression 24 hours after injury. Lipopolysaccharide administered 5 (but not 9) days before injury increased the expression of CD11b (233%) and of interferon β (500%) in uninjured mice, while it reduced the expression of CD68 (by 46%) and increased that of IL-6 (by 52%) in injured mice. Lipopolysaccharide preconditioning conferred a long-lasting neuroprotection after TBI, which was associated with a modulation of microglia/macrophages activity and cytokine production.
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. To date, 40% to 50% of moderate-to-severe TBI patients survive with residual sequelae resulting in enormous societal costs (Maas et al, 2008). In TBI, a primary biomechanical injury is followed by a secondary injury, which, in survivors to the initial insult, is a major determinant of brain damage and functional outcome. Posttraumatic cerebral inflammation is a pivotal component of the secondary injury and is characterized by glial activation, leukocyte recruitment, activation of complement system, and upregulation of inflammatory mediators such as cytokines and chemokines (Ziebell and Morganti-Kossmann, 2010). Although it was previously thought to be deleterious, neuroinflammation is now considered to have both beneficial and detrimental roles. Clear benefits can be achieved if inflammation is controlled in a regulated manner and for a defined period of time (Ziebell and Morganti-Kossmann, 2010).
Preconditioning, defined as a brief noninjurious stimulus that is able to protect the brain from a subsequent severe insult (Dirnagl et al, 2009; Marsh et al, 2009), has been widely studied in the setting of stroke. Modulation of the inflammatory response, as that induced by a preconditioning stimulus, is effective in inducing neuroprotection. In particular, endotoxin (lipopolysaccharide, LPS), a surface component of Gram-negative bacteria and a potent trigger of the innate immune response, when administered in low doses is able to confer neuroprotection against a subsequent ischemia (Stenzel-Poore et al, 2007). The rationale of investigating the field of preconditioning is to explore and understand the protective endogenous mechanisms that the brain has evolved to tolerate noxious stimuli or deficits in substrate delivery, with the goal of learning how to therapeutically modulate them. In the setting of TBI, preconditioning has been scarcely investigated so far. However, the observation that endogenous neuroprotection can be elicited by transient ischemia (Perez-Pinzon et al, 1999) or by heat acclimation before the injury (Shein et al, 2007, 2008; Umschwief et al, 2010) indicates that this may be a relevant mechanism to induce a protective phenotype in this condition too.
We tested the hypothesis that a low dose of LPS could attenuate the neurobehavioral sequelae and histological damage of a subsequent TBI. We observed that LPS acted as a preconditioning stimulus and attenuated the sequelae of TBI by modulating microglia/macrophages response to the injury and by increasing neuroprotective cytokines.
Materials and methods
Animals
Male C57Bl/6 mice (8 weeks old, 20 to 25 g, Harlan Laboratories, Bresso, Italy) were used. All procedures that were conducted conform the institutional guidelines that are in compliance with national (D.L. n.116, G.U. suppl. 40, 18 February 1992) and international laws and policies (EEC Council Directive 86/609, OJL 358,1; 12 December 1987; NIH Guide for the Care and Use of Laboratory Animals, US National Research Council 1996). The study was reviewed and approved by the Local Ethics Committee of the Mario Negri Institute. Before beginning any procedure, mice were housed for at least 1 week in their home cages at a constant temperature, with a 12-hour light-dark cycle, and
Lipopolysaccharide Administration
Mice were subjected to either intraperitoneal injection of LPS (from
Experimental Traumatic Brain Injury
Mice, anesthetized with sodium pentobarbital (65 mg/kg intraperitoneally), were placed in a stereotactic frame and subjected to craniectomy followed by induction of controlled cortical impact brain injury. This model uses a 3-mm rigid impactor driven by a pneumatic piston rigidly mounted at an angle of 20° from the vertical plane and applied perpendicularly to the exposed dura mater over the left parieto-temporal cortex, between bregma and lambda, at a velocity of 5 m/s and depth of deformation of 1 mm (Longhi et al, 2004, 2009; Ortolano et al, 2009). Sham-operated mice received identical anesthesia and surgery without brain injury. This model of controlled cortical impact brain injury is usually associated with no mortality (Longhi et al, 2004, 2009; Ortolano et al, 2009).
Experimental Design and Blinding
Mice were assigned to the following experimental groups: (1) mice receiving LPS or saline injection at different time points before TBI (1, 3, 5, 7, and 9 days interval) and killed 1 week after injury for evaluation of time window of LPS preconditioning on functional (Neuroscore) and histological outcomes,
Mice were allocated to surgery and treatment groups distributing surgery and treatment equally across cages and days. The intraperitoneal LPS/saline injections were performed by an investigator not involved in the surgery/outcome evaluations that assigned a code to each mouse. To minimize the variability, all surgeries were performed by the same investigator, blinded to the experimental groups. All subsequent behavioral, histological, and molecular evaluations were done by masked investigators.
Assessment of Neurologic Motor Function
Neurologic motor function was assessed by performing the composite Neuroscore and the Beam walk test. The Neuroscore evaluates animals scoring them from 4 (normal) to 0 (severely impaired) for each of the following indices: (1) forelimb function, (2) hindlimb function, and (3) resistance to lateral right and left pulsion as previously described (Longhi et al, 2004, 2009). The maximum score per animal is 12. The Beam walk test evaluates balance and coordination of movement (Fujimoto et al, 2004). The numbers of footfaults of the right hindlimb were counted, while the mice walked twice along a 5-mm wide, 1-m long (60 steps) beam.
Assessment of Cognitive Function
Evaluation of cognitive function was performed using the Morris water maze. Our Morris water maze is a pool (1 m diameter) that is filled with water (18°C to 20°C) made opaque by adding nontoxic water-soluble white paint. The learning task requires that animals learn how to locate a submerged platform placed 0.5 cm under the surface of the water using external visual cues. Latencies to reach and climb onto the platform are recorded for each trial, with a maximum of 60 seconds per trial. The learning task consists of 8 trials/day for 3 consecutive days for a total of 24 trials. The cognitive performance of all animals was obtained by averaging the latencies of 24 trials over 3 days (Longhi et al, 2004, 2009).
Tissue Processing
At 1 and 5 weeks after injury, mice were killed for histological analysis and brains removed and frozen as previously described (Longhi et al, 2009; Ortolano et al, 2009). Twenty-micron-thick serial sections were cut using a cryostat from bregma + 1 mm to bregma −4 mm. Finally, eight of the sections (bregma + 0.6, 0, −0.8, −1.5, −2.25, −2.65, −3.25, and −4 mm) were stained with neutral red (Neutral Red Sigma-Aldrich, St Louis, MO, USA).
Contusion Volume
Traumatic brain damage in the injured hemisphere was quantified at 1 and 5 weeks after injury by acquiring images on a computer and using the image analyzer Analytical Image System (Imaging Research Inc., Brock University, St Catharines, Ontario, Canada). The ipsilateral and contralateral hemispheres were manually outlined (Figure 1B). Subsequently, the injured area was calculated by subtracting the contralateral hemisphere minus the ipsilateral hemisphere. Finally, the contusion volume was calculated integrating the injured area according to the following formula:
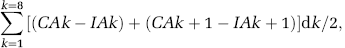
where
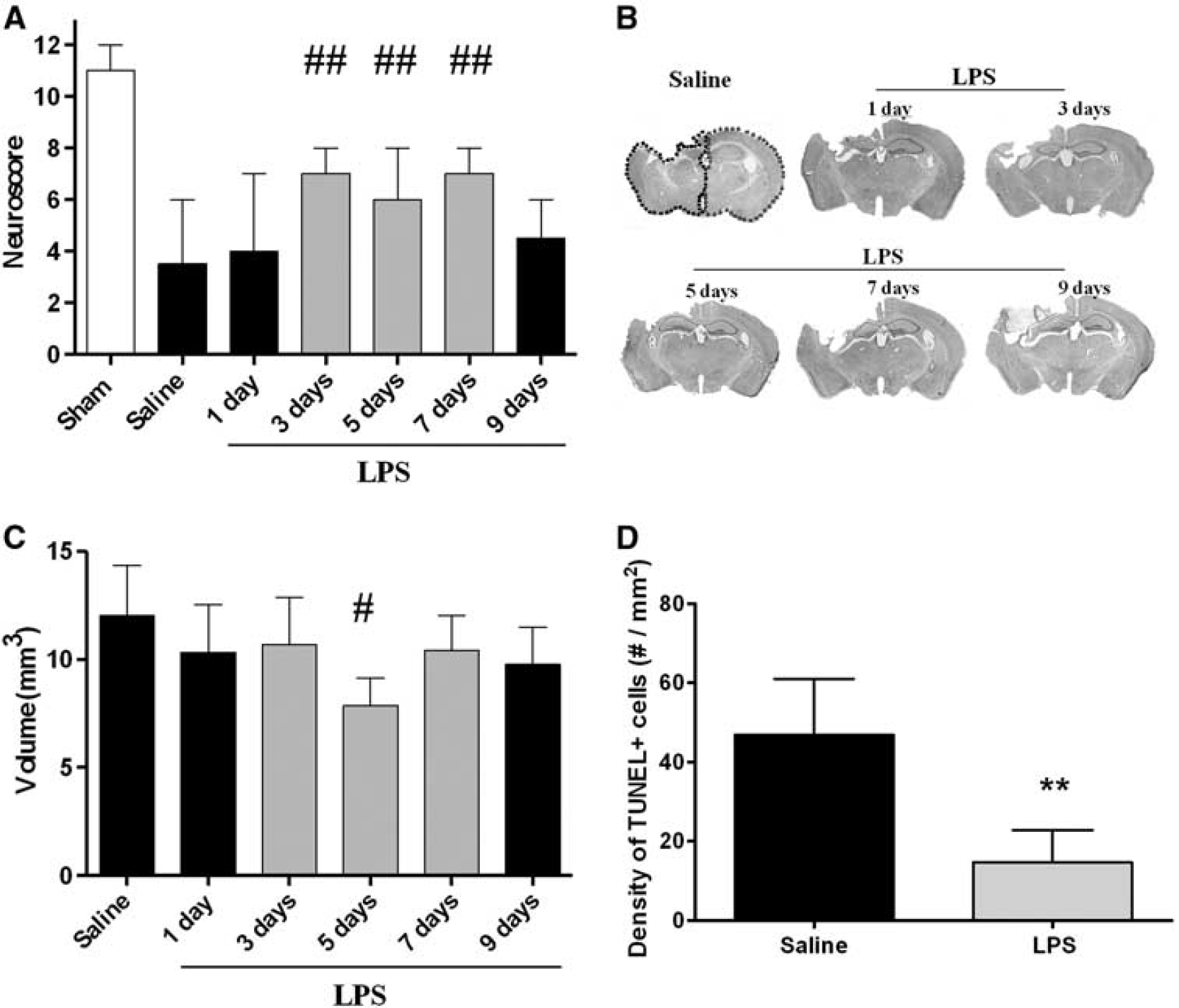
Time window of lipopolysaccharide (LPS) preconditioning. (
Transferase-Mediated dUTP Nick End Labeling Staining
To assess the presence of injured cells showing DNA damage, terminal deoxynucleotidyl TUNEL staining was performed on 20 µm sections by
RNA Isolation, cDNA Synthesis, and Real-Time Polymerase Chain Reaction
Twenty-four hours after TBI, mice were killed and ipsilateral cortices dissected for gene expression analysis as previously described (Storini et al, 2005; Troglio et al, 2004). Briefly, total RNA was obtained from tissue specimens using the Trizol reagent (Gibco BRL, Kidlington, MD, USA). Samples of total RNA (1.5 µg) were reverse transcripted with random hexamer primers using multiscribe reverse transcriptase (TaqMan reverse transcription reagents, Applied Biosystems, Foster City, CA, USA). Realtime polymerase chain reaction was conducted according to manufacturer's instructions by using a GeneAmp 5700 Sequence Detection System (Applied Biosystems) (Capone et al, 2007). The amplified transcripts were quantified using the comparative cycle threshold method (Applied Biosystems users bulletin #2). β-Actin was used as reference gene and relative gene expression levels were determined according to the manufacturer's ΔΔCt method (Applied Biosystems). Primers were designed using Primer Express 3.0 software (Applied Biosystems) (Table 1).
Primers used for semiquantitative real-time polymerase chain reaction analysis of β-actin, GFAP, CD11b, CD68, TNF, IFNβ, IL-6, and IL-10 transcript levels

GFAP, glial fibrillary acidic protein; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Forward.
Reverse.
Immunohistochemistry
Mice receiving saline or LPS injection 5 days before TBI were killed 1 week after injury. Immunohistochemistry was performed on 20 µm brain coronal sections using rat anti-CD11b (1:1000, kindly provided by Dr Doni (Capone et al, 2007), rat anti-CD68 (1:200, Serotec, Kidlington, UK) to measure microglia/macrophages activation and phagocytic activity, respectively. Positive CD11b and CD68 cells were stained by reaction with 3,3 diaminobenzidine tetrahydrochloride (Vector laboratories, Burlingame, CA, USA). For each reaction, adequate negative controls were performed. Three brain coronal sections per mouse at −0.8, −1.5, and −2.6 mm from bregma were used to quantify CD11b- and CD68-stained area. Thirty-three fields per mouse were acquired at × 40 magnification over a cortical region proximal to the lesion. An Olympus BX61 microscope equipped with a motorized stage and managed with AnalySIS software (Olympus) was used for image acquisition. Immunostained area for each marker was measured using ImageJ software (http://rsbweb.nih.gov/ij/) and expressed as positive pixels/total assessed pixels and indicated as staining percentage area for subsequent statistical analysis (Storini et al, 2005).
Statistical Analysis
Neuroscore is reported as median and the comparisons between different groups at each time point were performed using the nonparametric Kruskall-Wallis analysis of variance followed by Mann-Whitney
Results
Time Window of Lipopolysaccharide Preconditioning
Histopathology
At 1 week after injury, TBI induced a macroscopic area of cortical tissue loss extending rostro-caudally from bregma + 0.6 mm to bregma −4 mm with an associated shrinkage of the ipsilateral hippocampus. The core of the lesion was observed from bregma −1.5 mm to −2 mm (Figure 1B). Mice receiving LPS 5 days before TBI showed a contusion volume of 7.9 ± 1.3 mm3 that was significantly smaller compared with that of mice receiving saline (12 ± 2.3 mm3,
At 1 week after injury, we observed intense TUNEL-positive labeling in the injured cortex, suggestive of the presence of dying cells with damaged DNA. Quantification of TUNEL-positive labeling showed that mice receiving LPS 5 days before TBI had significantly reduced numbers of damaged cells compared with mice receiving saline (Figure 1D).
Long-Term Effects of Lipopolysaccharide Preconditioning
Based on the results obtained during the first set of experiments, a 5-day interval was chosen to assess the persistence of preconditioning protective effects on motor deficits and contusion volume and to determine its effects on cognitive performance.
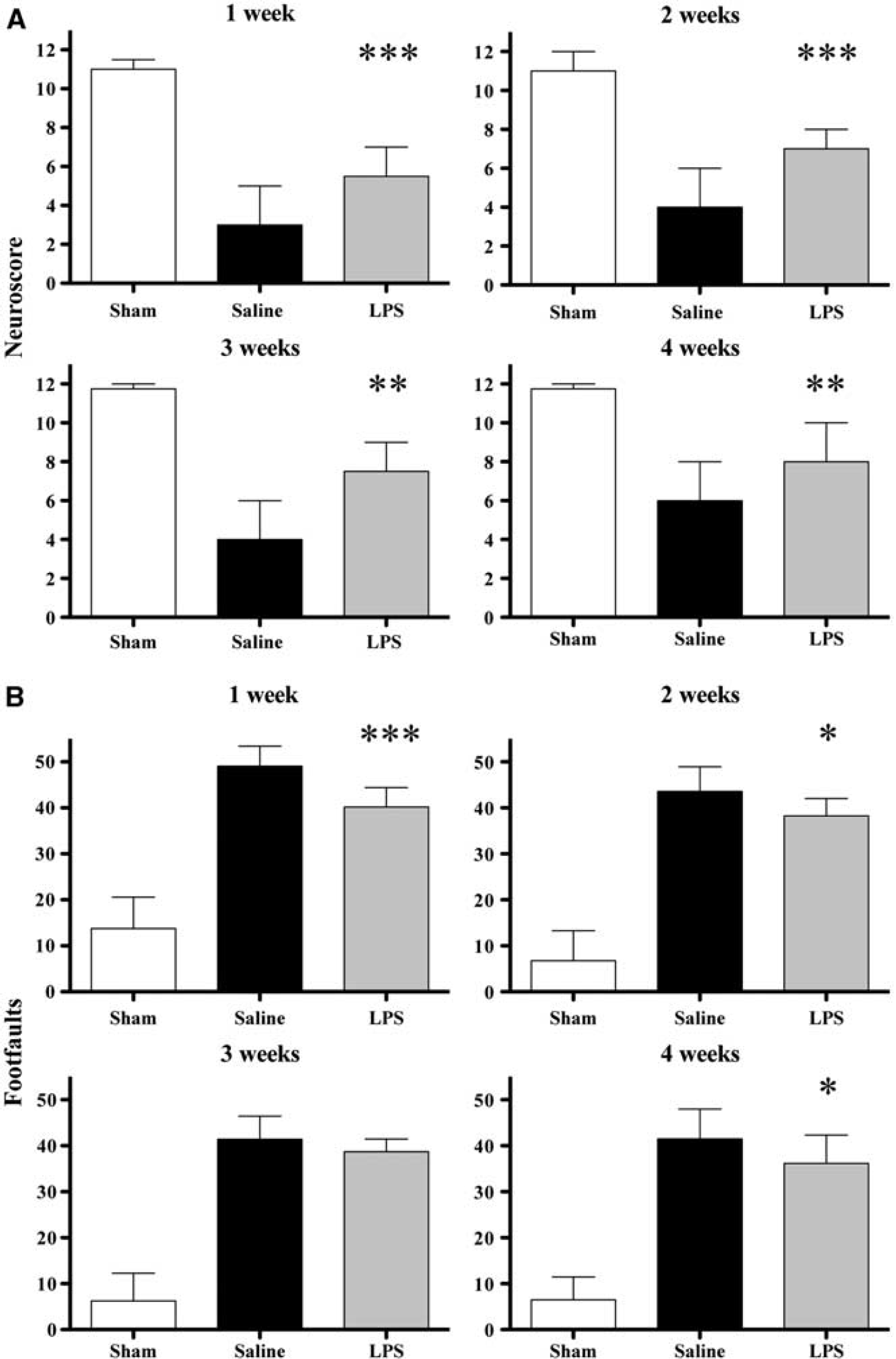
Long-term effects of lipopolysaccharide (LPS) preconditioning. Neuromotor function over 4 weeks after injury after either saline or LPS administration 5 days before traumatic brain injury (TBI). (
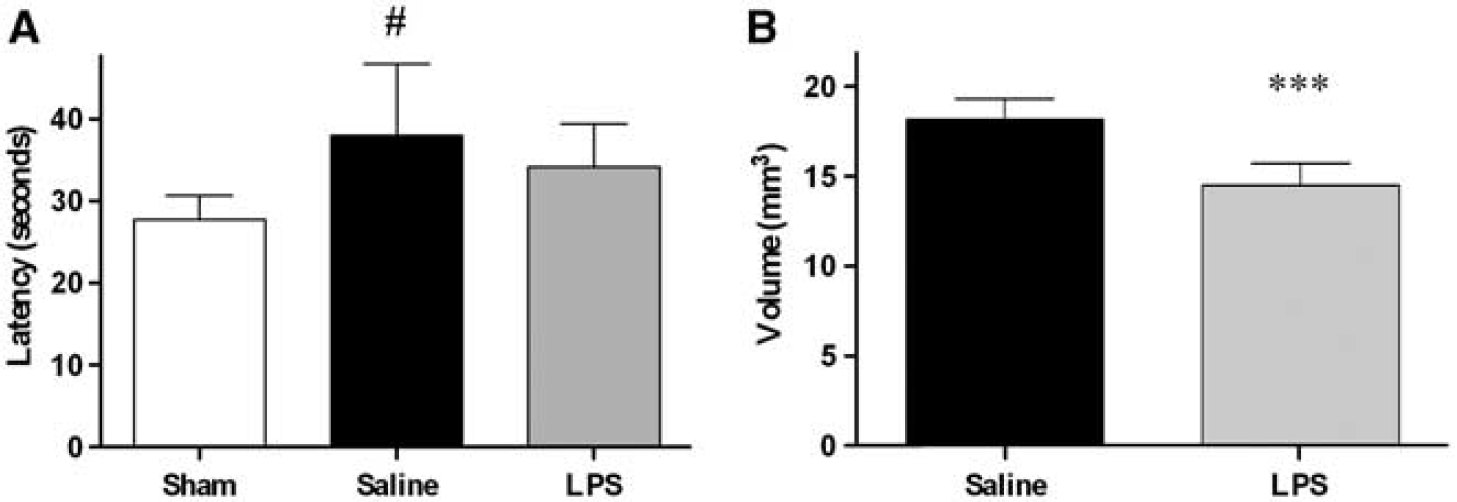
Long-term effects of lipopolysaccharide (LPS) preconditioning. (
Gene Expression
To relate molecular changes with behavioral results, gene expression analysis was evaluated 24 hours after TBI in mice receiving LPS at 5 days before injury, an interval associated with protection and at 9 days before injury, an interval that was not associated with protection. We analyzed the mRNA expression of few genes known to be modulated by acute brain injury and/or preconditioning (Marsh and Stenzel-Poore, 2008; McIntosh et al, 1998): glial fibrillary acidic protein, a marker for astrocytes, CD11b for microglia/macrophages, and CD68 for phagocytic microglia/macrophages, and that of inflammatory cytokines tumor necrosis factor (TNF)-α, interferon (IFN)β, interleukin (IL)-10, and IL-6. Traumatic brain injury significantly increased the mRNA expression of CD11b, CD68, and of IL-6 as well as that of glial fibrillary acidic protein, TNF-α and IL-10 compared with uninjured mice. When LPS was administered 5 days before TBI, it induced a significant increase in the expression of CD11b and IFNβ in uninjured mice and a significant decrease of CD68 and an increase of IL-6 mRNA expression in injured mice. Furthermore, the overall significant interaction (analysis of variance
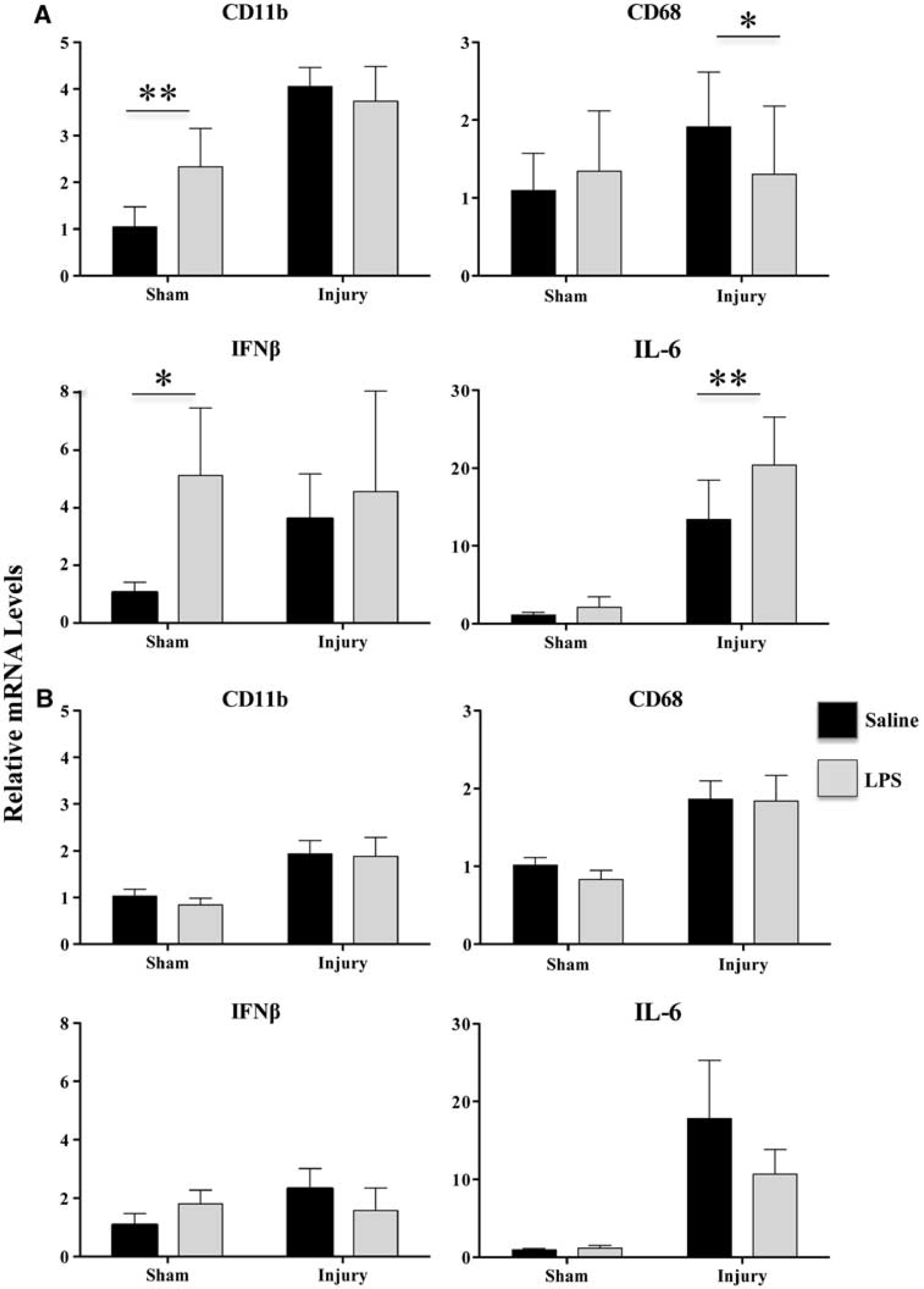
Gene expression. (
Immunohistochemistry
At 1 week after injury, mice receiving LPS or saline treatment 5 days before injury displayed a similar level of CD11b immunoreactivity (Figures 5A and 5B). In contrast, injured mice receiving LPS displayed a reduced level of CD68 immunoreactivity compared with those receiving saline (Figures 5C and 5D). These findings confirm gene expression data.
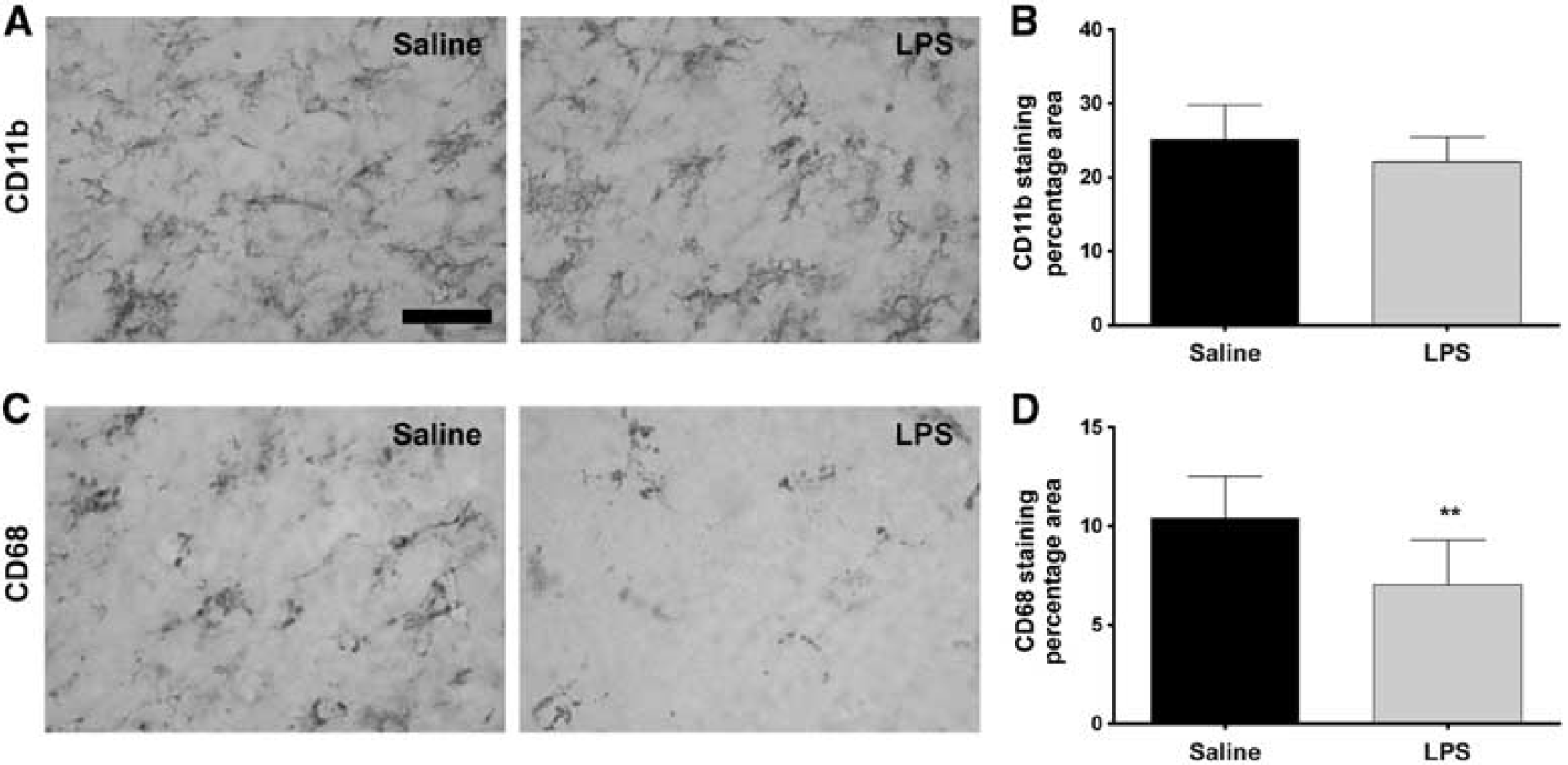
Immunohistochemistry. Representative micrographs of CD11b (
Discussion
This study shows that pretreatment with a low dose of LPS acts as a preconditioning stimulus and attenuates the neurobehavioral sequelae and histological damage of TBI. Notably, preconditioning-induced protective effects were robust and persisted up to 1 month after injury. Exposure to LPS was followed by an increase in IFNβ and an early modulation of microglial/macrophages activity. This resulted in a decrease of phagocytic activity and increase in IL-6 gene expression in protected mice.
Preconditioning is a phenomenon whereby a subthreshold insult is applied to the brain to activate cellular pathways that can help attenuating damage due to a subsequent severe injury (Dirnagl et al, 2003; Gidday, 2006; Pignataro et al, 2009). In the setting of ischemia, systemic administration of a low dose of LPS has been shown to confer robust attenuation of infarct volume after transient middle cerebral artery occlusion (Rosenzweig et al, 2004, 2007). To investigate the mechanisms involved in preconditioning in TBI, we first tested the hypothesis that TBI could be a condition susceptible to LPS-induced protection and explored the time window of such protection using functional and histological outcomes at 1 week after injury. We observed an attenuation of functional deficits when LPS was administered 3, 5, and 7 days before TBI. Notably, the 5-day interval was the best time interval in our model, as it was associated with both neurologic and histological recovery. Protection was not present at 1 day and was lost at 9 days, confirming the hypothesis that LPS-associated neuroprotection is a transient phenomenon that requires time to develop and disappears over time, suggestive of a ‘delayed tolerance’ that requires new gene expression and protein synthesis (Gidday, 2006; Stenzel-Poore et al, 2007).
We then evaluated whether the protection afforded by LPS was long lasting. We chose the 5-day interval and observed that the improvements of functional and histological outcomes were sustained up to 1 month after injury, indicating that LPS preconditioning confers a sustained neuroprotection, rather than delaying degenerative processes of secondary injury after TBI. To our best knowledge, this is the first study documenting a robust and sustained long-lasting behavioral and histological improvement associated with LPS preconditioning after acute brain injury (Ahmed et al, 2000; Bastide et al, 2003; Dawson et al, 1999; Furuya et al, 2005; Kunz et al, 2007; Rosenzweig et al, 2004, 2007).
Notably, we as well as earlier studies addressing preconditioning in TBI (Perez-Pinzon et al, 1999; Shein et al, 2007, 2008; Umschwief et al, 2010) have been using models of cerebral contusions in which a core lesion is surrounded by an area (traumatic penumbra) of reduced cerebral blood flow (Assaf et al, 1999; Kochanek et al, 2002; Yamakami and McIntosh, 1989). We do not know whether the target of preconditioning neuroprotection after TBI is the area around the contusion characterized by reduced cerebral blood flow and thus similarly to ischemic tissue. As TBI in humans is a heterogeneous disease associated also with diffuse axonal injury, it would be important to test preconditioning using also models of axonal injury.
In clinical practice, the cumulative deleterious effects of repetitive mild TBI are well known. We have previously shown that after a single mild TBI, there is a time window of vulnerability to a second mild TBI that lasts up to 5 days and leads to functional sequelae and increased histological damage (Longhi et al, 2005). To date, we have not been able to show that mild TBI induces tolerance to a subsequent TBI or crosstolerance to another insult. A difference between TBI and LPS (as preconditioning stimulus) might depend on the fact that, by definition, a preconditioning stimulus is subthreshold. In our previous study, the occurrence of a mild TBI was associated with brain damage indicating a stimulus above the threshold of damage.
Systemic LPS pretreatment has been shown to induce a genetic reprogramming of the response to ischemia by modulating the inflammatory response. Relevant data come from Marsh
We also evaluated the expression level of IL-6, a pleiotropic cytokine that has been shown to be neuroprotective in several types of brain injury including stroke and TBI (Morganti-Kossmann et al, 2002; Suzuki et al, 2009; Ziebell and Morganti-Kossmann, 2010). Interleukin-6 may actually attenuate oxidative damage, exert antiapoptotic actions, and participate in brain repair processes (Penkowa et al, 1999; Stahel et al, 2000). We found that LPS-preconditioned mice at 5 days interval had increased IL-6 mRNA expression after TBI compared with saline-treated mice, supporting the hypothesis that stimulation of production of neuroprotective factors (IFNβ and IL-6) is part of the mechanisms by which LPS preconditioning confers neuroprotection.
We also observed that LPS preconditioning modulated the microglia/macrophage phenotype. Actually, preconditioned uninjured mice showed increased mRNA expression of CD11b, while preconditioned injured mice showed a significant reduction of CD68 compared with nonpreconditioned mice. These findings were further confirmed by immunohistochemical analysis of protein expression. We can thus hypothesize that LPS preconditioning causes a ‘priming’ of microglia/macrophage that can ultimately lead to a reduction of phagocytic activity in protected mice.
Interestingly, the changes in gene expression that we could observe in protected mice, i.e., in TBI mice receiving LPS 5 days before injury, were not present in mice receiving LPS 9 days before injury, a treatment that did not elicit to either functional or histological protection. These data indicate that LPS induced a transient ‘protective molecular phenotype’ that vanished over time.
It should be acknowledged that we analyzed the inflammatory genes only at a single time point after TBI; therefore, we cannot exclude that relevant changes in other molecules may have occurred at earlier time points. This may be true in particular for TNF-α, whose modulation has a major role in preconditioning (Nawashiro et al, 1997; Rosenzweig et al, 2004). Indeed, LPS preconditioning induced a transient upregulation of circulating TNF-α before cerebral ischemia and a reduction of circulating TNF-α (coupled with reduced neutrophilia and monocyte activation) after ischemia (Rosenzweig et al, 2004, 2007). Importantly, mice lacking TNF are not protected by LPS preconditioning (Rosenzweig et al, 2007). Thus, it is at all possible that also in case of TBI the preconditioning action of LPS, which is an effective inducer of TNF-α synthesis, is mediated by this cytokine. Future work needs to be performed to specifically address this point.
The experimental approach of preconditioning has interesting potential clinical applications. First, it allows the identification of endogenous protective/regenerative mechanisms that the brain has evolved to tolerate noxious stimuli such as injury, hypoxia/ischemia, seizures, infections, etc. It is conceivable that once these pathways have been clarified, their potentiation after the occurrence of brain injury may be used as therapeutic tool (Dirnagl et al, 2009). Interestingly, in the setting of TBI, all monodrugs approaches have failed when translated from a laboratory setting to the clinical stage. It has been advocated that therapies with multipotential effects might represent a new promising strategy for TBI (Faden and Stoica, 2007) and preconditioning-based strategies might have the potential advantage of modulating different aspects of the after injury cascades. Possible therapies associated with preconditioning include cytokine administration, induction of immunological tolerance, and remote conditioning, which has recently been tested with positive results in patients with acute myocardial infarction (Botker et al, 2010). An additional relevant application to the clinical practice is the implementation of preconditioning to induce protection in those people that are at known risk of brain injury, i.e., people engaged in contact sports/soldiers that are at high risk of TBI (Shein et al, 2007).
In conclusion, we have shown that low dose of LPS acts as preconditioning stimulus and confers a robust and long-lasting neuroprotection toward a subsequent TBI leading to modulation of microglia/macrophages action and increased production of protective cytokines. In spite of the different pathogenetic pathways involved in stroke and TBI, our present data and those on ischemic preconditioning suggest that LPS may induce similar pathways of protection and help tease out the relevant endogenous protective pathways that may be targeted by novel therapeutic strategies in acute brain injury.
Footnotes
Acknowledgements
The authors thank Tracy McIntosh, PhD, for his valuable advice.
The authors declare no conflict of interest.