
Abstract
There is now extensive evidence to show that the cytokine interleukin-1 (IL-1) contributes directly to reversible and permanent ischemic brain damage in rodents. Because interleukin-18 (IL-18) shares many structural and functional similarities with IL-1, the authors tested the hypothesis that IL-18 contributes directly to ischemic brain damage in mice exposed to focal, reversible (15-minute or 30-minute) middle cerebral artery occlusion. IL-18 expression was not induced acutely by middle cerebral artery occlusion, and deletion of the IL-18 gene (IL-18 knockout mice) did not affect infarct volume. The present results suggest that IL-18 does not contribute to acute ischemic brain damage.
Interleukin-1 (IL-1) is a prominent proinflammatory cytokine that is strongly expressed in acute and chronic brain injury, such as brain trauma, focal cerebral ischemia, and Alzheimer disease (for reviews see Allan and Rothwell 2001; Touzani et al., 1999). Administration of IL-1 exacerbates experimentally induced ischemic brain damage, whereas acute inhibition of IL-1, by administration of IL-1 receptor antagonist or blocking antibodies, reduces infarct size in rodents by ∼50% (Touzani et al., 1999). We have also shown a reduction in infarct volume (∼80%) in response to focal cerebral ischemia in mice lacking both IL-1α and IL-1β (Boutin et al., 2001).
Interleukin-18 (IL-18) was originally named IL-1γ because of its sequence homology with IL-1β. IL-18 shares several features with IL-1β, including similar three-dimensional conformations, and both are synthesized as inactive precursors that lack a signaling sequence and are cleaved by caspase-1 (Fantuzzi and Dinarello, 1999). In addition, IL-18 and IL-1 bind to related heterodimeric receptor complexes to activate common signaling pathways (Fitzgerald and O'Neill, 2000). Interleukin-18 is a key mediator of peripheral inflammation and host defense responses (for review, see Nakanishi et al., 2001), and although its role in the brain is not clear, there is evidence that IL-18 may be involved in central inflammatory responses. Interleukin-18 messenger RNA (mRNA) is expressed constitutively in the brain and is upregulated during the acute phase of experimental autoimmune encephalomyelitis or neuritis in rats (Culhane et al., 1998; Jander and Stoll 1998, 2001). Furthermore, IL-18 protein expression is increased in activated microglia/macrophages during Wallerian degeneration after nerve crush in rats, in response to influenza virus infection in mice, and in the late stages (3 to 6 d) of focal cerebral ischemia in rats (Jander et al., 2002; Menge et al., 2001; Mori et al., 2001).
The aim of the present study was to test the hypothesis that IL-18, like IL-1, contributes to ischemic damage. We compared the severity of focal cerebral ischemia in wild-type (WT) and IL-18–deficient mice and examined the effect of this insult on IL-18 expression.
MATERIALS AND METHODS
Mice
Adult, male mice, in which the IL-18 gene had been deleted by homologous recombination (Takeda et al., 1998), were raised on C57BL/6 background and bred in the local animal facilities. Genotyping was achieved by polymerase chain reaction analysis. All knockout (KO) mice were born healthy, and no major differences in growth or weight between the IL-18–KO and WT mice were found (Takeda et al., 1998, and present study). C57BL/6 WT mice were supplied by Harlan Olac (U.K.). The animals were housed in a controlled environment of 12-h light/dark cycles (08:00/20:00 h) at 22°C. All experiments were performed in accordance with U.K. legislation under the 1986 Animals (Scientific Procedures) Act.
Surgical procedures
The animals were studied at 3 to 6 months of age and weighed between 22 and 31 g at the time of use. Anesthesia was induced by inhalation of 4% halothane in a NO2/O2 (70%/30%) mixture and maintained by inhalation of approximately 1.5% to 2% halothane in a NO2/O2 (70%/30%) mixture. Body temperature was monitored throughout surgery (via rectal probe), and animals were maintained normothermic (37.2°C ± 0.2°C) by a heating blanket (Homeothermic Blanket Control Unit; Harvard Apparatus Ltd., Edenbridge, Kent, U.K.).
Temporary middle cerebral artery occlusion
Laser Doppler flowmetry (Moor Instruments Ltd., Axminster, U.K.) was used to monitor cerebral blood flow (CBF) during and after middle cerebral artery occlusion (MCAO). A small incision was made in the skin overlying the temporal muscle, and a 0.7-mm flexible laser-Doppler probe (model P10) was positioned on the superior portion of the temporal bone (6-mm lateral and 2-mm posterior from bregma) and secured with glue. This position corresponded to the center of the ischemic territory (Connolly et al., 1996a, b ).
Focal cerebral ischemia was induced by occlusion of the right middle cerebral artery (MCA) using the intraluminal filament technique (Clark et al., 1997) using modifications that have been described previously (Boutin et al., 2001). Fifteen or 30 minutes after induction of MCAO, the occluding filament was gently withdrawn back into common carotid to allow reperfusion. In sham-operated mice, the same surgical procedure was carried out, except that the filament was not advanced to occlude the MCA. Mice subjected to 15-minute MCAO were maintained under anesthesia until reperfusion. Mice exposed to 30-minute MCAO were allowed to recover consciousness after MCAO to reduce time under anesthesia and were reanesthetized for reperfusion. CBF was monitored to ensure that MCAO was effective.
Measurement of infarct volume
Twenty-four hours after MCAO, mice were killed by anesthetic overdose with halothane and decapitated. Brains were removed and frozen in cooled (−40°C) isopentane. Frozen brains were cut by cryostat and coronal brain sections (20-μm sections at 300-μm intervals) were stained with cresyl fast violet to identify viable tissue. Blind analysis was performed in which infarcted areas were delineated by the relative paleness of histologic staining in the ischemic tissue. Infarct volumes were calculated by the integration of infarcted areas on each brain slice, using a computer-assisted image analyzer (SigmaScan 5.0; SPPS Inc., Chicago, IL, U.S.A.). To correct for the effect of edema, the total infarcted area was also determined indirectly by subtracting the area of the healthy tissue in the ipsilateral hemisphere from the area of the contralateral hemisphere on each section. Edema was then estimated by the difference between volumes corrected and not corrected for edema (Lin et al., 1993; Osborne et al., 1987).
Detection of immunoreactive interleukin-18
Animals were killed 4 or 24 h after MCAO induction and perfused with diethyl pyrocarbonate–saline by cardiac puncture. The brains were removed, the frontal and occipital lobes were removed, and the hemispheres were separated, snap-frozen, and stored at −80°C until use. Samples were homogenized in phosphate-buffered saline (PBS) containing protease inhibitor cocktail (0.2-mmol/L AEBSF, 1-μg/mL aprotinin, 1-mmol/L benzamide, 1-mmol/L ethylenediamine tetraacetic acid, 10-μg/mL leupeptin, 10-μg/mL pepstatin in 1× PBS; 50 μL/mg tissue), centrifuged at 14,000g (20 minutes), and the supernatant was harvested.
Interleukin-18 protein was measured in diluted samples using a commercial mouse IL-18 enzyme-linked immunosorbent assay (ELISA; MBL Co. Ltd., Nagoya, Japan) that had greater affinity for mature IL-18 than the inactive proform of IL-18 (Wheeler et al., in press), according to the manufacturer's instructions.
For Western blot analysis, proteins (40-μg samples) were resolved on 12% sodium dodecyl sulfate–polyacrylamide gels and transferred to a nitrocellulose membrane. Nonspecific binding was blocked by incubating the membrane in 10% low-fat milk/0.02% Tween/PBS for 1 h at room temperature. The membranes were then probed with goat, anti–mouse IL-18 polyclonal antibody (1/500; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), or rabbit anti-actin (1/10,000; Sigma-Aldrich Company Ltd., Gillingham, U.K.) in 5% low-fat milk/0.02% Tween/PBS overnight at 4°C, followed by the appropriate horseradish peroxidase–conjugated secondary antibody (1/4,000; Autogen Bioclear U.K. Ltd., Calne, U.K.) in 5% low-fat milk/0.02% Tween/PBS for 1 h at room temperature. Proteins were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, U.K.).
Statistical analyses
All data are presented as mean ± SD. In order to determine differences between groups, unpaired Student t-tests were performed to compare infarct volumes and CBF. ELISA results were analyzed by one-way ANOVA with Tukey post hoc test. Western blot results were quantified by densitometry (Northern Eclipse version 6.0; Empix Imaging Inc., North Tonawanda, NY, U.S.A.) and analyzed by one-way ANOVA with Tukey post hoc test.
RESULTS
Body weight and rectal temperature were not statistically different between WT and IL-18–KO mice (body weight, 15-minute MCAO groups: 28.5 ± 2.2 g vs. 28.1 ± 1.7 g; 30-minute MCAO groups: 26.4 ± 2.2 g vs. 24.8 ± 1.0; core temperature, 15-minute MCAO: 37.1°C ± 0.2°C vs. 37.2°C ± 0.2°C; 30-minute MCAO: 37.1°C ± 0.1°C vs. 37.1°C ± 0.2°C, WT vs. IL-18 KO respectively). CBF measurement ensured that focal cerebral ischemia was of similar severity in both strains (reduction in CBF measured by laser Doppler expressed as a percentage of the baseline: −87% ± 6% vs. −88% ± 3% in 15-minute MCAO groups and −81.7% ± 8.7% vs. −75.3% ± 13.5% in 30-minute MCAO groups, respectively). Reperfusion was monitored after 15-minute MCAO and was similar in both strains (expressed as percentage of baseline value; 73% ± 17% in WT and 78% ± 22% in IL-18–KO mice). Fifteen or 30 minutes of MCAO was chosen to induce moderate or severe focal cerebral ischemia based on our previous studies in WT and various KO animals (Boutin et al., 2001; Boutin and Rothwell 2001; Touzani et al., 2002). In the present study, and as we previously observed, ischemic brain damage was limited to the striatum and piriform cortex after 15 minutes of MCAO, whereas 30-minute MCAO induced much larger damage including striatum and piriform, frontoparietal and temporal cortical areas. Analysis of infarct volume showed that there were no significant differences between WT and IL-18–KO mice after either moderate (15-minute) or severe (30-minute) focal cerebral ischemia (Fig. 1). Edema volumes were not statistically different between WT and IL-18 KO (15-minute MCAO: 2.15 ± 2.11 mm3 vs. 2.64 ± 1.33 mm3 and 30-minute MCAO: 20.3 ± 16.6 mm3 vs. 19.2 ± 14.9 mm3, respectively).
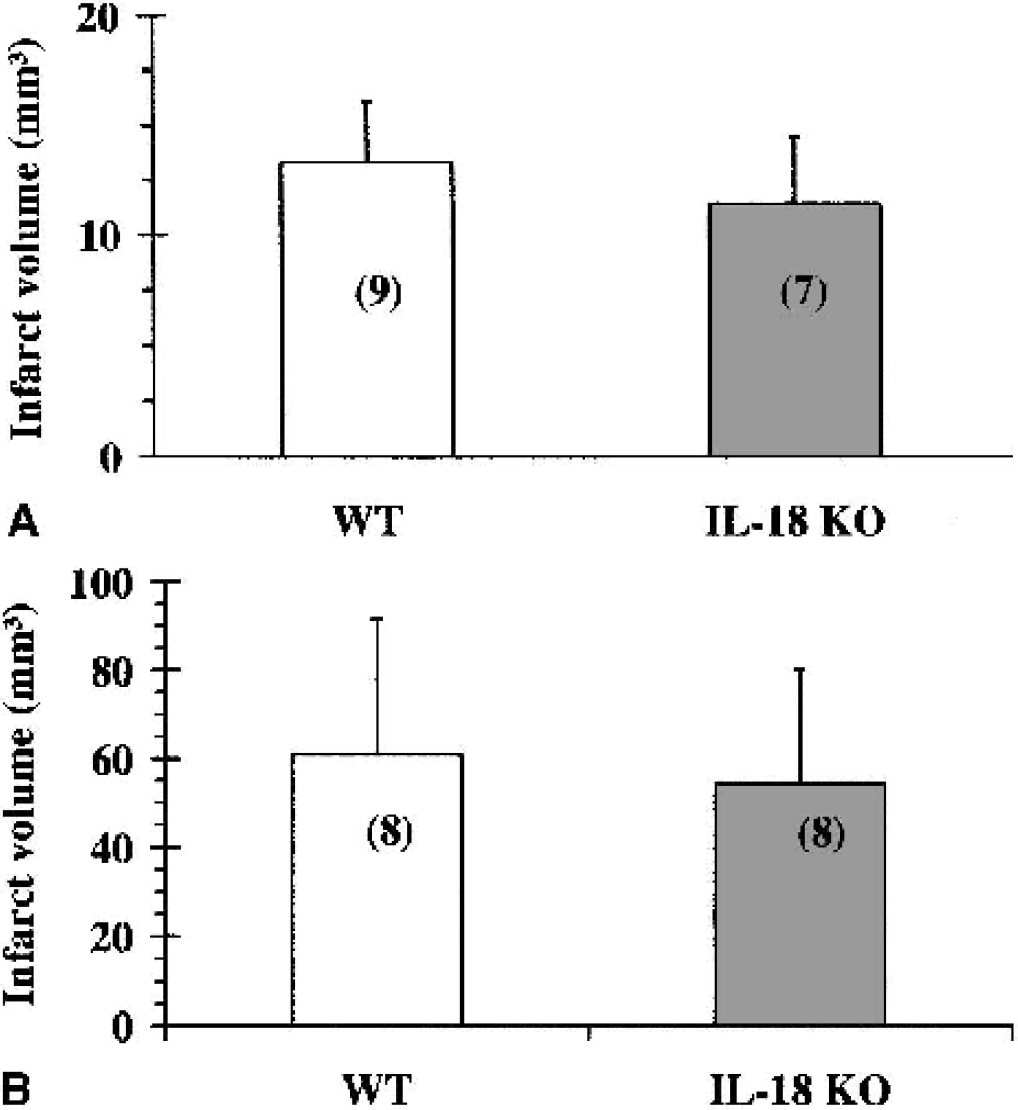
Total infarct volume in IL-18–KO mice compared with WT mice, after 15-minute
In WT mice, IL-18 protein expression was significantly reduced in the ipsilateral hemisphere 24 h after MCAO compared with sham-operated animals (2.2 ± 0.5 pg/mg and 3.3 ± 0.5 pg/mg, respectively, P < 0.05), but neither the 4-h nor 24-h MCAO group was significantly different from naive animals (Fig. 2A). There was no significant difference between any of the other treatment groups. To distinguish between the pro- and mature forms of IL-18, brain samples were analyzed by Western blot. In all the samples tested, only proIL-18 was detected (Fig. 2B), though the antibody used detected mature IL-18 loaded as a control. Quantification of these Western blot results revealed no significant differences between any of the treatment groups (Fig. 2C).
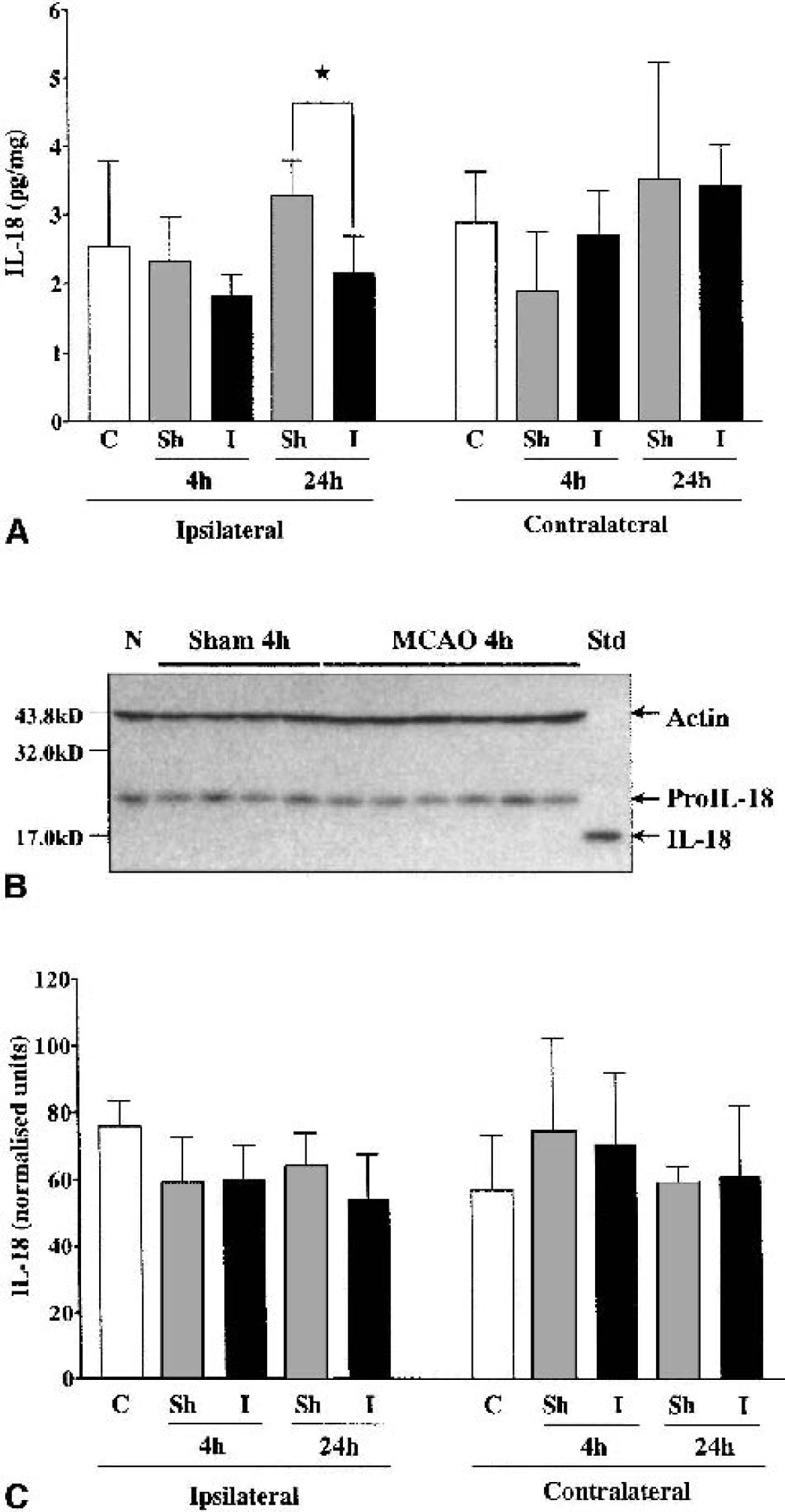
DISCUSSION
The present results show that deletion of the IL-18 gene did not influence infarct size 24 h after 15-minute (moderate) or 30-minute (severe) focal cerebral ischemia in mice. However, a significant reduction in IL-18 protein expression was observed in the ipsilateral hemisphere 24 h after MCAO, compared with sham-operated animals. This is unlikely to be physiologically relevant because IL-18 levels between either treatment group and naive animals were not significantly different. In addition, the levels of IL-18 detected by ELISA were very low, and Western blot analysis revealed only the precursor form of IL-18. Quantification of the Western blots did not reveal significant differences between any of the treatment groups. It is possible that mature IL-18 was present (but at levels below the detection limit of Western blot analysis), or that mature IL-18 was rapidly degraded, or both.
Overall, these results suggest that IL-18 plays little or no role in acute ischemic processes. A recent report suggests that immunoreactive IL-18 levels correlate with the localization of phagocytic microglia or infiltrated macrophages in brain parenchyma at a later stage (between day 3 and 6) after focal cerebral ischemia in rats (Jander et al., 2002). Similarly, Hedtjärn et al. (2002) have also shown that IL-18 is induced later (12 h to 14 d) after hypoxic–ischemic brain injury in neonatal rats, and that infarct volume and neuropathologic score are moderately reduced in IL-18–deficient mice after hypoxic–ischemic injury. A further study (Yatsiv et al., 2002) reported neuroprotective effects of IL-18 binding protein after experimental closed head injury. Thus, injury measured later after MCAO in IL-18–KO mice may have been altered compared with WT animals. It was not possible to study delayed infarction in these mice owing to restrictions under the U.K. Animals (Scientific Procedures) Act. The published results, together with those presented here, suggest that IL-18 could contribute to delayed neuronal injury and/or repair, whereas IL-1 is induced early after injury (Jander et al., 2002; Liu et al., 1993; Zhang et al., 1998; for review, see Touzani et al., 1999) and contributes to acute neurodegeneration (Boutin et al., 2001; Garcia et al., 1995; Relton and Rothwell 1992; for review, see Touzani et al., 1999).
The data presented here suggest that IL-18 is not required for the development of ischemic brain damage after temporary MCAO and that IL-18 expression is not regulated by this insult. Thus, IL-18 is not a potential therapeutic target for acute treatment of stroke. However, it may be induced during the later phase of brain infarction and modify repair and recovery.
Footnotes
Acknowledgments:
The authors thank Dr. Ros LeFeuvre for her expert comments, and Anthea Hughes and Sally Shepherd for their technical assistance to this work.