
Abstract
Cytokines are important mediators of intracranial inflammation following traumatic brain injury (TBI). In the present study, the neurological impairment and mortality, blood-brain barrier (BBB) function, intracranial polymorphonuclear leukocyte (PMN) accumulation, and posttraumatic neuronal cell death were monitored in mice lacking the genes for tumor necrosis factor (TNF)/lymphotoxin-α (LT-α) (TNF/LT-α−/−) and interleukin-6 (IL-6) and in wild-type (WT) littermates subjected to experimental closed head injury (total n = 107). The posttraumatic mortality was significantly increased in TNF/LT-α−/− mice (40%; P < 0.02) compared with WT animals (10%). The IL-6−/− mice also showed a higher mortality (17%) than their WT littermates (5.6%), but the difference was not statistically significant (P > 0.05). The neurological severity score was similar among all groups from 1 to 72 hours after trauma, whereas at 7 days, the TNF/LT-α−/− mice showed a tendency toward better neurological recovery than their WT littermates. Interestingly, neither the degree of BBB dysfunction nor the number of infiltrating PMNs in the injured hemisphere was different between WT and cytokine-deficient mice. Furthermore, the analysis of brain sections by in situ DNA nick end labeling (TUNEL histochemistry) at 24 hours and 7 days after head injury revealed a similar extent of posttraumatic intracranial cell death in all animals. These results show that the pathophysiological sequelae of TBI are not significantly altered in mice lacking the genes for the proinflammatory cytokines TNF, LT-α, and IL-6. Nevertheless, the increased posttraumatic mortality in TNF/LT-α-deficient mice suggests a protective effect of these cytokines by mechanisms that have not been elucidated yet.
Cytokines seem to play an important pathophysiological role in inflammatory diseases of the central nervous system (Rothwell and Hopkins, 1995). Accumulating evidence has demonstrated that local synthesis of cytokines by resident cells in the central nervous system is initiated in response to traumatic brain injury (TBI) (for review, see Ott et al., 1994; Morganti-Kossmann et al., 1997; Ghirnikar et al., 1998; Feuerstein et al., 1998a; Shohami et al., 1999). Local induction of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) mRNA expression as well as intrathecal release of these cytokines have been demonstrated in various head trauma models such as experimental cortical contusion (Shohami et al., 1994, 1997; Holmin et al., 1997), surgical brain injury (Woodroofe et al., 1991; Yan et al., 1992; Tchelingerian et al., 1993), fluid percussion trauma (Taupin et al., 1993; Fan et al., 1996; Knoblach et al., 1999), and experimental axonal injury (Hans et al., 1999a). In accordance with these data derived from animal experiments, clinical studies have demonstrated elevated levels of TNF, IL-6, and soluble IL-6 receptor in cerebrospinal fluid and serum from head trauma patients (Goodman et al., 1990; McClain et al., 1991; Ott et al., 1994; Ross et al., 1994; Kossmann et al., 1995, 1996; Morganti-Kossmann et al., 1997; Bell et al., 1997; Hans et al., 1999b). However, the potential pathophysiological role of these cytokines in brain injury remains controversial (Rothwell and Hopkins, 1995; Gruol and Nelson, 1997; Feuerstein et al., 1998a; Shohami et al., 1999).
TNF and lymphotoxin-α (LT-α), the soluble form of TNF-β, which shares the 55- and 75-kDa TNF receptors with similar affinity (Ruddle, 1992; Bazzoni and Beutler, 1996), have long been attributed a neurotoxic role, based on in vitro studies and experimental models of ischemic, traumatic, and autoimmune-mediated brain injury (Merrill, 1992; Selmaj et al., 1991; Shohami et al., 1996, 1997; Barone et al., 1997; Meistrell et al., 1997; Suen et al., 1997; Riminton et al., 1998; Yang et al., 1998; Lavine et al., 1998; Raine et al., 1998; Feuerstein et al., 1998b; Knoblach et al., 1999). Furthermore, evidence from in vivo and in vitro studies shows that administration of recombinant TNF can induce intracranial inflammation and a breakdown of the blood-brain barrier (BBB), a pathophysiological hallmark of neurotrauma (Ramilo et al., 1990; Kim et al., 1992; Megyeri et al., 1992; de Vries et al., 1996). In contrast to TNF and LT-α, IL-6 has been characterized mainly as a neuroprotective cytokine (for review, see Gruol and Nelson, 1997), for example, by inducing intracerebral neurotrophin production in response to brain injury (Kossmann et al., 1996; Ebadi et al., 1997). However, these premises were recently rejected by studies that revealed an antiinflammatory and neuroprotective role for TNF and LT-α (Cheng et al., 1994; Barger et al., 1995; Bruce et al., 1996; Probert et al., 1997; Nawashiro et al., 1997; Liu et al., 1998; Gary et al., 1998) and demonstrated that IL-6 contributes to adverse outcome in experimental autoimmune encephalomyelitis (Eugster et al., 1998). In addition, experimental studies on transgenic mice have demonstrated that chronic intracerebral overexpression of either TNF or IL-6 mediates neurotoxicity by induction of a neurodegenerative inflammatory encephalopathy (Campbell et al., 1993; Stalder et al., 1998). On account of these controversial findings, a dual role of cytokines in neuropathology and/or neuroprotection after brain injury has been recently proposed (Morganti-Kossmann et al., 1997; Shohami et al., 1999). The hypothesis of dual time-dependent actions of cytokines in neuropathology has been recently corroborated by a study on controlled cortical impact brain injury in TNF-deficient mice, demonstrating early detrimental and chronic beneficial functions of TNF (Scherbel et al., 1999).
Altogether, despite clear evidence that cytokines such as TNF, LT-α, and IL-6 are important mediators of neuroinflammation, their exact role in neurotrauma remains to be elucidated. In the present study, we investigated clinical and neuropathological parameters in TNF/LT-α-double-deficient and IL-6-deficient mice undergoing experimental TBI, using a previously characterized model of closed head injury (Chen et al., 1996).
MATERIALS AND METHODS
Animals
Generation and development of mice deficient in genes for IL-6 (IL-6−/−) or double-deficient in TNF and LT-α genes (TNF/LT-α−/−) were previously described (Kopf et al., 1994; Eugster et al., 1996). The TNF/LT-α−/− mice are on a mixed C57BL/6× 129Sv/Ev (B6×129) genetic background (Eugster et al., 1996), whereas the IL-6−/− animals have been back-crossed for 10 generations to a C57BL/6 (B6) background (M. Kopf, unpublished data). For assessment of posttraumatic neurological scores and mortality, genetically matching wild-type (WT) littermates were used as control animals: B6 mice (n = 18) as control for the IL-6−/− mice (n = 18) and B6×129 (n = 20) as control for the TNF/LT-α−/− mice (n = 20). Additional animals were killed 4 hours after TBI for assessment of posttraumatic BBB dysfunction (n = 18) and for immunohistochemical analysis of posttraumatic polymorphonuclear leukocyte (PMN) infiltration 24 hours after trauma (n = 13). For these studies, mice were decapitated under ether anesthesia, and the brains were immediately removed, frozen in liquid nitrogen, and stored at −70°C until analyzed. All animals used in this study were males aged 8 to 16 weeks, with an average weight of 28 to 32 g. They were bred in a specific pathogenfree environment, kept under standard conditions of temperature and light in cages of four to six mice, and fed with food and water ad libitum. The study was performed according to the guidelines of the Institutional Animal Care Committee of the Hebrew University of Jerusalem, Israel.
Experimental brain injury
Experimental closed head injury was performed in mice (n = 107) using a previously described model system (Chen et al., 1996). In brief, the animals were anesthetized with ether, and their skull was exposed by a longitudinal incision of the skin. Focal trauma was induced to the left hemisphere 2 mm lateral to the midline in the midcoronal plane, using an electric weight-drop device with a metal rod of 333 g falling from a height of 2 cm. A silicone tip of 3-mm diameter was fixed at the end of the impacting rod to avoid penetrating skull fractures. After trauma, the mice received supporting oxygenation with 100% O2 until fully awake and were then brought back to their cages, with food and water ad libitum. Sham-operated animals (n = 3 for each group) underwent anesthesia and scalp incision, but they were not subjected to experimental head trauma. These animals were killed 24 hours after skin incision, and the brains were removed for immunohistochemical analysis.
Neurological scores
A 10-point neurological severity score (NSS) was used for assessment of posttraumatic neurological impairment, as described elsewhere in detail (Beni-Adani et al., 1998). This new score is derived from a 25-point NSS, which was previously used for assessment of neurological impairment in the same model system (Chen et al., 1996). The original NSS was shortened to 10 “essential” parameters that are easy to assess, objective in interpretation, and fairly independent of the individual investigator's subjective appraisal (Beni-Adani et al., 1998). Furthermore, the new NSS was shown to correlate to severity of brain damage, as determined by in vivo magnetic resonance studies in mice (L. Beni-Adani et al., unpublished data). A brief description of the NSS is presented in Table 1. The score consists of 10 individual clinical parameters, including tasks on motor function, alertness, and physiological behavior, whereby 1 point is given for failure of the task and no point for succeeding (Table 1). A maximal NSS of 10 points indicates severe neurological dysfunction, with failure at all tasks. In the present study, the NSS was assessed at t = 1 hour, 24 hours, 48 hours, 72 hours, and 7 days after trauma. Evaluation of task performance was performed by two investigators who were blinded about the animal groups. The ΔNSS, calculated as the difference between the NSS at t = 1 hour and the NSS at any later time point, is a parameter that reflects the degree of spontaneous recovery following TBI, as described earlier (Chen et al. 1996).
Neurological Severity Score (NSS) for mice

Assessment of blood-brain barrier dysfunction
A previous study on experimental TBI in mice, using the same model system, revealed a dramatic disruption of the BBB function in the injured hemisphere, peaking at 4 hours after trauma (Chen et al., 1996). Thus, in this study, we analyzed the posttraumatic BBB dysfunction at the same time point (t = 4 hours). The integrity of the BBB was evaluated in B6 (n = 3), IL-6−/− (n = 3), B6×129 (n = 7), and TNF/LT-α−/− (n = 5) mice, according to the degree of Evans blue extravasation into brain tissue, as described by Uyama et al. (1988). In brief, 20 μL of a 2% solution of Evans blue (Sigma, St. Louis, MO, U.S.A.) in saline was injected intravenously into the tail vein at t = 3 hours after TBI and allowed to circulate for 60 minutes. Subsequently, the chest wall was opened under ether anesthesia, and the intravascular dye was removed by saline perfusion (40 to 50 mL) through the left heart ventricle. The brains were then removed and the two hemispheres separated and weighed before homogenization in 1 mL of 50% trichloroacetic acid in distilled H2O. Thereafter, the samples were centrifuged for 20 minutes at 10,000 rpm and the supernatants removed and diluted 1:4 in 100% ethanol. After vigorous mixing, an aliquot of each sample was diluted threefold in a solution of 50% trichloroacetic acid/100% ethanol. For fluorescence measurement, a Perkin-Elmer LS-5 fluorospectrophotometer (Norwalk, CT, U.S.A.) was used at an excitation wavelength of 620 nm and an emission wavelength of 680 nm. A serial dilution of 2% stock Evans blue was taken as a standard curve, which was linear from 25 to 500 ng/mL. The concentrations of the samples were normalized against the brain tissue weight and calculated as nanograms of Evans blue per gram of brain tissue.
Immunohistochemistry and polymorphonuclear leukocyte cell count
The posttraumatic intracerebral invasion of leukocytes, an important pathological consequence of head trauma, was analyzed by immunohistochemistry using a PMN-specific antibody. Three mice of each group (except B6×129; n = 4) were killed 24 hours after trauma, the time point of maximal PMN infiltration, as determined in previous studies (Biagas et al., 1992; Shapira et al., 1993; Clark et al., 1994). The brains were removed as described above, and both hemispheres were separated, immediately snap-frozen in liquid nitrogen, and stored at −70°C until used for experiments. Immunohistochemical analysis of 10-μm-thick cryosections of the left (contused) hemisphere was performed using a biotin/avidin/peroxidase technique and diaminobenzidine tetrahydrochloride as chromogen (Vector, Burlingame, CA, U.S.A.). The primary antibody, a monoclonal rat anti-mouse 7/4 antigen antibody specific for murine PMN (Biosource, Camarillo, CA, U.S.A.), was used at a dilution of 1:100. Nonimmunized rat IgG (Jackson, West Grove, PA, U.S.A.) was used as primary control antibody at the same concentration as the α-PMN antibody. Ten adjacent sections of 10 μm for each brain were stained, and positive PMNs were counted using a stereological grid in the ocular lens of an Olympus BH2 microscope (Hamburg, Germany). The counts were performed in five randomly selected fields of 0.4 mm2 for each brain section (left contusion hemisphere). The density of cellular infiltration was calculated as the number of PMNs per 1 mm3 of brain tissue.
TUNEL histochemistry
Intracranial cell death was assessed by the detection of DNA fragmentation with the TUNEL technique (Gavrieli et al., 1992). Cryosections (10 μm) of the left brain hemisphere of cytokine-deficient and WT mice were analyzed after closed head injury (t = 24 hours, n = 8; t = 7 days, n = 8) or sham operation (t = 24 hours, n = 4) by TUNEL fluorescence staining (In situ cell death detection kit; Boehringer-Mannheim, Germany). In negative controls, the terminal deoxynucleotidyl transferase enzyme was omitted. Simultaneously, cell nuclei were visualized with bisbenzimide Hoechst 33342 fluorochrome (HO33342; Calbiochem, Lucerne, Switzerland), which stains the nuclei of all (live and dead) cells (Pollack and Ciancio, 1990). HO33342-positive cells were detected by fluorescence microscopy using an ultraviolet filter with an excitation wavelength of 400 nm, and TUNEL-positive cells were visualized by a blue light filter with an excitation wavelength of 500 nm, using an Olympus BH2 microscope. The TUNEL-positive cells were counted in fields of 0.4 mm2, and the total number of cells in the same optic fields was counted by switching the filter for visualization of HO33342-positive cells. Only nuclei showing bright fluorescence signals were defined as positive cells, whereas weak cellular background signals were not counted. The extent of cell death was calculated as follows: % dead cells = number of TUNEL-positive cells/number of HO33342-positive cells (per 0.4-mm2 optic field). The percent-age of dead cells in the whole hemisphere was calculated as the mean ± SD of all percentages per 0.4-mm2 field. The differentiation of TUNEL-positive cells (neurons versus neuroglia) was performed according to the morphological characteristics and localization of the cells.
Statistical analysis
Statistical analysis of the data was performed on StatView software (version 4.5; Abacus Concepts, Berkeley, CA, U.S.A.). The nonparametric Mann-Whitney U test was used for analysis of data that were not normally distributed, such as NSS and ΔNSS scores. Data with normal distribution (Evans blue extravasation, PMN infiltration, and TUNEL data) were analyzed by the unpaired Student's t-test. Differences in post-traumatic mortality were evaluated on a contingency table using χ2 and Fisher's exact test, where appropriate. A probability of P < 0.05 was considered statistically significant.
RESULTS
Neurological scores and mortality
The clinical status of the injured mice was assessed by the 10-point NSS (Table 1) at 1 hour, 24 hours, 48 hours, 72 hours, and 7 days after experimental TBI. Figure 1A shows the median NSS and the according percentiles (5 to 95%) of each animal group at these time intervals. Noninjured control mice, WT and knockouts (KOs), as well as sham-operated animals (n = 3 for each group), had an average NSS of 0 to 1 point and never had a score that exceeded 2 (data not shown). In contrast, the median NSS of B6×129 (n = 20) and B6 (n = 18) WT animals subjected to experimental head trauma increased to a score of 8 and 7.5, respectively, at 1 hour after injury. In the further course, the NSS decreased to a median score of 5 in both WT animal groups by 7 days after TBI. No significant differences of the NSS were seen between the two WT groups at any time point (B6×129 versus B6; P > 0.05). The median NSS of the TNF/LT-α−/− (n = 20) and the IL-6−/− (n = 18) mice at 1 hour was 7.5 and 7, respectively, decreasing to a median score of 3 (TNF/LT-α−/−) and 4.5 (IL-6−/−) by 7 days after injury. The comparison of the NSS kinetics between the KO animals and their corresponding WT littermates revealed no significant difference in NSS at any time point (B6×129 versus TNF/LT-α−/−, B6 versus IL-6−/−; P > 0.05). In addition, when the 10 individual neurological parameters (as listed in Table 1) were split up for analysis of the single task performance, no significant difference between the WT and KO animals was detected either (data not shown). Nevertheless, the TNF/LT-α−/− mice had a trend toward better neurol ogical recovery than their WT littermates, as shown by a lower NSS at 7 days after trauma.
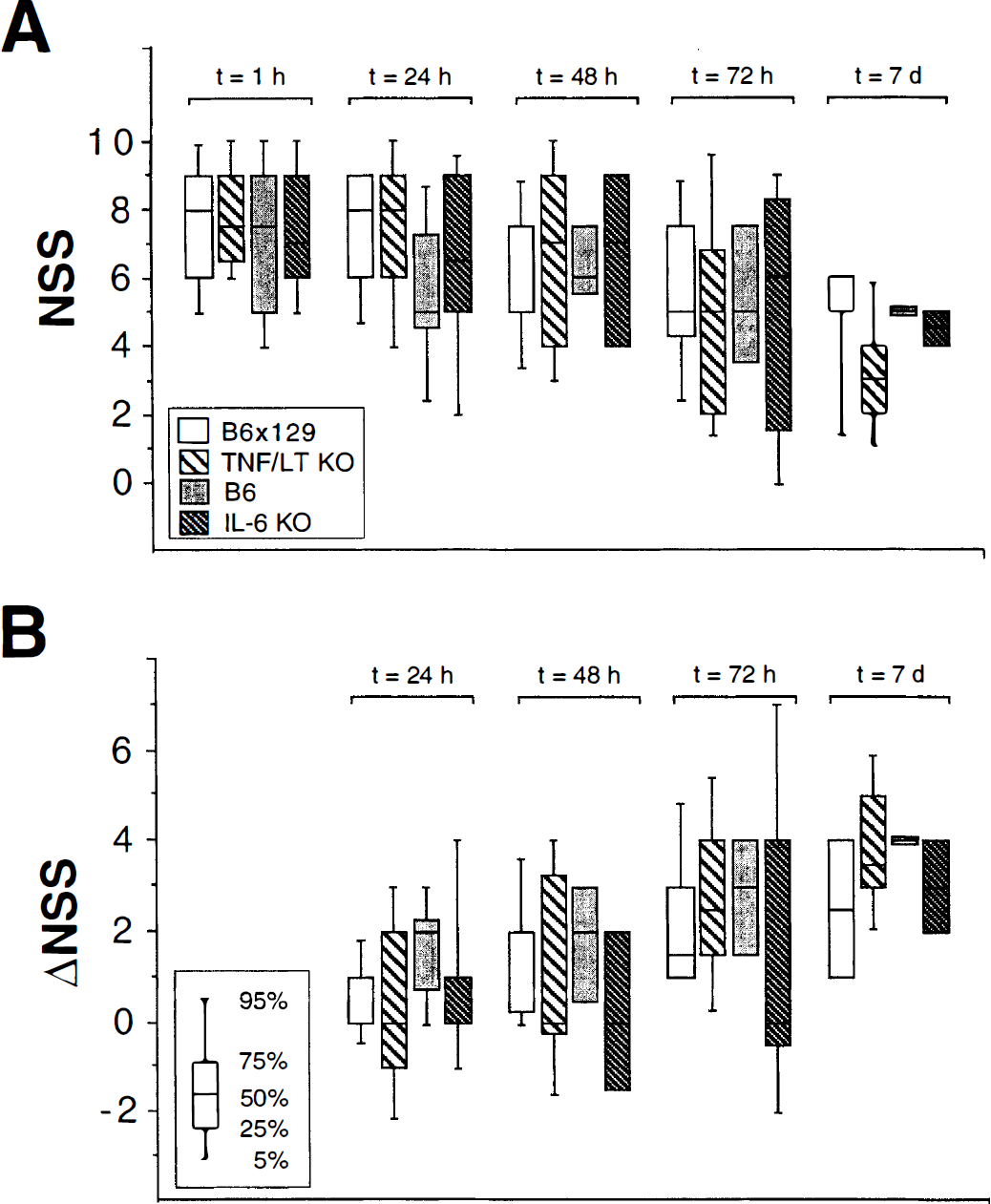
Kinetics of neurological severity score (NSS)
With regard to the ΔNSS, a progressive increase of the median score was observed from 24 hours to 7 days after head injury in WT and cytokine-deficient mice, reflecting the spontaneous recovery after trauma (Fig. 1B). In the WT groups, the median ΔNSS improved from 1 (B6×129) and 2 (B6) at 24 hours to 2.5 (B6×129) and 4 (B6), respectively, after 7 days. As with the NSS, the differences in ΔNSS between the WT groups were not statistically significant (P > 0.05). Similarly, no significant difference in the median ΔNSS was detected by comparison of the WT animals and the corresponding cytokine KO mice, which had a progressing median ΔNSS from 0, at t = 24 hours (both KO groups), to 3.5 (TNF/LT-α−/−) and 3 (IL-6−/−) after 7 days. Although the TNF/LT-α−/− mice showed a tendency toward better recovery at 7 days after trauma compared with the WT and the IL-6−/− animals (that is, lower NSS, higher ΔNSS), the differences were not statistically significant (P > 0.05).
Figure 2 shows the overall posttraumatic mortality of WT and cytokine-deficient mice within 7 days after closed head injury. During the first 24 hours after trauma, 2 of 38 WT mice died (5.3%), and only 1 additional WT mouse died between 24 hours and 7 days (overall WT mortality: 7.9%). The deceased WT mice were B6×129 (n = 2) and B6 (n = 1), and the inter-group mortality was in a similar range (B6×129: 10%; B6: 5.6%; P > 0.05). In contrast, the early (24 hours) and late (7 days) mortality was increased in cytokine-deficient mice. In the TNF/LT-α−/− group, 5 of 20 mice died within 24 hours (25%), and the overall mortality was 40% by 7 days (P < 0.05, compared with B6×129). The mortality of the IL-6−/− mice was also increased compared with the WT group, but the difference was not statistically significant (24-hour mortality: 11%; overall mortality: 17%; P > 0.05).
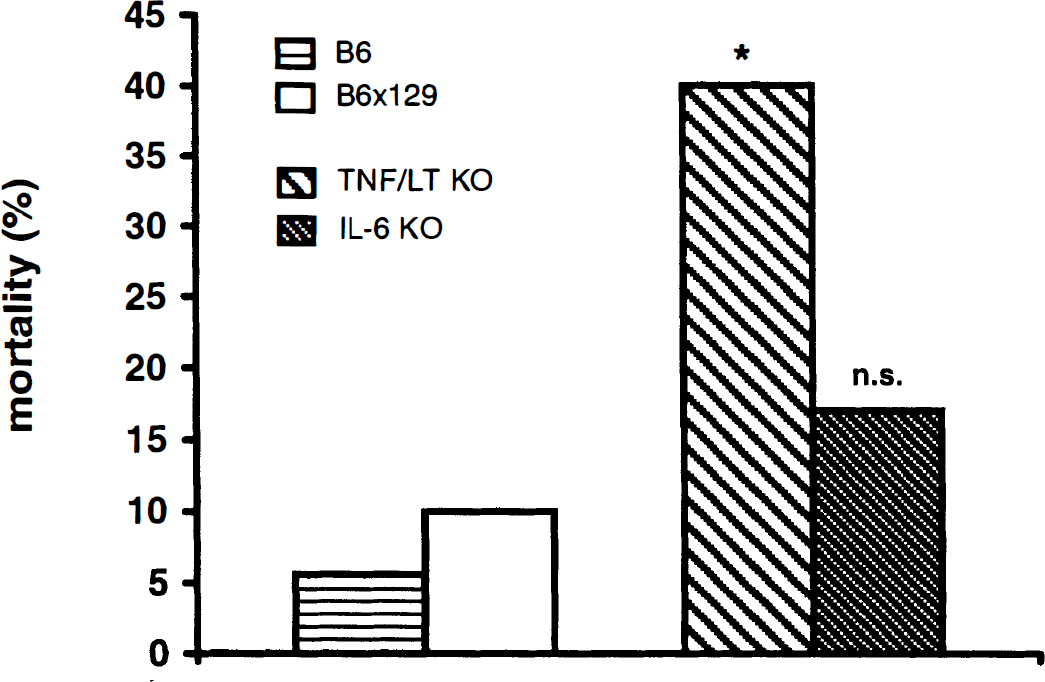
Mortality of wild-type (WT) and cytokine-deficient mice in the course of 7 days after experimental head trauma. Tumor necrosis factor/lymphotoxin-α−/− (TNF/LT KO) and B6×129: n = 20; interleukin-6−/− (IL-6 KO) and B6: n = 18. Statistical analysis was performed using Fisher's exact test. *P < 0.05 for TNF/LT-α−/− versus B6×129 mice; n.s., not significant for IL-6−/− versus B6 mice, P > 0.05.
Posttraumatic blood-brain barrier dysfunction
The amount of Evans blue dye extravasation into the injured brain tissue at t = 4 hours after TBI, as a correlate of damaged BBB integrity after head trauma (Uyama et al., 1988), is shown in Fig. 3. In accordance with previous findings on mice and rats, with use of the same experimental model system (Chen et al., 1996; Shapira et al., 1993), focal trauma to the left hemisphere induced a significant increase of Evans blue extravasation in the ipsilateral hemisphere of WT and KO animals at 4 hours after injury compared with the contralateral side (left versus right hemisphere, P < 0.02; Fig. 3). Interestingly, the quantitative analysis of Evans blue extravasation revealed significantly higher Evans blue levels in the injured left hemisphere of B6 WT mice (1,012 ± 567 ng/g of tissue; mean ± SD) than B6×129 WT mice (421 ± 251 ng/g of tissue; P < 0.03) and TNF/LT-α−/− mice, originating from the B6×129 background (374 ± 160 ng/g of tissue; P < 0.03). The same results were seen in IL-6−/− mice (B6 genetic background) when compared with B6×129 WT and TNF/LT-α−/− animals (794 ± 350 versus 421 ± 251 and 374 ± 160 ng/g of tissue, respectively; P < 0.05). No significant differences in intracerebral Evans blue concentrations were detected when the cytokine KO mice were compared with their corresponding WT animals (IL-6−/− versus B6 and TNF/LT-α−/− versus B6×129; P > 0.05).
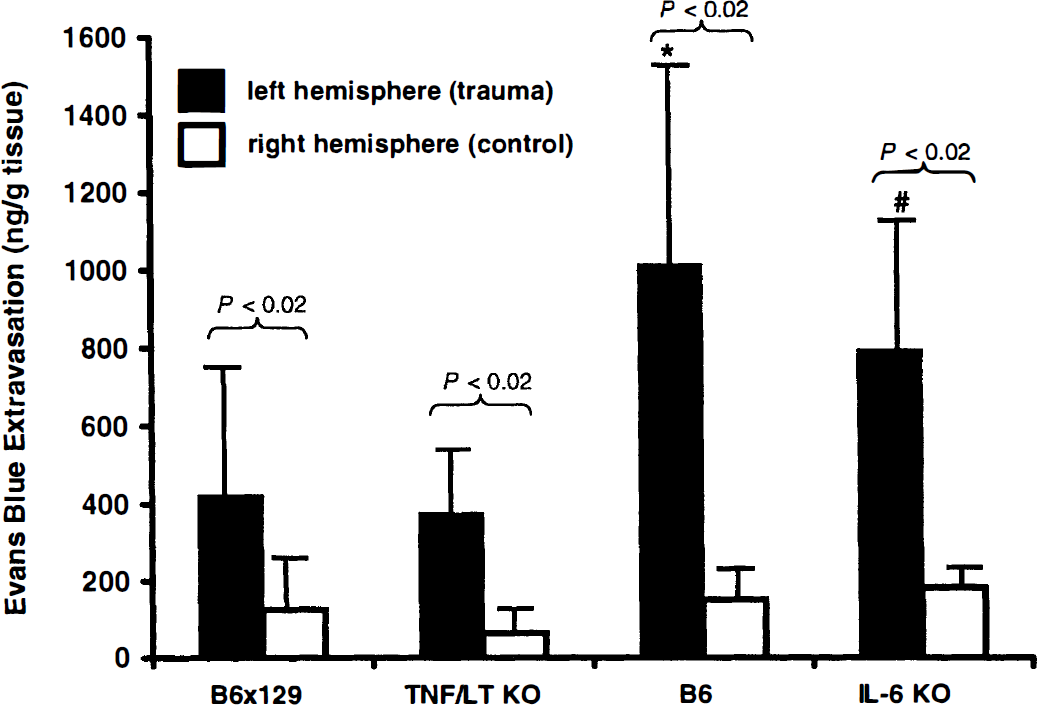
Posttraumatic blood-brain barrier (BBB) dysfunction, as assessed by Evans blue extravasation into brain tissue at t = 4 hours after experimental closed head injury. Tumor necrosis factor/lymphotoxin-α−/− (TNF/LT KO): n = 5; B6×129: n = 7; interleukin-6−/− (IL-6 KO) and B6: n = 3. The data are presented as means ± SD. Statistical analysis was performed using the unpaired Student's t-test. *P < 0.03 for B6 versus B6×129 or TNF/LT-α−/− mice; #P < 0.05 for IL-6 knockout (KO) versus B6×129 or TNF/LT-α−/− mice.
Intracerebral polymorphonuclear leukocyte infiltration
Experimental closed head injury induced a massive intracranial accumulation of PMNs within 24 hours in the injured left hemisphere of WT and cytokine KO mice, as evaluated by microscopic cell count in serial adjacent brain sections stained by immunohistochemistry using an antibody specific for murine PMNs (Fig. 4A to Fig. 4D). The mean count of infiltrating PMNs in the WT group was slightly higher in B6×129 mice than in B6 mice, but the difference was not statistically significant (716 ± 161 versus 594 ± 146 cells/mm3; P > 0.05; Table 2). As presented in Table 2, the PMN cell numbers within the injured brain hemisphere of the cytokine-deficient mice were in a similar range as their corresponding WT animals (TNF/LT-α−/−: 700 ± 156 cells/mm3; IL-6−/−: 617 ± 76 cells/mm3; P > 0.05). In addition, no significant differences in the number of infiltrating PMNs at the lesion site were observed between the KO animal groups either (P > 0.05). The immunohistochemical analysis of tissue sections from brain-injured mice using preimmune rat IgG as primary control antibody did not reveal any nonspecific cellular staining (Fig. 4E).
PMN cell counts in the injured hemisphere

Posttraumatic infiltration of polymorphonuclear (PMN) leukocytes within the injured left hemisphere of wild-type and cytokine-deficient mice at 24h after closed head injury. Cell counts were performed as described in Materials and Methods. The data are represented as mean ± SD. No statistically significant differences were seen between the cytokine-deficient mice and their according wild-type littermates (P > 0.05, unpaired Student's t-test).
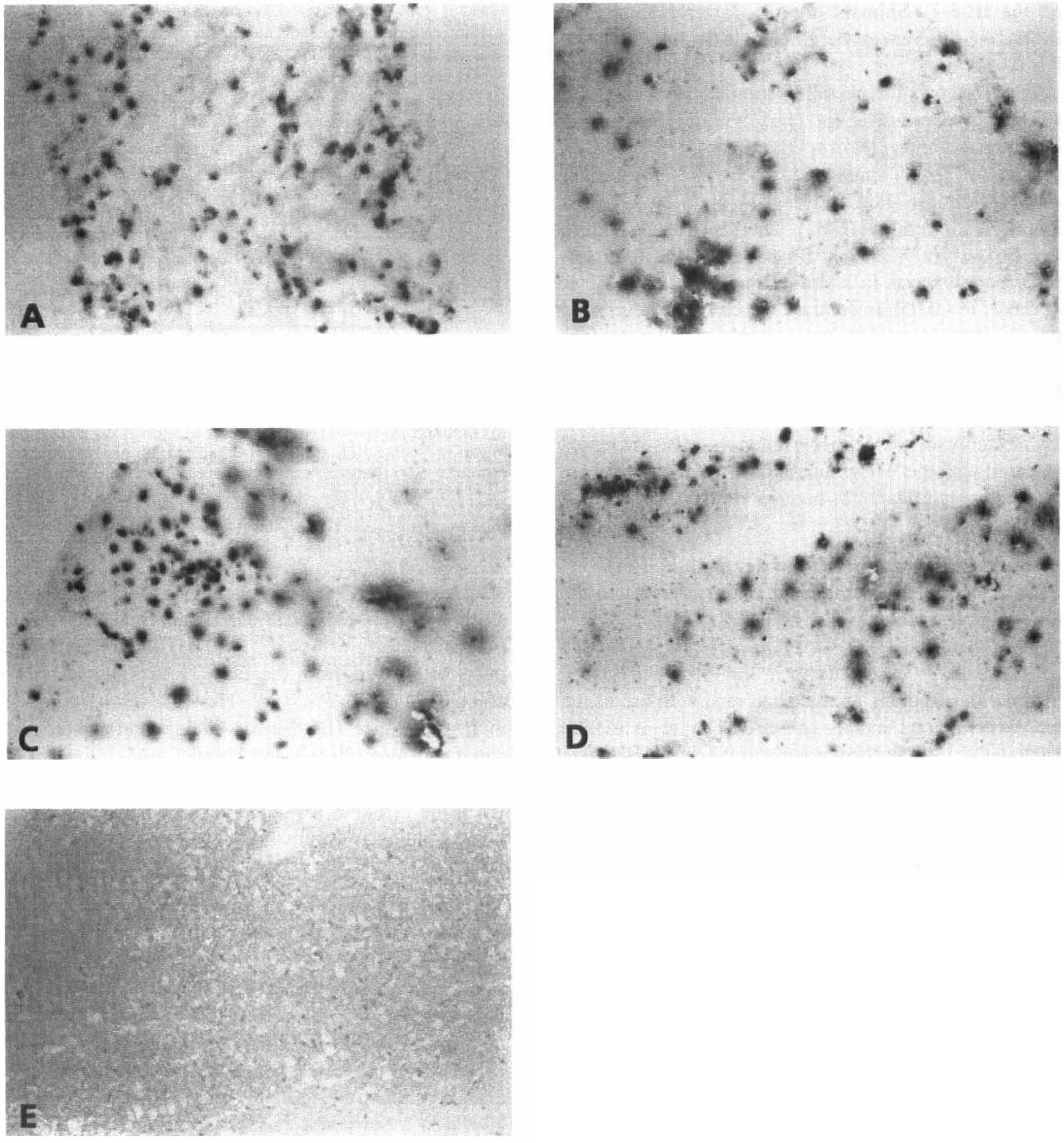
Infiltration of polymorphonuclear leukocytes (PMNs) in the injured hemisphere of mice subjected to experimental closed head injury at t = 24 hours after trauma. Ten-micron-thick cryosections were analyzed by immunohistochemistry using a monoclonal rat anti-mouse PMN antibody
Analysis of posttraumatic cell death
To assess the extent of posttraumatic intraparenchymal cell death after closed head injury, tissue sections of brain-injured mice were analyzed by TUNEL histochemistry (Fig. 5). Experimental closed head injury induced a high level of intracerebral cell death in WT and KO mice by 24 hours. As shown in Fig. 5A, the mean ± SD percentage of dead cells in the injured hemisphere at 24 hours was similar in KO and corresponding WT mice (B6×129: 47 ± 17%; TNF/LT-α−/−: 53 ± 9%; B6: 42 ± 10%; IL-6−/−: 45 ± 8%). By 7 days after closed head injury, the percentage of dead cells was significantly decreased in all animal groups (B6×129: 8 ± 5%; TNF/LT-α−/−: 7 ± 2%; B6: 5 ± 2%; IL-6−/−: 3 ± 1%) as compared with 24 hours after trauma (P < 0.05, unpaired Student's t-test). However, when the KO and corresponding WT mice (that is, TNF/LT-α−/− and B6×129; IL-6−/− and B6) were compared at equivalent time points (24 hours or 7 days), no statistically significant difference was found (P > 0.05). Only very few TUNEL-positive cells were detected in the left hemisphere of sham-operated mice after 24 hours, as shown representatively for B6×129 and TNF/LT-α−/− mice (Figs. 5B and 5D). The left column in Fig. 5 shows, furthermore, TUNEL-positive cells below the contusion site in the cortical layer III of B6×129 and TNF/LT-α−/− mice at 24 hours (Figs. 5F and 5H, respectively) and 7 days (Figs. 5J and 5L, respectively) after closed head injury. The total cell number in these tissue sections is shown by detection of HO33342-positive cells (Fig. 5, right; identical views as for the TUNEL stainings in the left column). No signals were evident when the terminal nucleotidyl transferase enzyme was omitted in TUNEL experiments as negative control (data not shown). The morphological analysis and localization (cortical layers II to IV below the contusion site) of TUNEL-positive cells revealed that neurons represent the predominant cell type with DNA fragmentation (Figs. 5F, 5H, 5J, and 5L). However, we cannot exclude a few TUNEL-positive glial cells (microglia, astrocytes) in these sections.
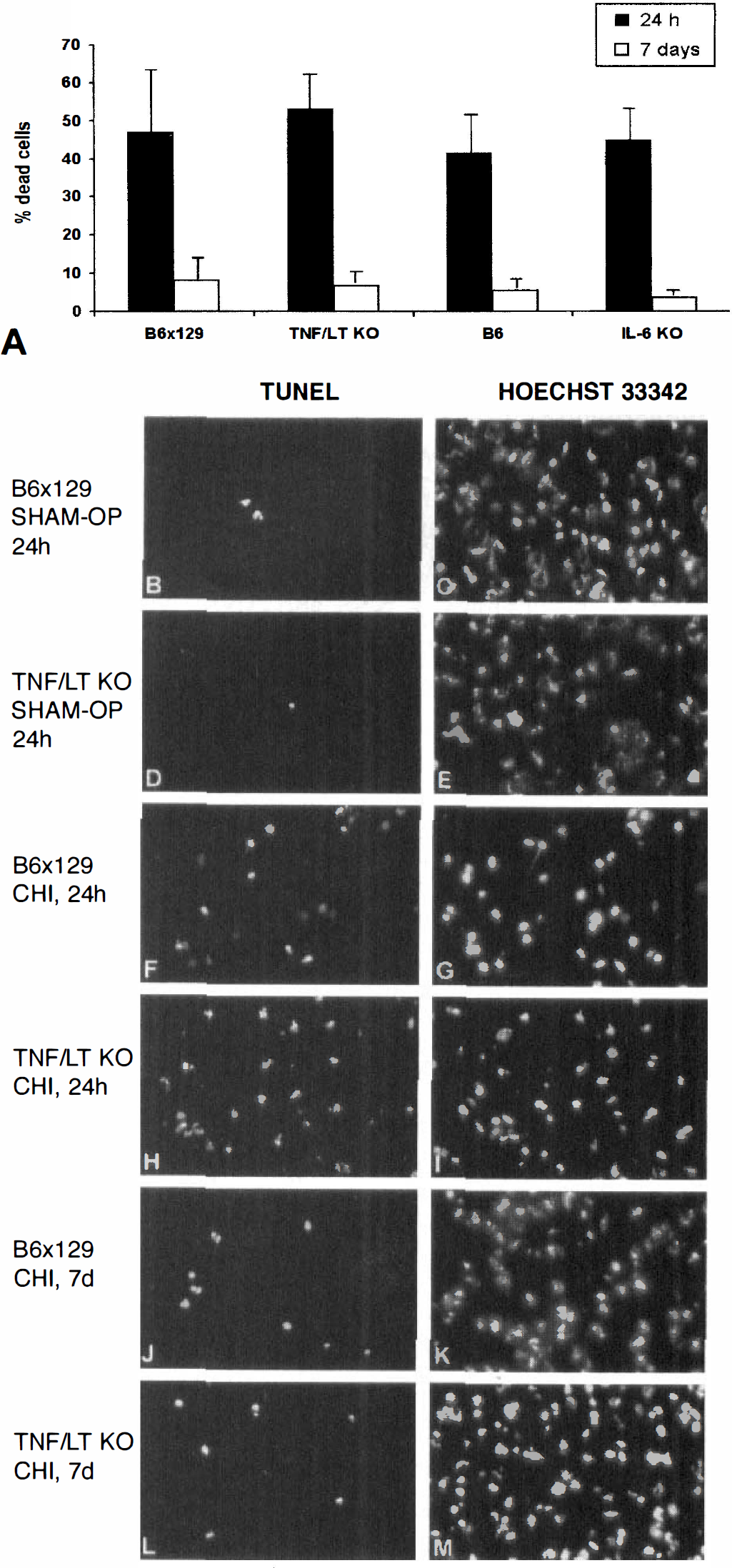
Posttraumatic cell death in the injured hemisphere of mice after experimental closed head injury. Cryosections of 10 μm were analyzed by TUNEL histochemistry, and the percentage of dead cells was calculated as the ratio of TUNEL-positive cells divided by the total number of cells in optic fields of 0.4 mm2, as determined by Hoechst 33342 fluorescence.
DISCUSSION
In the present study, we investigated the role of the pro-inflammatory cytokines TNF, LT-α, and IL-6 in the pathophysiology of TBI. For this purpose, TNF/LT-α-double-deficient and IL-6-deficient mice, as well as their WT littermates, were subjected to experimental focal closed head injury, and the clinical outcome, based on the neurological deficit and mortality, as well as the post-traumatic BBB dysfunction, the accumulation of PMNs, and the extent of neuronal cell death within the injured hemisphere of these animals were assessed. Interestingly, despite previous data that suggest either a deleterious or a beneficial role of cytokines such as TNF and IL-6 in central nervous system pathology (Gruol and Nelson, 1997; Ghirnikar et al., 1998; Eugster et al., 1998; Feuerstein et al., 1998a; Shohami et al., 1999; Penkowa et al., 1999), our present findings show that the neurological impairment in the posttraumatic course of experimental TBI does not differ in IL-6-deficient or TNF/LT-α-double-deficient mice compared with WT animals. The assessment of a 10-parameter NSS, which was previously shown to correlate to the extent of brain damage in mice with the same experimental closed head injury (Beni-Adani et al., 1998, and unpublished data), revealed a similar severe neurological impairment in all animals between 1 hour and 7 days after TBI. Furthermore, a similar degree of spontaneous recovery after trauma was seen in both WT and cytokine-deficient mice, as indicated by the changes in NSS and ΔNSS kinetics, showing a continuous decrease in NSS and a constant increase in ΔNSS in the course of 7 days after brain injury (Fig. 1). These data are in contrast to previous studies on experimental closed head injury in the rat, demonstrating that pharmacological inhibition of TNF improves post-traumatic recovery from 24 hours to 14 days after injury, as shown by an increased ΔNSS compared with vehicle-treated control animals (Shohami et al., 1996, 1997). However, despite the lack of significant differences in neurological impairment in the present study, the enhanced posttraumatic mortality of TNF/LT-α−/− mice suggests a protective role of these cytokines in the first week after experimental closed TBI (Fig. 2). Interestingly, a recent report on controlled cortical impact brain injury in TNF “single” KO mice has brought evidence of early deleterious (that is, within 48 hours after injury) and chronic beneficial (2 to 4 weeks after trauma) effects of TNF (Scherbel et al., 1999). These results differ from our present findings with regard to the outcome within the first week after TBI, as in our study the TNF/LT-α−/− mice had a higher posttraumatic mortality than WT littermates. Nevertheless, the observation that the survivors within the TNF/LT-α−/− group had a trend toward better neurological recovery (that is, lower NSS and higher ΔNSS than WT littermates) by 7 days after closed head injury suggests that TNF and LT-α may mediate early detrimental functions, similarly as in TNF “single” KO mice after controlled cortical impact brain injury (Scherbel et al., 1999). However, we should emphasize that results derived from studies using TNF “single” deficient mice (Scherbel et al., 1999) cannot be directly compared with data derived from experiments with TNF/LT-α−/− mice, even though both TNF and LT-α may have redundant functions due to binding to equal receptors (namely, TNF receptor types 1 and 2) with similar affinity (Ruddle, 1992; Bazzoni and Beutler, 1996). Supportive of this assumption are recent findings from animal models of experimental autoimmune encephalomyelitis showing differences in the extent of neuropathology between “single” TNF or LT-α KO mice versus TNF/LT-α-double-deficient animals, and surprisingly even between TNF−/− mice generated in different laboratories (Frei et al., 1997; Liu et al., 1998; Riminton et al., 1998; Eugster et al., 1999). More specifically, two independently generated TNF−/− mice on an apparently identical genetic background (B6) showed either an exacerbation (Liu et al., 1998) or a delayed onset of experimental autoimmune encephalomyelitis (Riminton et al., 1998). Even more pronounced differences in experimental autoimmune encephalomyelitis severity were observed with TNF/LT-α−/− mice on different genetic backgrounds. As such, TNF/LT-α−/− mice on the SJL background were highly susceptible to the autoimmune disease (Frei et al., 1997), whereas TNF/LT-α−/− mice on the B6×129 background showed only very mild disease symptoms (Eugster et al., 1999). These data suggest that as yet unknown polymorphic alleles may drastically influence the function of TNF and LT-α in the pathophysiology of experimental autoimmune encephalomyelitis. Thus, one has to be careful in comparing data from different gene KOs, different mouse strains, or different experimental models of neuropathology. As the increased mortality in TNF/LT-α−/− mice did not correlate to any of the pathophysiological parameters assessed in the present study (that is, BBB dysfunction, neutrophil infiltration, neuronal death), we speculate that other mechanisms such as changes in posttraumatic hemodynamics or other systemic alterations must be responsible for increased mortality. The mechanisms leading to enhanced mortality in TNF/LT-α−/− mice after closed head injury will be further investigated in future studies.
The posttraumatic mortality of the IL-6−/− animals was also elevated compared with that of WT animals, but the difference was not statistically significant. Up to now, only few data are available on the potential role of IL-6 in the course of central nervous system trauma. An experimental study of facial motor nucleus axotomy demonstrated attenuated activation of astrocytes and microglia in IL-6-deficient mice, suggesting a function for IL-6 in controlling posttraumatic cellular homeostasis (Klein et al., 1997). Other data on the same mice have revealed a crucial role for IL-6 in activation of astrocytes and recruitment of macrophages into the brain after experimental cryogenic lesioning (Penkowa et al., 1999). We have recently postulated that IL-6 released intrathecally in head trauma patients may induce astrocytic production of the neurotrophin nerve growth factor and may systemically mediate the hepatic acute-phase response after TBI (Kossmann et al., 1995, 1996). Our present results show that the deficiency of IL-6 and the potential concomitant attenuation of the acute-phase response (Kopf et al., 1994) do not seem to influence the clinical course and the mortality of experimental TBI. This is in contrast to data from a recent study on focal cryogenic brain injury in IL-6−/− mice, where a neuroprotective effect of IL-6 was postulated (Penkowa et al., 1999). However, the latter study was based on a distinct experimental model system (freeze lesion), and different end points were analyzed, such as activation of glial cells and brain macrophages, as well as the regulation of intracerebral growth factor and acute-phase protein expression, which makes a direct comparison of these divergent studies difficult.
In addition to clinical outcome and mortality, we have also monitored the posttraumatic BBB dysfunction in cytokine-deficient and WT mice after closed head injury. Increased BBB permeability was previously described in the contused hemisphere of mice after experimental closed head injury, with a peak at 4 hours after trauma (Chen et al., 1996). It is interesting to note that the kinetics of TNF activity in brain-injured rats, peaking at 4 hours after head injury, correlated to maximal BBB dysfunction in the same model system (Shohami et al., 1994). Furthermore, the intracranial application of recombinant TNF has been shown to induce a breakdown of the BBB within 3 to 4 hours, thus revealing a direct causative relationship between TNF and BBB dysfunction (Saukkonen et al., 1990; Kim et al., 1992). Similar results are derived from in vitro experiments demonstrating that the exposure of primary cerebral endothelial monolayers, as an in vitro BBB model, to recombinant TNF induced a rapid decline in trans-endothelial electrical resistance (de Vries et al., 1996). Thus, based on these data, it seems rational to assume that posttraumatic BBB dysfunction may be attenuated in TNF/LT-α−/− mice in the present model of experimental closed head injury. However, in our study, the degree of BBB dysfunction in the injured hemisphere at 4 hours after trauma was not different between TNF/LT-α−/− mice and their corresponding WT (B6×129) mice, with mean Evans blue levels of 374 and 421 ng/g of brain tissue, respectively. In accordance with previously published data (Chen et al., 1996), the degree of Evans blue extravasation into brain tissue was significantly higher in the ipsilateral hemisphere than in the contralateral hemisphere in all animals tested. In the present study, it was interesting to note that the mean Evans blue levels in the injured hemisphere were significantly elevated in both B6 WT and IL-6−/− mice (1,012 and 794 ng/g, respectively) compared with B6×129 WT and TNF/LT-α−/− mice (421 and 374 ng/g, respectively). These data suggest that mice on a “pure” B6 genetic background are more susceptible to posttraumatic BBB dysfunction than the “mixed” B6×129 animals. Supportive of these findings is a recent study that demonstrated significant differences in neurological impairment after neurotrauma depending on the background strain (for example, B6 versus 129Sv) of mice subjected to brain injury (Fox et al., 1999). In our study, the lack of significant differences in the extent of BBB damage between the cytokine KO and their corresponding WT littermates suggests that deficiency of these cytokines neither aggravates nor protects from BBB dysfunction after experimental closed head injury. In this regard, the genetic background of the mice seems to play a more important role in the pathogenesis of BBB damage after TBI than mediators of inflammation, such as TNF, LT-α, and IL-6.
We have furthermore analyzed posttraumatic leukocyte infiltration in the contused hemisphere by PMN cell count in serial adjacent tissue sections. As previously described in experimental TBI, a massive PMN infiltration was detected at 24 hours after trauma around the contusion site in WT animals (Biagas et al., 1992; Clark et al., 1994). In addition, the intrathecal injection of recombinant TNF was shown to induce recruitment of leukocytes into the intracranial compartment (Saukkonen et al., 1990; Liu et al., 1994; Feuerstein et al., 1998b), suggesting that the inactivation of TNF and LT-α genes may possibly attenuate the degree of intracranial leukocyte infiltration after experimental brain trauma. Neverthe-less, in the present study, no difference in PMN density was observed within the contused hemisphere of both cytokine KO mice when compared with their WT littermates (Table 2). It is very likely that the posttraumatic intracranial release of potent PMN chemoattractants such as C-X-C chemokines (Ransohoff and Tani, 1998) and complement-derived anaphylatoxins (Stahel et al., 1998) may compensate for the lack of cytokines in the KO animals in terms of leukocyte recruitment into the injured central nervous system. Another candidate molecule that may compensate for the lack of TNF or IL-6 in our study may be interleukin-1β, a potent mediator of intracranial inflammation (Ramilo et al., 1990; Quagliarello et al., 1991; Rothwell and Hopkins, 1995). However, the levels of interleukin-1β in the brains of TNF/LT-α−/− and IL-6−/− mice (n = 24) between 4 hours and 7 days after closed head injury were not altered in comparison with WT mice, as determined by enzyme-linked immunosorbent assay in brain homogenates (V. I. Otto et al., unpublished observations). For the correct interpretation of studies based on gene KO mice, it should also be held in mind that developmental changes due to the chronic lack of cytokines in KO mice (Durum and Muegge, 1998) may cause responses to brain injury that differ from those in experimental models in WT animals, where cytokine-mediated effects are blocked acutely, for example, by administration of antibodies (Lavine et al., 1998) or pharmacological agents (Shohami et al., 1996, 1997). This is also supported by a recent study on experimental controlled cortical impact brain injury, demonstrating that the histologic and functional outcome of mice lacking the intercellular adhesion molecule-1 was not different between WT and KO animals, whereas previous studies had demonstrated a significant reduction in the extent of brain injury, based on various model systems, after acute blocking of intercellular adhesion molecule-1 (Whalen et al., 1999).
Finally, the analysis of injured brains by TUNEL histochemistry revealed that cell death within the injured hemisphere was similar in all animal groups (KO and WT) at either 24 hours or 7 days after trauma (Fig. 5A). Whereas sham-operated mice had only occasional positive TUNEL signals in the brain parenchyma (Figs. 5B and 5D), experimental closed head injury induced high levels of posttraumatic intracranial cell death by 24 hours (Figs. 5F and 5H), ranging from 42 to 53% dead cells within the ipsilateral hemisphere (Fig. 5A). These cells were characterized as neurons, based on the localization in cortical layers II to IV and their cellular morphology. By 7 days after trauma, the extent of TUNEL-positive cells dropped in KO and WT animals, with a residual percentage of dead cells ranging from 3 to 8% (Figs. 5A, 5J, and 5L). These results do not support a critical role for TNF, LT-α, or IL-6 in mediating posttraumatic neuronal death or cerebroprotection after experimental head trauma.
Altogether, this study on experimental closed head injury in mice shows that the deficiency of genes for the pro-inflammatory cytokines TNF, LT-α, and IL-6 neither exacerbates deleterious effects such as induction of BBB dysfunction and accumulation of inflammatory cells within the brain nor influences the degree of post-traumatic neurological impairment. Nevertheless, the finding of enhanced posttraumatic mortality in TNF/LT-α-deficient mice suggests that these cytokines may mediate protective effects after closed head injury, which are regulated by mechanisms that have not been elucidated yet. In this regard, we plan to further explore the molecular events occurring in cytokine-deficient mice after experimental closed head injury.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Ms. Emerita Ammann for technical assistance and Drs. Horst Bluethmann and Manfred Kopf for the supply of IL-6−/− and WT mice.