
Abstract
Pre-B-cell colony-enhancing factor (PBEF) (also known as nicotinamide phosphoribosyltransferase) is a rate-limiting enzyme in the salvage pathway for mammalian biosynthesis of nicotinamide adenine dinucleotide (NAD+). By synthesizing NAD+, PBEF functions to maintain an energy supply that has critical roles in cell survival. Cerebral ischemia is a major neural disorder with a high percentage of mortality and disability. Ischemia leads to energy depletion and eventually neuronal death and brain damage. This study investigated the role of PBEF in cerebral ischemia using a photothrombosis mouse model. Using immunostaining, we initially determined that PBEF is highly expressed in neurons, but not in glial cells in the mouse brain. To study the role of PBEF in ischemia in vivo, we used PBEF knockout heterozygous (Pbef+/−) mice. We showed that these mice have lower PBEF expression and NAD+ level than do wild-type (WT) mice. When subjected to photothrombosis, Pbef+/− mice have significantly larger infarct volume than do age-matched WT mice at 24 hours after ischemia. Higher density of degenerating neurons was detected in the penumbra of Pbef+/− mice than in WT mice using Fluoro-Jade B staining. Our study shows that PBEF has a neuronal protective role in cerebral ischemia presumably through enhanced energy metabolism.
Introduction
Human pre-B-cell colony-enhancing factor (PBEF) was first cloned in activated T lymphocytes (Samal et al, 1994). It was further identified as a rate-limiting nicotinamide phosphoribosyltransferase to convert nicotinamide to nicotinamide mononucleotide to nicotinamide adenine dinucleotide (NAD+) in the salvage pathway of NAD+ biosynthesis (Revollo et al, 2007; Rongvaux et al, 2002). Pre-B-cell colony-enhancing factor is highly conserved during evolution and exists widely in various tissues (Martin et al, 2001). Intracellular PBEF was found to exist in the nuclei, mitochondria, and cytosol (Kitani et al, 2003). Nicotinamide adenine dinucleotide is known as a major coenzyme for numerous redox reactions in energy metabolism. Pre-B-cell colony-enhancing factor is a link for NAD+ biosynthesis, metabolism, and diseases (Imai, 2009a). It was found to be a longevity protein that can extend the lifespan of human smooth muscle cells (van der Veer et al, 2007). In the bone marrow, PBEF is essential for early B-lineage precursor cell growth and granulocyte-colony stimulating factor-induced myeloid through the NAD+-dependent pathway for differentiation (Skokowa et al, 2009). Pre-B-cell colony-enhancing factor is important in cell apoptosis and survival. Increased PBEF protects hypoxia-induced cell death through an intact mictochondrial NAD+ salvage pathway (Yang et al, 2007). Pre-B-cell colony-enhancing factor was found to be involved in multiple disorders, including acute lung injury (Ye et al, 2005), rheumatoid arthritis (Brentano et al, 2007), chronic kidney disease (Axelsson et al, 2007; Song et al, 2008), and malignant astrocytoma (Reddy et al, 2008).
However, little is known about the roles of PBEF in the central nervous system. Although PBEF was found in human and murine brains using western blotting (Kitani et al, 2003; Samal et al, 1994), neither the cell-specific distributions of PBEF in the brain nor whether PBEF is involved in brain disorders is known. Among many brain diseases, cerebral ischemia is a major neural disorder with a high percentage of mortality and disability. Ischemia leads to energy depletion and eventually neuronal death and brain damage (Dirnagl et al, 1999; Lo et al, 2003). As PBEF is a rate-limiting enzyme of NAD+ biosynthesis, we reasoned that PBEF might have a beneficial role in brain ischemia through enhanced energy metabolism. To test this hypothesis, we used knockout mice that express a reduced level of PBEF, i.e., PBEF heterozygous knockdown (Pbef+/−) mice to determine the effect of PBEF on neuronal death and brain damage after photothrombosis-induced cerebral ischemia.
Materials and methods
Animals
The Pbef+/− mice were generated using C57BL/6J background as described previously (Hong et al, 2008). Throughout the study, Pbef+/− and wild-type (WT) (Pbef+/+) mice aged 10 to 13 weeks were used for ischemia study. All procedures were performed according to the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Missouri Animal Care Quality Assurance Committee.
Photothrombosis-Induced Brain Ischemia Model
We used a photothrombosis-induced brain ischemia model, which was established in our previous studies (Ding et al, 2009; Wang et al, 2010). Briefly, a circular craniotomy (2.0 mm in diameter) was made using a high-speed drill over the somatosensory cortex at the center of −0.8 mm from the bregma and 2.0 mm lateral to the midline after mice were anesthetized. The dura was carefully removed using fine forceps. The photosensitive dye rose Bengal dissolved in artificial cerebral spinal fluid was then injected through the tail vein at a dose of 30 mg/kg. To induce photothrombosis, an area of 1 mm diameter was focally illuminated for 2 minutes with a green light through a × 10 objective to activate the dye.
Transcardial Perfusion and the Measurement of Infarct Volume
The procedure was similar to that of our previous studies (Ding et al, 2009; Wang et al, 2010). Coronal sections (with a thickness of 20 μm) of the brain were cut by using a cryostat (CM1900, Leica, Bannockburn, IL, USA) and were serially placed on gelatin-coated glass slides or in a 48-well plate with 0.01 mol/L phosphate-buffered saline. To measure the infarct volume, every fifth brain slice on the glass slide was stained with 0.25% cresyl violet (Nissl staining). The infarct volume was determined by measuring the areas showing the loss of Nissl staining in the brain sections (Schroeter et al, 2002; Vendrame et al, 2004). The areas of cerebral infarction were delineated and quantified using ImageJ software (NIH, Bethesda, MD, USA). The total volume of ischemic tissue was calculated by multiplying the individual infarct area by the total thickness of the five slices.
Immunostaining and Fluoro-Jade B Staining
Floating method was used for immunohistochemical staining as described previously (Ding et al, 2009; Wang et al, 2010). Briefly, for neuronal nuclei (NeuN) staining, the brain sections were incubated overnight with mouse anti-NeuN monoclonal antibody (1:200, Chemicon, Billerica, MA, USA) after being blocked with 10% donkey serum; subsequently, they were incubated with fluorescein isothiocyanate- or rhodamine-conjugated goat anti-mouse IgG secondary antibody (1:200, Chemicon). Similar procedures were used for glia fibrillary acidic protein (GFAP) staining using a mouse anti-GFAP monoclonal antibody (1:800, Chemicon), for S100β staining using a mouse anti-S100β monoclonal antibody (1:500, Sigma, St Louis, MO, USA), for Iba1 staining using a rabbit anti-Iba1 polyclonal antibody (1:600, Wako Pure Chemical Industries, Richmond, VA, USA), for PBEF staining using a rabbit anti-human PBEF polyclonal antibody (1:300, Bethyl Laboratories, Montgomery, TX, USA), and for CD31 staining using a mouse anti-CD31 antibody (1:50, BD Pharmingen, San Diego, CA, USA). Finally, the brain sections were mounted with an antifade medium containing DAPI (Invitrogen, Carlsbad, CA, USA). The stained sections were observed under an Olympus FV1000 confocal microscope (Olympus, Center Valley, PA, USA) and analyzed by off-line MetaMorph software (Molecular Device, Sunnyvale, CA, USA).
For detecting degenerating neurons, brain sections were stained with Fluoro-Jade B (FJB) as described previously (Ding et al, 2007; Wang et al, 2010). Briefly, brain sections on glass slides were washed with ddH2O and 0.06% potassium permanganate, and then immersed into 0.0004% FJB in 0.1% acetic acid solution for 60 minutes. Images of the stained sections were acquired with MetaMorph Imaging software (Molecular Device) using a Nikon epi-fluorescence microscopy (Nikon, Melville, NY, USA) equipped with a CoolSNAP-EZ CCD-camera (Photo-metrics, Tucson, AZ, USA). The number of positive cells in the penumbra from each brain section was counted using the MetaMorph Imaging software. Cells were counted if they contained a whole-cell body (Liu et al, 2009c) and were presented as per mm2. The data from slices of each mouse brain were averaged and expressed as mean ± s.e.m.
Immunoblot and Western Blot Analyses of Pre-B-Cell Colony-Enhancing Factor
Immunoblot was used for determining the PBEF level in blood. Blood serum (2 μL) was separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membranes were blocked by 5% bovine serum albumin and incubated subsequently with a rabbit anti-human PBEF polyclonal antibody (1:300, Bethyl Laboratories) and horseradish peroxidase-conjugated secondary antibody. The protein band was visualized by enhanced chemiluminescent reagent. The integrative density for the Ponceau values was measured. The expression was represented as percentages relative to the control.
We used western blot analysis to determine PBEF protein in the brain. Total protein was extracted from the freshly harvested brain cortex and hippocampus using a lysis buffer plus protease inhibitor (Pierce Biotechnology, Rockford, IL, USA), and phosphatase inhibitor cocktails (Sigma), pH 8.2. The homogenized tissue was centrifuged at 12,000 X g for 30 minutes at 4°C. The supernatant fluid is the total cell lysate. The protein concentration of the cell lysate was determined using a BCA protein assay kit (Pierce Biotechnology). Equivalent amounts of protein from each sample were diluted with Laemmli buffer, boiled for 5 minutes, subjected to electrophoresis in 10% SDS-polyacrylamide gels at 100 mV, and subsequently transferred to polyvinylidene fluoride membranes. Membranes were blocked for 1 hour with 5% (w/v) nonfat dry milk in Tris-buffered saline containing 0.1% (v/v) Tween 20 and were incubated overnight at 4°C in 3% (w/v) bovine serum albumin with 0.02% (w/v) sodium azide in Tris-buffered saline Tween-20 with anti-human PBEF antibody (1:4,000) (Bethyl Laboratories) or monoclonal anti-β-actin antibody (1:2,000; Cytoskeleton, Denver, CO, USA). The membranes were incubated with goat horseradish peroxidase-conjugated anti-rabbit IgG (1:5,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted in 5% (w/v) nonfat dry milk in Tris-buffered saline Tween-20 for 1 hour at room temperature. The membranes were then exposed to SuperSignal West Pico Chemiluminescent detection reagents (Pierce Biotechnology) and autoradiograph films (ISC BioExpress, Kaysville, UT, USA) to visualize bands. The density of bands was measured using the Quantity One software (Bio-Rad, Hercules, CA, USA). Relative fold difference of brain tissue Pbef protein between WT and Pbef+/− mice was presented by dividing the integrated density value of the Pbef protein over that of β-actin.
Nicotinamide Adenine Dinucleotide Measurement of Brain Tissue
The concentration of NAD+ was measured using NAD+ assay kit according to the manufacturer's instructions (Bioassay Systems, Hayward, CA, USA). Freshly harvested cortical tissues (∼20 mg) were placed into a 1.5 mL tube with 100 μL NAD+ extraction buffer and homogenized. The extracts were heated at 60°C for 5 minutes, and then 20 μL assay buffer and 100 μL of the opposite extraction buffer were added to neutralize the extracts. The samples were briefly mixed and spanned down at 14,000 r.p.m. for 5 minutes. The supernatant was used for NAD+ enzymatic assay according to the manufacturer's instructions. For NAD+ measurement in brain tissue after ischemia, we induced photothrombosis in a region of 3 mm in diameter in the barrel cortex, so that enough tissue from ischemic region could be collected. We measured NAD+ levels in the acute phase of ischemia, i.e., 6 hours after photothrombosis. The NAD+ concentration was obtained from calibration curve using standard NAD+ from the kit and converted to picomole/milligram tissue.
Definition of the Penumbra
Theoretically, the penumbra is the region of the brain tissue that is damaged but not yet dead after focal ischemia (Lo, 2008). Therefore, the transition region from the normal tissue to the ischemic core is considered as the penumbra in experiments. For NeuN staining, the transition region from the normal NeuN staining to the region with reduced NeuN reactivity was considered as a penumbra (Figures 4A and 4B). For identification of glial activation in the penumbra, we took images in the region that is close to astrocyte and microglia with normal morphology. For FJB+ cell counting, as there is a sharp transition from the FJB– region to the FJB+ region, we counted FJB+ cells in the region next to the FJB– region (Figure 4C).
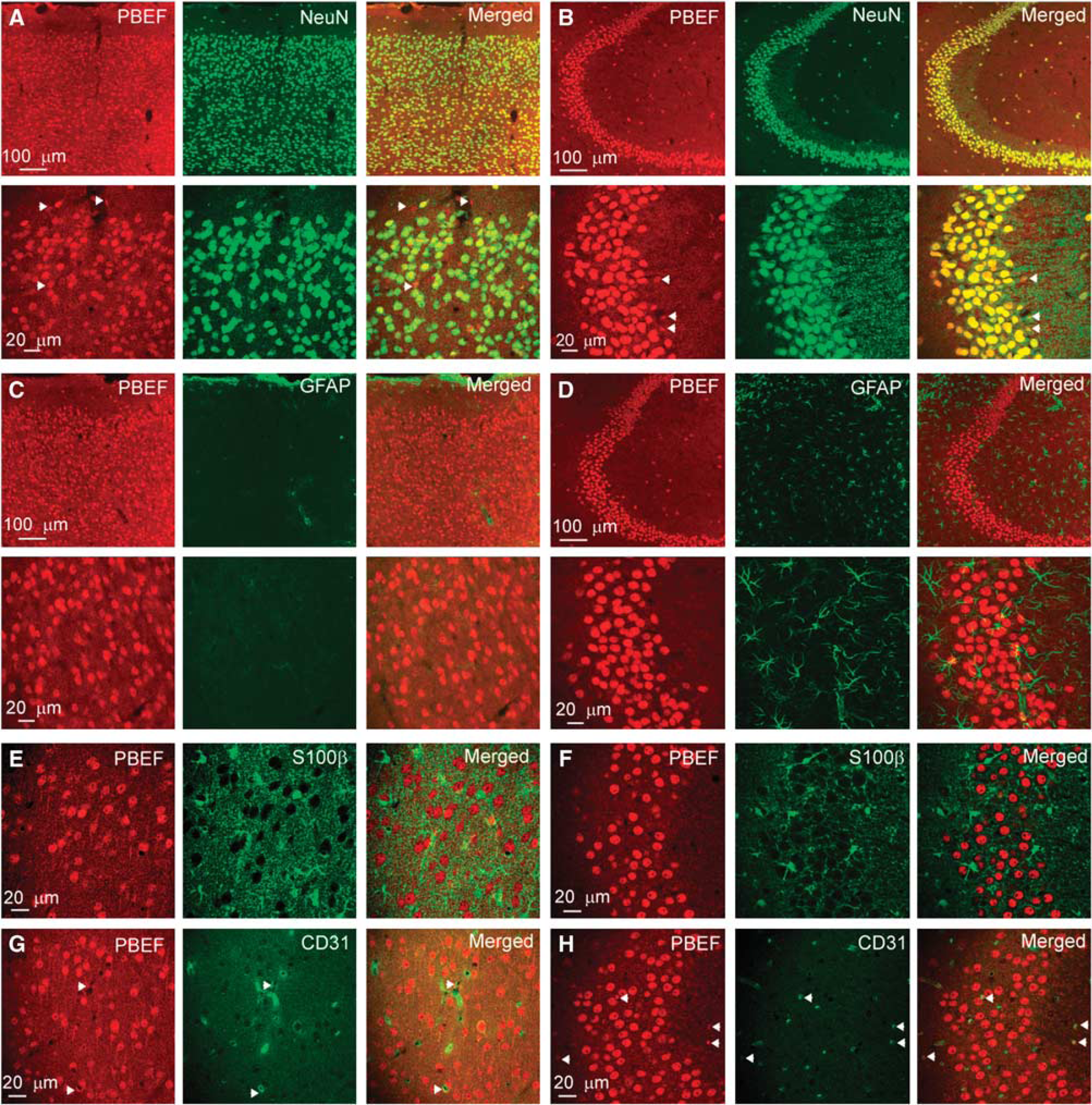
Cellular expression of PBEF in the mouse brain. (
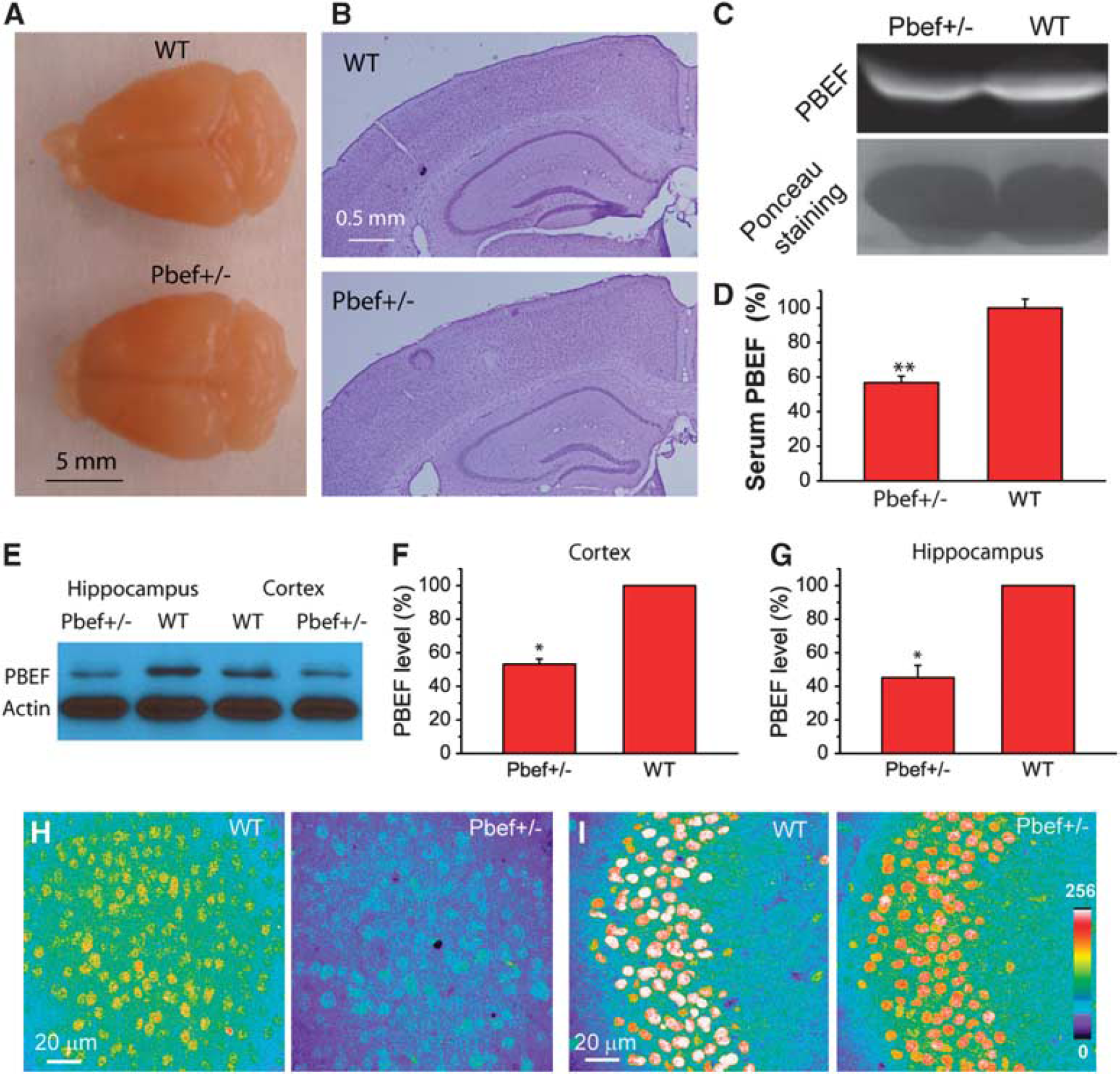
Characterization of Pbef+/− mice. (
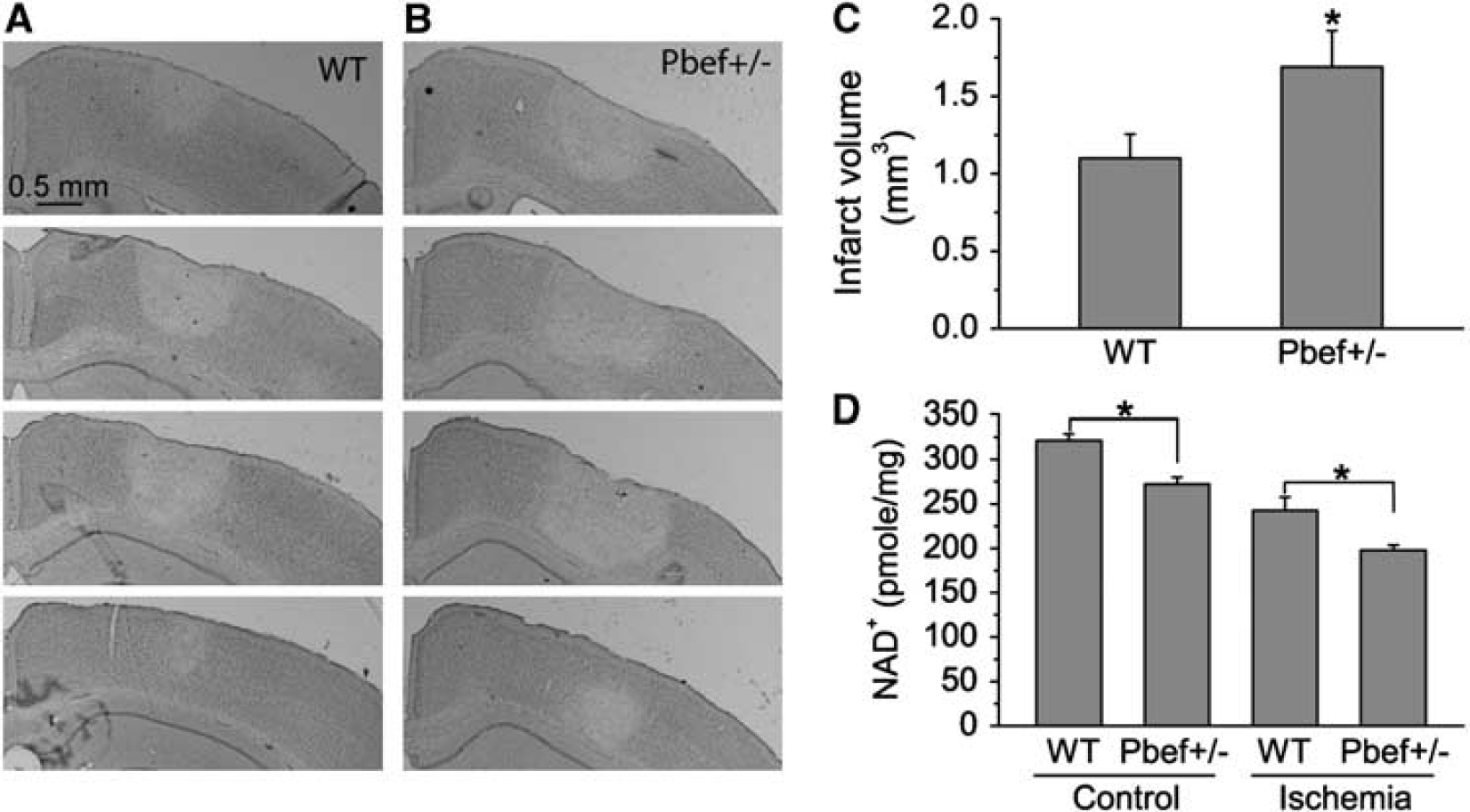
Pbef+/− mice have more severe photothrombosis-induced brain damage than do WT mice. (
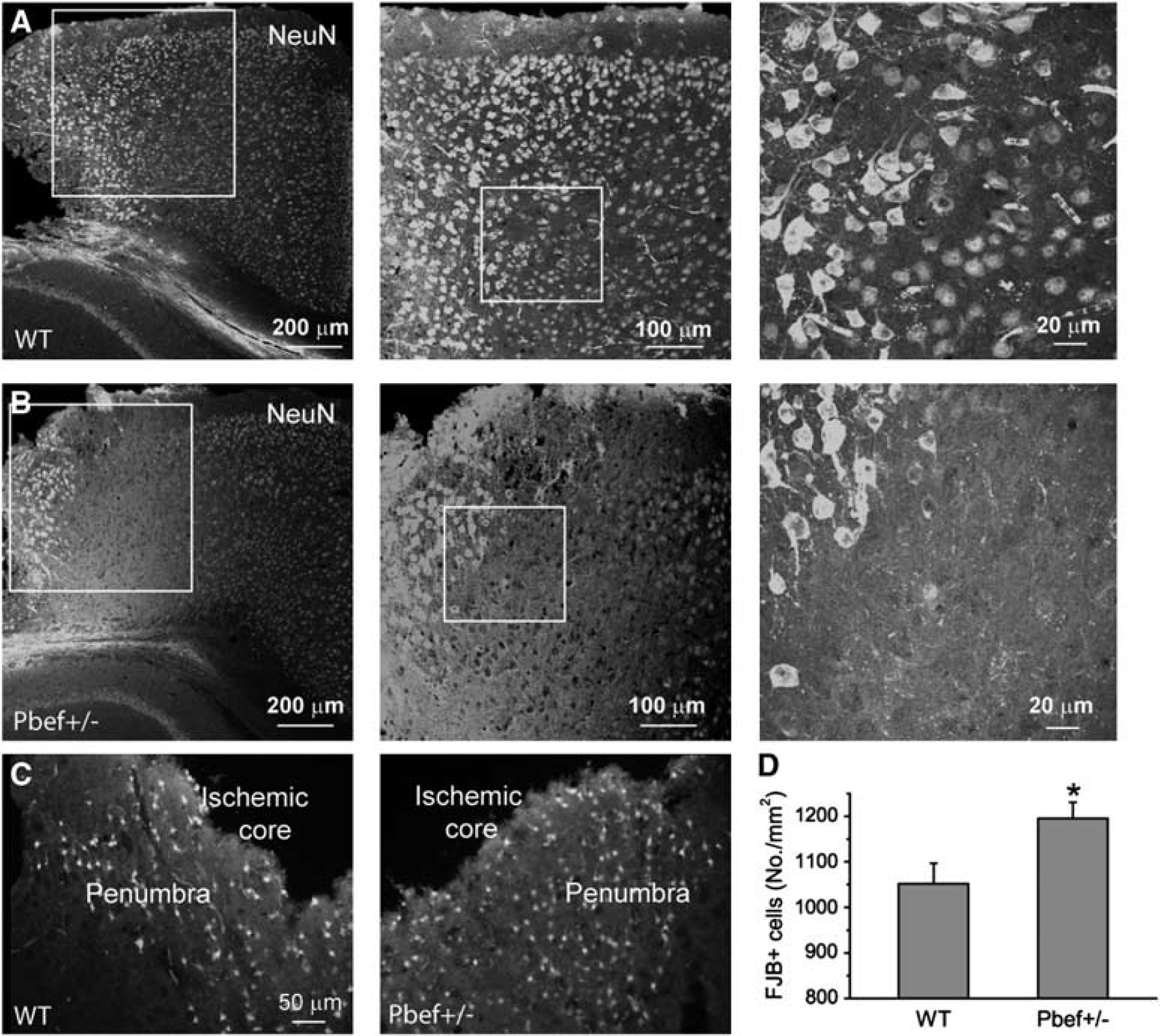
Neuronal responses and degeneration in WT and Pbef+/− mice after ischemia. (
Statistical Analysis
Data are reported as mean ± s.e.m. Statistical comparisons of data between WT and Pbef+/− mice were made by Student's t-test. P < 0.05 was considered to be statistically significant.
Results
Pre-B-Cell Colony-Enhancing Factor is Exclusively Expressed in Neurons in the Mouse Brain
To determine the cellular distributions of PBEF in the mouse brain, we performed double immunostaining of PBEF with neuronal marker NeuN and glial markers. Our data showed that PBEF is expressed both in the cortex and in the hippocampus of the mouse brain (Figures 1A and 1B). The majority of PBEF+ cells are colocalized with NeuN signals (Figures 1A and 1B). To test whether PBEF is expressed in astrocytes, we first performed double staining of PBEF and GFAP (Figures 1C and 1D). In the hippocampal region, there is no overlapping between PBEF and GFAP signals. As astrocytes in the cortex express very weak GFAP, we performed additional double immunostaining of S100β and PBEF. S100β is another astrocyte-specific maker that is expressed in astrocytes in the cortex and hippocampus. Our results show that there is no colocalization between S100β and PBEF (Figures 1E and 1F). Thus, our study shows that astrocytes both in the cortex with weak GFAP expression and in the hippocampus with strong GFAP expression do not express PBEF based on assays with a polyclonal rabbit anti-PBEF peptide antibody. These PBEF+ cells are not likely expressed in the microglia either, based on their morphology and the population of NeuN+/PBEF+ cells. As NeuN is a neuron-specific marker, which is evenly expressed in the nuclei of neurons, the colocalization of PBEF with NeuN shows that PBEF is largely expressed in neuronal nuclei. This can be further shown by the colocalization of PBEF and nuclear-stained DAPI fluorescence (data not shown). In the hippocampal region, all fields CA1, CA2, CA3, and the dentate gyrus exhibited higher signal intensity than did the cortex (see CA3 region in Figure 1B).
Notably, in a very small fraction of cells, the PBEF signal is not colocalized with NeuN (i.e., PBEF+/NeuN– cells, arrows in Figures 1A and 1B). We counted these cells as the percentage of total PBEF+ cells. We found that 4.9% ± 0.3% and 4.5% ± 0.5% of PBEF+ cells in the cortex and hippocampus, respectively, are NeuN– cells. In another words, 95.1% and 95.5% NeuN+ cells are PBEF+ in the cortex and hippocampus, respectively (Supplementary Figure 1). As these cells are morphologically different from NeuN+/PBEF+ neurons and GFAP+/S100β+ astrocytes, and some of them are located at the walls of the blood vessel, we further performed double staining of PBEF and CD31, an endothelial marker, to investigate whether PBEF is expressed in endothelial cells in blood vessels. A small number of PBEF+ cells were identified as CD31+ cells (Figures 1G and 1H, see arrows). Thus, we confirmed that PBEF is also expressed in endothelial cells. As they account for a very small percentage in PBEF+ cells and do not belong to neurons and glial cells, we concluded that PBEF is exclusively expressed in the neurons in the central nervous system.
Characterization of Pbef+/− Mice
To examine the roles of PBEF in brain ischemia, we used Pbef+/− mice in our experiment because Pbef−/− mice are embryonically lethal as reported previously (Hong et al, 2008; Revollo et al, 2007). Pbef+/− mice are viable and fertile like WT (Pbef+/+) mice. These mice display no gross anatomic abnormalities of the whole brain (Figure 2A) and brain cytoarchitecture in the cortex and hippocampus revealed by Nissl staining (Figure 2B). To confirm the reduction of PBEF level in Pbef+/− mice, we first performed an immunoblot to measure the PBEF level in blood serum. We found that PBEF expression levels in Pbef+/− mice were decreased by 43% in blood serum as compared with WT mice (Figures 2C and 2D). As our current study focuses on the role of PBEF in brain injury, we further analyzed PBEF expression levels in the brain and neurons of Pbef+/− and WT mice. Western blot analysis showed that PBEF expression levels in the hippocampus and the cortex were reduced by 55% and 47%, respectively, in Pbef+/− mice as compared with WT mice (Figures 2E–2G). Under the same conditions of immunostaining and imaging acquisition, we found that the intensity of the PBEF signal in the cortex and hippocampus in Pbef+/− mice was much lower than that in WT mice (Figures 2H and 2I). Similar to WT mice, PBEF is mainly expressed in the neurons in the cortex and hippocampus in Pbef+/− mice. These results confirmed that Pbef+/− mice indeed have reduced PBEF expression levels in the brain and neurons. Furthermore, Pbef+/− mice exhibited a significant reduction in NAD+ level in the cortex as compared with WT mice under normal conditions (Figure 3D), suggesting the critical role of PBEF in energy metabolism.
Pbef+/− Mice have Exacerbated Ischemic Brain Damage Compared with Wild-Type Mice After Photothrombosis
As PBEF is involved in energy metabolism and ischemic injury is induced by energy depletion, we postulated that PBEF levels in the neurons might affect brain injury after ischemia. To test this hypothesis, we used a photothrombosis-induced mouse model of cerebral ischemia (Ding et al, 2009; Wang et al, 2010) and Pbef+/− mice to determine whether knock out of PBEF would increase brain damage after ischemia. Photothrombosis was induced unilaterally in the barrel cortex of the brain through craniotomy. One day after ischemia, animals were perfused and sectioned. We evaluated the brain damage in Pbef+/− and WT mice. Nissl staining clearly showed a brain infarct area with sharp transition between the normal and ischemic regions in the cortex (Figures 3A and 3B). The damage was limited to the cortical region under this ischemic condition. Infarct volumes were measured in each mouse to assess the effect of PBEF on brain damage. Our data show that Pbef+/− mice (N = 10) have much larger infarct area and volume than do WT mice (N = 9) (Figure 3C). The data show that PBEF has a brain-protective role in photothrombosis-induced ischemia.
We further tested whether reduced PBEF expression affects energy metabolism after ischemia by comparing cortical NAD+ level in WT and Pbef+/− mice after ischemia. At 6 hours after ischemia, NAD+ levels in both WT and Pbef+/− mice are reduced as compared with the normal condition as expected, and furthermore, NAD+ level is significantly lower in Pbef+/− mice than that in WT mice (Figure 3D), suggesting that PBEF protects neurons through increased NAD+ production in ischemia.
Neuronal Degeneration After Photothrombosis-Induced Ischemia
Neuronal nuclei protein is located in the nuclei of neurons in the normal brain and is widely used for detecting the injury of neurons, including those from ischemic brains (Unal-Cevik et al, 2004). Neuronal nuclei staining showed that there are three different regions showing the different patterns of NeuN immunoreactivity (Figures 4A and 4B). The NeuN+ cells close to the ischemic core had NeuN expression in the cytosol and nuclei. These cells might represent the degenerating neurons. The second region shows reduced NeuN reactivity, suggesting the loss of NeuN expression. The decrease in NeuN reactivity may represent minor injury caused by ischemia, but not necessarily neuronal cell loss (Unal-Cevik et al, 2004). Neurons far from the ischemic area had normal NeuN staining in nuclei, suggesting they are located in the uninjured region. This NeuN-reactivity pattern is the same for WT and Pbef+/− mice, but the loss of NeuN reactivity in the second region is much more severe in Pbef+/− mice than in WT mice. The data suggest that WT and Pbef+/− mice have different degrees of neuronal degeneration in the penumbra. To further compare neurodegeneration in the ischemic regions in Pbef+/− and WT mice, we performed FJB staining, which specifically detects the degenerating neurons (Liu et al, 2009b). The FJB+ cells were located in the penumbra with a distinct transition to a normal region (Figure 4C). We counted FJB+ cells in the penumbra and found that the density of FJB+ cell is significantly higher in Pbef+/− mice than in WT mice (Figure 4D). These results indicate that the knockout of PBEF exacerbated ischemia-induced neuronal degeneration.
Responses of Glial Cells (Microglia and Astrocytes) After Ischemia
Iba1 can stain both resting and activated microglia, as well as monocytes/macrophages (Voskuhl et al, 2009). We found that Iba1+ cells in the contralateral side of ischemia (Supplementary Figures 2A and 2C), and in the region distant from the ischemic region of the ipisilateral side (Supplementary Figures 2B and 2D, left) of WT and Pbef+/− mice show extensive branching and are ramified, a typical morphology of resting microglia. However, in the penumbra regions of WT and Pbef+/− mice, many Iba1 + cells exhibit either a globoid morphology (i.e., an enlarged cell body with shorter and fewer processes, see arrows) in the region adjacent to ischemic core or elongated processes (see arrows) adjacent to the normal region (Supplementary Figures 2B and 2D, right). These cells might be activated microglia and infiltrated monocytes and macrophages. Moreover, Iba1+ cells with elongated processes lost Iba1 reactivity in the cell body compared with resting microglia. Nevertheless, it is not evident that the knockout of PBEF affects the morphology of Iba1+ cells at 24 hours after ischemia. Using GFAP staining, we found that GFAP expression is very weak in both the contralateral side and the ipisilateral side of the cortex at this time point (Supplementary Figures 2E–H). The Pbef+/− mice showed similar GFAP expression pattern to WT mice. The data suggest that PBEF does not affect glial cell activation.
Discussion
In this study, we showed that PBEF is exclusively expressed in neurons in the mouse brain, but surprisingly not expressed in glial cells, which outnumber neurons. In addition, a small set of PBEF+ cells are endothelial cells. Pre-B-cell colony-enhancing factor has been firmly established as a NAD+ synthetic enzyme (Garten et al, 2009; Revollo et al, 2004). The brain is an organ that consumes a large amount of energy, and among the various cell types, neurons especially demand a high energy level to perform their functions (Hertz, 2008). The highly specific expression of PBEF in neurons may provide the required energy to neurons through synthesis of NAD+.
When subjected to photothrombosis-induced ischemia (Ding et al, 2009; Wang et al, 2010), Pbef+/− mice exhibit more severe brain damage and neuronal degeneration than do WT mice. The loss of NeuN in the penumbra was much more pronounced in Pbef+/− mice than in WT mice, which may represent a mild injury. Consistent with this observation, we found that Pbef+/− mice have higher density of FJB+ neurons in the penumbra than WT mice. Thus, we conclude that neuronal PBEF has a brain-protective role in cerebral ischemia.
There are multiple mechanisms that can interpret our results. As ischemic injury results from energy depletion, a compensation for an energy deficit might ameliorate acute neuronal death and brain damage through reduced glutamate excitotoxicity, a common mechanism of acute neuronal damage in the mouse model of ischemia (Dirnagl et al, 1999). Intracellular PBEF has been established as a rate-limiting enzyme in the mammalian salvage pathway of NAD+ biosynthesis (Revollo et al, 2004, 2007). Nicotinamide adenine dinucleotide is a major coenzyme for numerous redox reactions in energy metabolism and performs multiple biologic functions in regulating metabolism, stress response, and aging (Imai, 2009a, b ). It is possible that knockout of PBEF expression in Pbef+/− mice reduced the energy metabolism due to the reduction of NAD+ production, and consequently exacerbated brain damage in Pbef+/− mice. Consistent with this hypothesis, NAD+ levels in WT mice are indeed significantly higher than those in Pbef+/− mice at 6 hours after photothrombosis, i.e., in the acute phase of ischemia. Although it is conceivable that NAD+ level is time dependent after ischemia induction, 6 hours after ischemia is a critical time when neuronal death can be detected. Recent studies have suggested that replenishing NAD+ significantly enhanced neuronal survival after oxygen–glucose deprivation insult on primary cultured neurons (Wang et al, 2008). In addition, administration of nicotinamide, a substrate of PBEF, can prevent NAD+ depletion and reduce neuronal excitotoxicity and cerebral ischemic injury (Liu et al, 2009a). Taken together, our studies support the hypothesis that knockout of PBEF in Pbef+/− mice exacerbates ischemic brain injury through reduced energy metabolism resulting from the decreased production of NAD+ levels.
The activation of NAD+-consuming enzymes such as poly(ADP-ribose) polymerases (PARPs) and deacetylase sirtuins might also contribute to ischemic injury. It has been reported that the inhibitor of PARP-1 suppresses inflammation and neuronal death in ischemia (Hamby et al, 2007; Kauppinen et al, 2009), suggesting that PARP inhibition produces a brain-protective effect in ischemia. Poly (ADP-ribose) polymerase activation can deplete NAD+, subsequently leading to neuronal death by energy depletion. Knockout of PARP-1 renders mice more resistant to cerebral ischemia (Eliasson et al, 1997). Consumption of NAD+ by PARP-1 might further inhibit NAD+-dependent energy metabolism (production), thus increasing ischemia-induced brain damage in Pbef+/− animals.
Sirtuins are a family of deacetylase that use NAD+ as a cosubstrate to remove acetyl groups from proteins and are critical regulators of metabolism that can increase organism and cell survival through modification of the biologic function of the targets. SIRT1, a nuclear deacetylase of sirtuin family, has a critical role in neuronal disorders and energy homeostasis in the central nervous system (Araki et al, 2004; Qin et al, 2006; Ramadori et al, 2008). As SIRT1 deacetylase activity is dependent on the cosubstrate NAD+, the knockout of PBEF in Pbef+/− mice might reduce the deacetylase activity of SIRT1 and thus attenuate its protective role after ischemia through reduced NAD+ production.
In summary, we found that PBEF is exclusively expressed in the neurons in the mouse brain and is neuroprotective against brain ischemia. This novel protective role of PBEF in ischemia might be important in finding a therapeutic target for brain ischemia. Future studies would be required to generate PBEF-overexpressing mice or, using viral transduction, to overexpress PBEF in neurons and to test whether those molecular manipulations ameliorate brain damage and neuronal death after ischemia. In addition, further study on the mechanisms of the brain-protective role of PBEF is warranted.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.