
Abstract
Minocycline is protective in models of transient middle cerebral artery occlusion (MCAO). We studied whether minocycline and doxycycline, another tetracycline derivative, provide protection in permanent MCAO. Because minocycline inhibits matrix metalloprotease-9 (MMP-9), we also compared minocycline's protective effect in wild type (wt) and MMP-9 knock-out (ko) mice. Wt FVB/N, Balb/C, and two lines of MMP-9 ko and their wt C57Bl/6 control mice were subjected to 24- or 72-hour permanent MCAO. Drug administration was started either 12 hours before or 2 hours after the onset of MCAO. Infarct size was determined by triphenyltetrazolium staining or T2-weighted MRI. Zymography was used to study the expression of MMPs. In wt strains, tetracycline treatments started before MCAO reduced the infarct size by 25% to 50%, whereas the treatment started after MCAO was not protective. Minocycline inhibited ischemia-provoked pro-MMP-9 induction in wt mice, but was not protective in MMP-9 ko mice. Pro-MMP-2 was induced by MCAO in wt and MMP-9 ko mice. MCAO-induced pro-MMP-2 was downregulated by minocycline treatment in wt mice but remained in MMP-9 ko mice at the same level as in saline-treated wt mice. Tetracyclines are protective in permanent MCAO when the treatment is started before the insult. Minocycline may provide protection by interfering with MMPs.
Introduction
Minocycline and doxycycline are tetracyclines, which have antiinflammatory effects independent of their antimicrobial action. Recent data have shown that minocycline prevents microglial activation (Tikka and Koistinaho, 2001; Tikka et al, 2001, 2002; Yrjänheikki et al, 1998, 1999) and has notable beneficial effects in animal models of global and transient focal cerebral ischemia (Wang et al, 2003; Xu et al, 2004; Yrjänheikki et al, 1998, 1999) and other brain injuries (Brundula et al, 2002; Chen et al, 2000; Du et al, 2001; Zhu et al, 2002). Although inhibition of a number of cellular targets, including caspase-1 (Chen et al, 2000; Yrjänheikki et al, 1998, 1999), caspase-3 (Chen et al, 2000), cyclooxygenase-2 (Yrjänheikki et al, 1999), inducible nitric oxide synthase (Yrjänheikki et al, 1998, 1999), p38 mitogen-activated protein kinase (Du et al, 2001; Tikka and Koistinaho, 2001; Tikka et al, 2002), cytochrome c release (Zhu et al, 2002), and matrix metalloprotease-9 (MMP-9) (Brundula et al, 2002) have been reported to be associated with the neuroprotective effects of minocycline, the mechanism of protection in stroke is still unclear.
A growing body of evidence, starting from the reports on dental injury, indicate that tetracyclines suppress the levels and activities of MMPs (Golub et al, 1998; Paemen et al, 1996; Sorsa et al, 1998; Van den Steen et al, 2002), enzymes capable of degrading extracellular matrix (ECM) components (del Zoppo and Mabuchi, 2003; Fukuda et al, 2004; Van den Steen et al, 2002). Cerebral ischemia triggers a series of pathways that result not only in intracellular proteolytic cascades, but also extracellular proteolytic processes, in which major components of the basal lamina are degraded, leading to the breakdown of the blood–brain barrier and loss of microvascular integrity (Barone and Feuerstein, 1999; del Zoppo and Mabuchi, 2003; Fukuda et al, 2004; Rosenberg et al, 1998). Vascular injury results in edema, activation of resident microglial cells, infiltration of circulating inflammatory cells into the brain, and, finally, neuronal death (Barone and Feuerstein, 1999; del Zoppo and Mabuchi, 2003; Van den Steen et al, 2002). Cytokine-inducible MMP, MMP-9 (gelatinase B, 92-kDa type IV collagenase), is thought to be the terminal enzyme in the ECM remodeling cascade (Van den Steen et al, 2002). Recent studies have shown that inhibition of MMP-9 by genetic, immunologic or pharmacologic approaches reduces infarct volumes in mice (Asahi et al, 2000, Romanic et al, 1998), suggesting a deleterious role for MMP-9 in ischemic brain injury. In addition, expression of pro-MMP-2, at least in nonhuman primate models of stroke, is directly related to neuronal injury and its role in ischemic injury is supported by the findings that both activation of pro-MMP-2 and its protease activity are triggered after brain ischemia (Chang et al, 2003; Fukuda et al, 2004).
The aim of this study was to investigate whether minocycline or doxycycline are protective also in permanent focal cerebral ischemia. Because minocycline inhibits MMPs (Brundula et al, 2002; Golub et al, 1998; Paemen et al, 1996; Sorsa et al, 1998; Van den Steen et al, 2002) and MMP-9 inhibition is thought to be beneficial in the early phase of developing brain injury (Asahi et al, 2000; Gasche et al, 1999; Romanic et al, 1998; Rosenberg et al, 1998), we also hypothesised that minocycline may provide protection in cerebral ischemia through inhibition of MMP-9. Several mouse strains were studied to exclude the possibility that the effect of minocycline is restricted to particular strains.
Materials and methods
Animals
The numbers of animals of each strain used are indicated in Table 1. Male 2-month-old FVB/N mice and male 2-month-old Balb/C mice were from the National Laboratory Animal Center, Kuopio, Finland. To investigate the role of MMP-9, we used 2- to 3-month-old male wt and age-matched MMP-9 mutant mice from two different strains (Dubois et al, 1999; Opdenakker et al, 2003). In the first experiment, MMP-9 ko1 mice, backcrossed 10 times onto C57Bl/6 background, were compared with wt controls. We observed leakiness of a truncated MMP-9 in the brain of this strain (unpublished). Therefore, we used mice from another ko strain (MM-9 ko2), which completely lack MMP-9 expression, and their littermate controls (75% C57Bl/6) in consecutive experiments (Opdenakker et al, 2003). MMP-9 ko1 and ko2 mice and their wt controls were bred at the Rega Institute for Medical Sciences, University of Leuven, Belgium. The experiments were performed according to NIH guidelines for animal care and use, and were approved by the Animal Care and Use Committee of Kuopio University.
Strains, numbers, treatment and usage of experimental animals
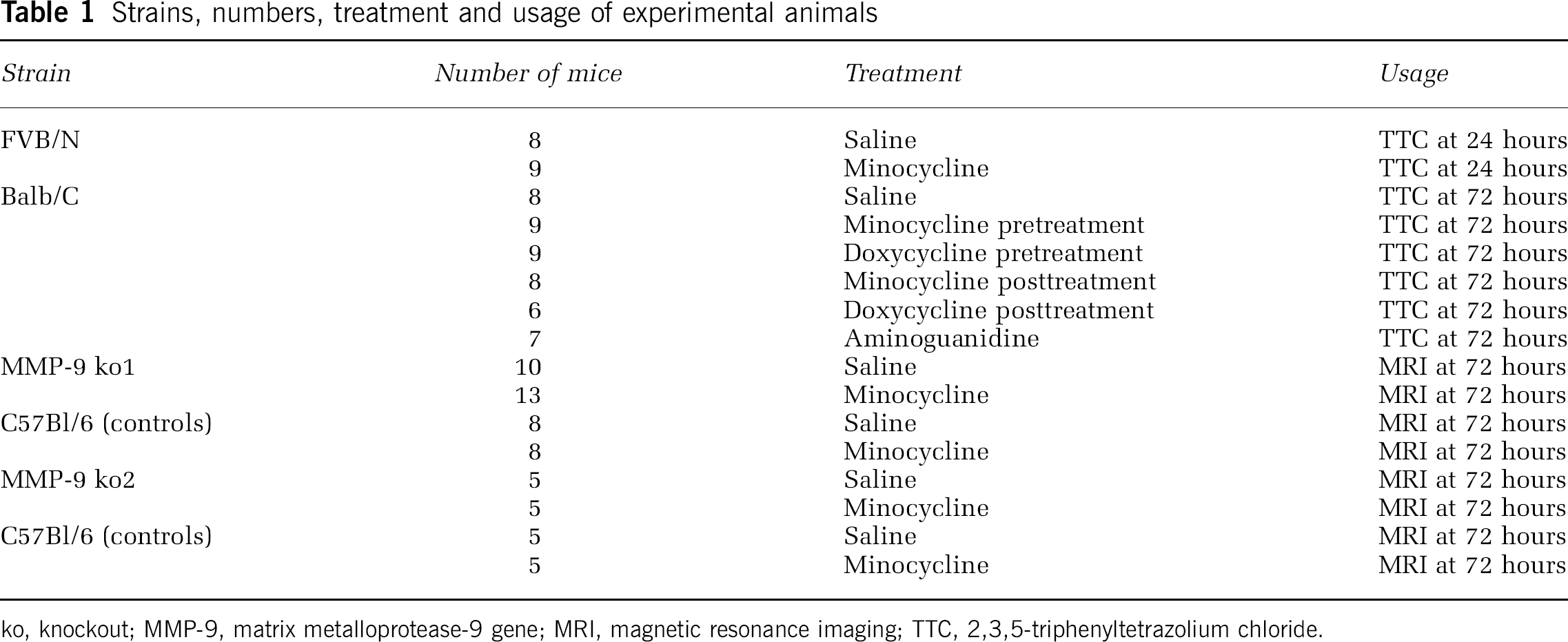
ko, knockout; MMP-9, matrix metalloprotease-9 gene; MRI, magnetic resonance imaging; TTC, 2,3,5-triphenyltetrazolium chloride.
Permanent Focal Cerebral Ischemia
Mice were anesthetized with 5% halothane (70% N2O, 30% O2) for induction and 1.25% halothane for maintenance. The rectal temperature was maintained at 36°C to 37°C with a heating pad during the surgery. The left MCA was exposed and electrocoagulated at the level of the inferior cerebral vein, as described previously (Koistinaho et al, 2002).
Drug Treatments
In the pretreatment groups, intraperitoneal minocycline hydrochloride (Sigma, St. Louis, MO, USA) or doxycycline hydrochloride (Sigma) injections were started 12 hours before MCAO at a dose of 45 mg/kg. The mice were injected at 12-hour intervals at a dose of 60 mg/kg during the first 24 hours and thereafter at 45 mg/kg until killed. Posttreatment with minocycline was performed similarly, except that the injections were started 2 hours after ischemia. Control mice received equal volumes of saline. Aminoguanidine (Sigma, 200 mg/kg at 12-hour intervals after 24 hours), an inhibitor of inducible nitric oxide synthase, which provides protection against permanent ischemic damage (Zhang and Iadecola, 1998), was used as a positive control treatment.
Measurement of Infarct Volume
FVB/N and Balb/C mice were killed 24 and 72 hours after MCAO, respectively. Eight 1-mm-thick coronal sections per brain were stained with 2.0% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma) at 37°C for 20 mins. The stained sections were scanned and images were opened in Image ProPlus program (Media Cybernetics). An indirect method that excludes the confounding effects of brain swelling was used to quantify the infarcted area in the cortex (Lin et al, 1993). Both strains of MMP-9-deficient mice and their wt controls were anesthetized with a subcutaneous injection of 1:4 dilution of fentanyl-fluanisone (Hypnorm, Janssen-Cilag) and midazolame (Dormicum, Roche) in sterile H2O administered 0.05 mL/10 g and externally fixed to a custom-built animal holder for high-resolution MRI. Warm air was blown through the magnet bore during MRI. An s.m.i.s. console (Surrey Medical Imaging Systems) interfaced to a 9.4-T vertical magnet (Oxford Instruments) was used for MRI with a single-loop surface coil (diameter 27 mm) in transmit/receive mode. Multislice T2-weighted images were acquired using a single-echo spin-echo method (time-to-repetition 3000 ms, time-to-echo 45 ms, 4 scans/line, field of view 25.6 × 12.8 mm2, matrix size 256 × 64 and slice thickness 1 mm). Quantitation of infarct volumes was performed as described above.
Determination of Physiologic Variables and Preparation of Cortical Tissue Samples
A polyethylene catheter was placed into the femoral artery, 100-μL blood samples were withdrawn and analyzed for arterial pO2, pCO2, pH (ABL-5, Radiometer), and glucose (One Touch FastTake, Lifescan). Mice were decapitated and the similarity of cerebrovascular anatomy in the MCA territory was confirmed by observing and photographing the contralateral hemisphere using the preparation microscope. For the analysis of MMPs, the ischemic area in the cortex and the corresponding area in the contralateral cortex were dissected out, snap-frozen in liquid nitrogen and stored at −80°C until used.
Zymography
Gelatin zymography was based on the previously published method with modifications (Hibbs et al, 1985). Sodium dodecyl sulfate (SDS)-polyacrylamide gels (8%) were copolymerized with 0.15% porcine skin gelatin, casein or casein plasminogen (Sigma). Enzyme samples (10-μL), corrected for protein, were mixed with 10 μL of 2 × nonreducing sample buffer (0.125 mol/L Tris–HCl, 20% glycerol, 4% SDS, and 0.003% bromophenol blue) and simultaneously loaded on a gel. The gels were subjected to electrophoresis at 4°C. Samples containing 2 ng bacterial collagenase (Sigma) and molecular weight markers were included on each gel. After electrophoresis, the gels were washed, renatured, and incubated in collagenase buffer (21 mmol/L Tris–HCl, pH 7.6, 10 mmol/L CaCl2, and 0.04% NaN3) for 20 hours at 37°C. Thereafter, the gels were single-step stained (Phast-Gel Blue R, Pharmacia) and zymolysis was quantitated with GelDoc flatbed scanner (BioRad) and ImageQuaNT software.
Statistical Analysis
All analyses were performed using SPSS for Windows software (SPSS, Chicago, IL, USA). Quantitative data are expressed as mean±s.e.m. Two-group comparisons were evaluated by two-tailed Student's t-tests. Multiple comparisons were analyzed by one-way ANOVA followed by Tukey HSD post hoc test. Significance was assumed if P≤0.05.
Results
Pretreatment with minocycline or doxycycline protects in permanent focal cerebral ischemia
In the first experiment, the ability of minocycline to protect against permanent ischemic injury in FVB/N mice was studied. When the treatment was started 12 hours before the onset of MCAO and the mice were killed at 24 hours after the ischemic insult, minocycline was shown to reduce the infarct volume by 33% (P<0.01, t-test, Figures 1A and 1B).
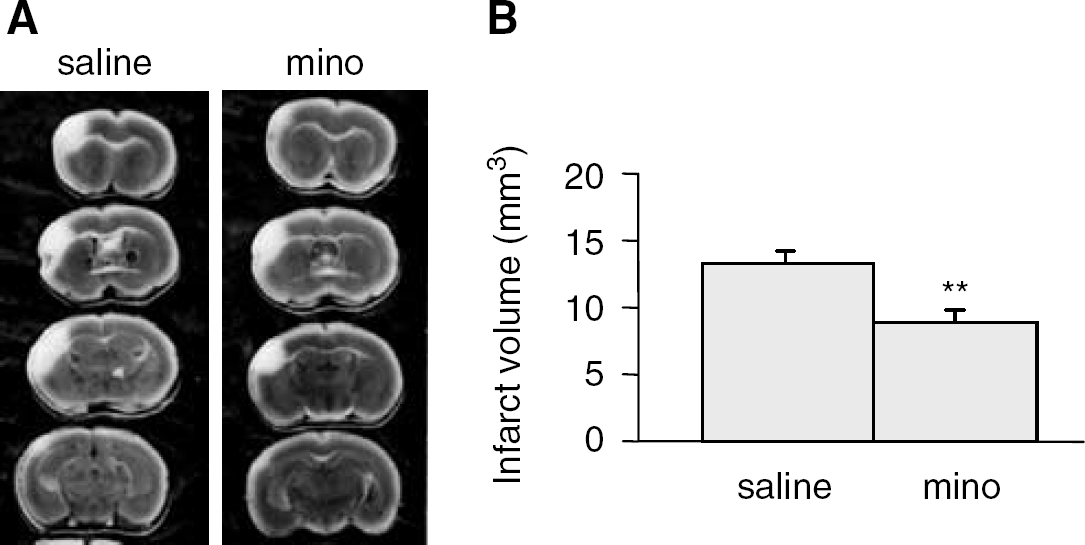
Minocycline protects against permanent focal ischemic damage. (
The second set of experiments was performed to confirm this finding using another mouse strain and also to study the effect of minocycline and doxycycline in a longer treatment and survival protocol. As a positive control, 20 mg/kg of aminoguanidine provided significant (−33% versus saline treated controls, P≤0.05) protection in Balb/C mice at 72 hours. When Balb/C mice were subjected to permanent MCAO 12 hours after the start of minocycline or saline treatment and infarcts quantified 72 hours later, mice treated with minocycline showed significantly smaller infarcts than saline-treated mice (Figures 2A and 2B, P⩽0.0001). Administration of doxycycline starting 12 hours before the onset of MCAO also significantly reduced the ischemic volumes at 72 hours (Figures 2A and 2B). When minocycline treatment was started 2 hours after MCAO, no significant protection was achieved (Figures 2A and 2B).
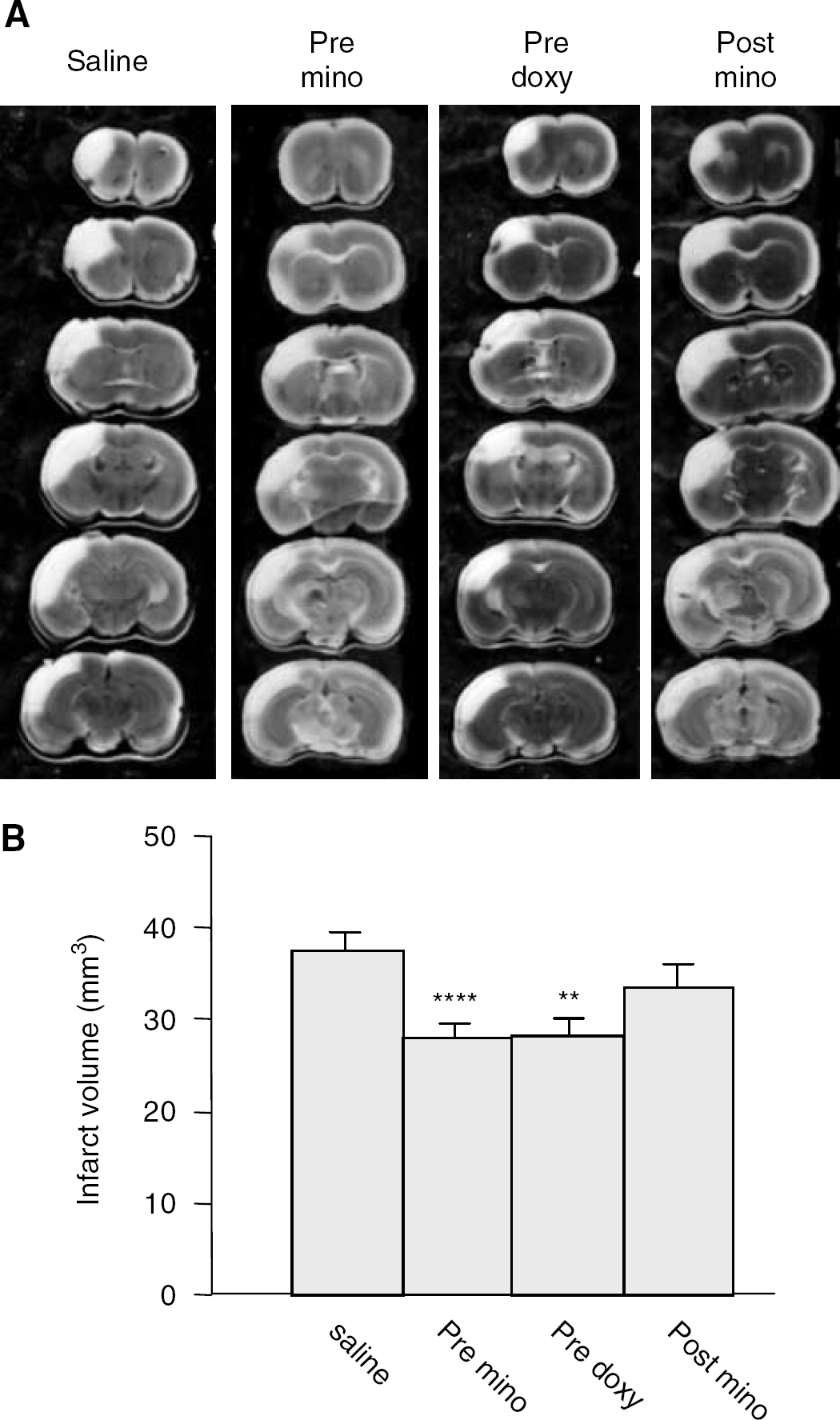
Pretreatment with either minocycline or doxycycline is protective in permanent ischemia. (
Minocycline's Protective Effect is Abolished in MMP-9 ko1 and MMP-9 ko2 Mice
MMP-9 ko1 and MMP-9 ko2 strains and their wt controls were divided into minocycline and saline pretreatment groups and subjected to focal permanent ischemia. MRI data at 72 hours revealed that minocycline treatment in wt C57Bl/6 mice started 12 hours before the onset of MCA occlusion resulted in 50% reduction in infarct volumes (Figures 3A and 3B). In contrast, minocycline treatment did not reduce the infarct size in MMP-9 ko2 (Figures 3A and 3B) or MMP-9 ko1 (data not shown). Importantly, infarct volumes did not differ between saline-treated MMP-9 ko and wt mice (Figures 3A and 3B). Neither MMP-9 deficiency nor minocycline treatment affected ischemic physiologic variables (Table 2). Cerebrovascular anatomy is a strong determinant in vulnerability to focal brain ischemia. However, the MCA territory was of similar size and had approximately the same distribution in MMP-9 ko2 and wt mice (Figure 4).
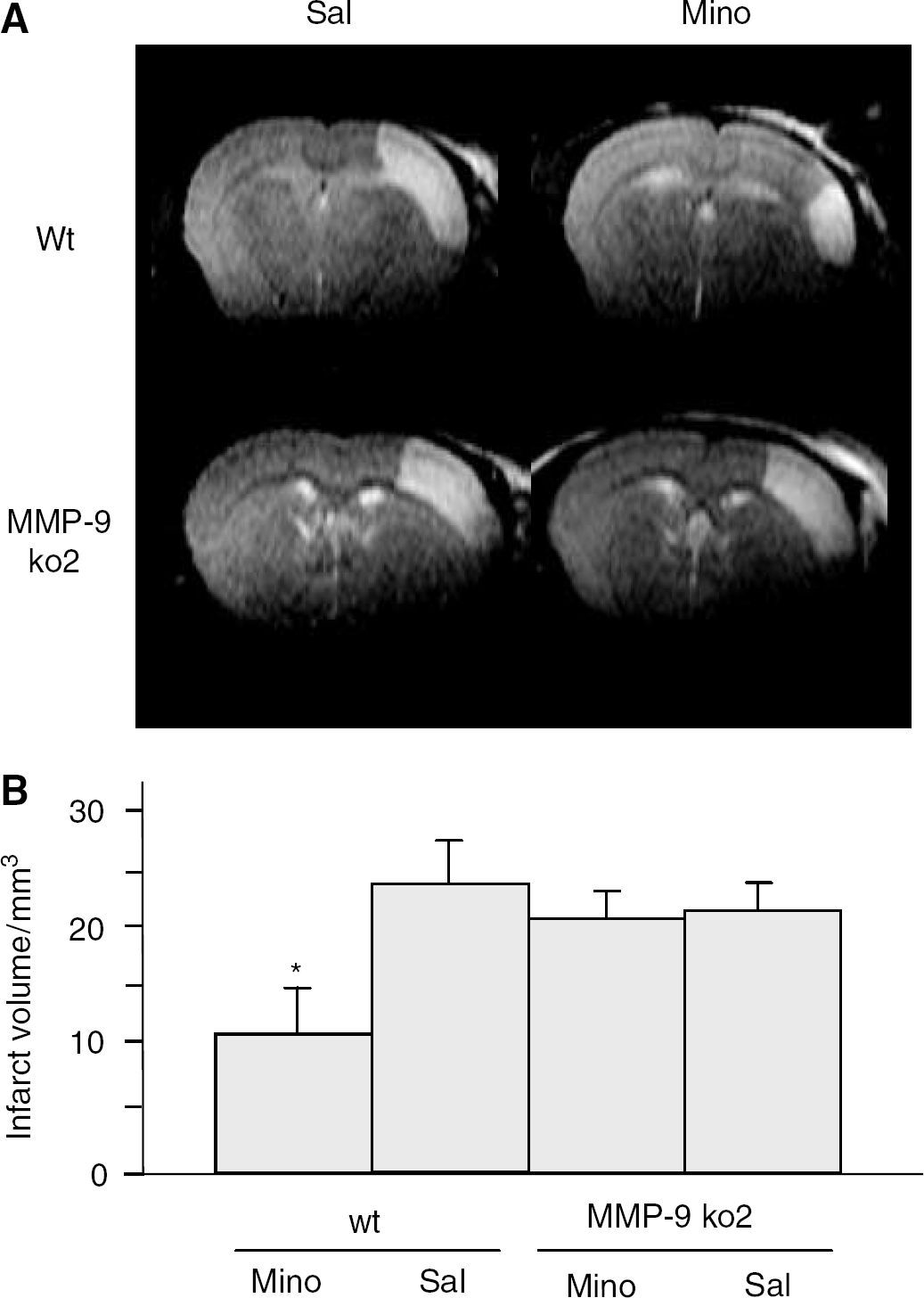
Minocycline treatment reduces the infarct size in wt but not in MMP-9 ko2 mice. (
The distribution of the contralateral MCA territory was similar in MMP-9 ko2 (
Physiologic variables of ischemic mice
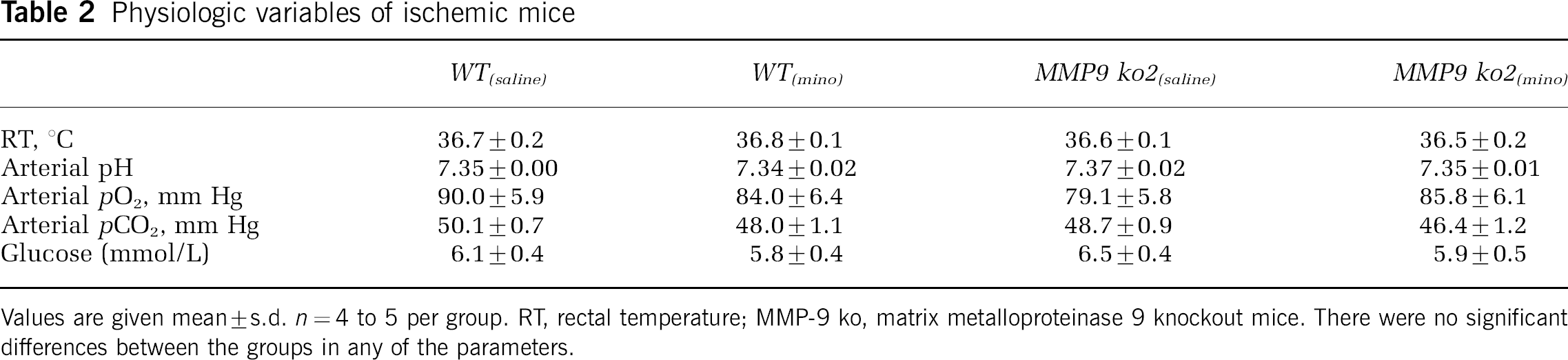
Values are given mean±s.d. n=4 to 5 per group. RT, rectal temperature; MMP-9 ko, matrix metalloproteinase 9 knockout mice. There were no significant differences between the groups in any of the parameters.
Minocycline Downregulates pro-MMP-2 Expression in Wt but not in MMP-9 ko Mice
Zymography showed that, in wt mice, pro-MMP-9 and pro-MMP-2 were induced in the ischemic brain regions 72 hours after MCAO and that minocycline inhibited both pro-MMP-9 and pro-MMP-2 to the levels which did not significantly differ from sham-operated mice (Figures 5A and 5B). In the MMP-9 ko1, expression of a truncated MMP-9 was unaltered by ischemia or minocycline (not shown). In the other MMP-9 ko strain, no MMP-9 expression was detected. Pro-MMP-2 was induced in MMP-9 ko2 mice, and after minocycline treatment pro-MMP-2 levels remained at a significantly higher level than in minocycline-treated wt mice and corresponded to the levels detected in saline-treated wt mice (Figure 5B). MMP-3 was barely detectable in casein zymography and no alterations in plasmin activity were seen in casein plasminogen zymography (data not shown).
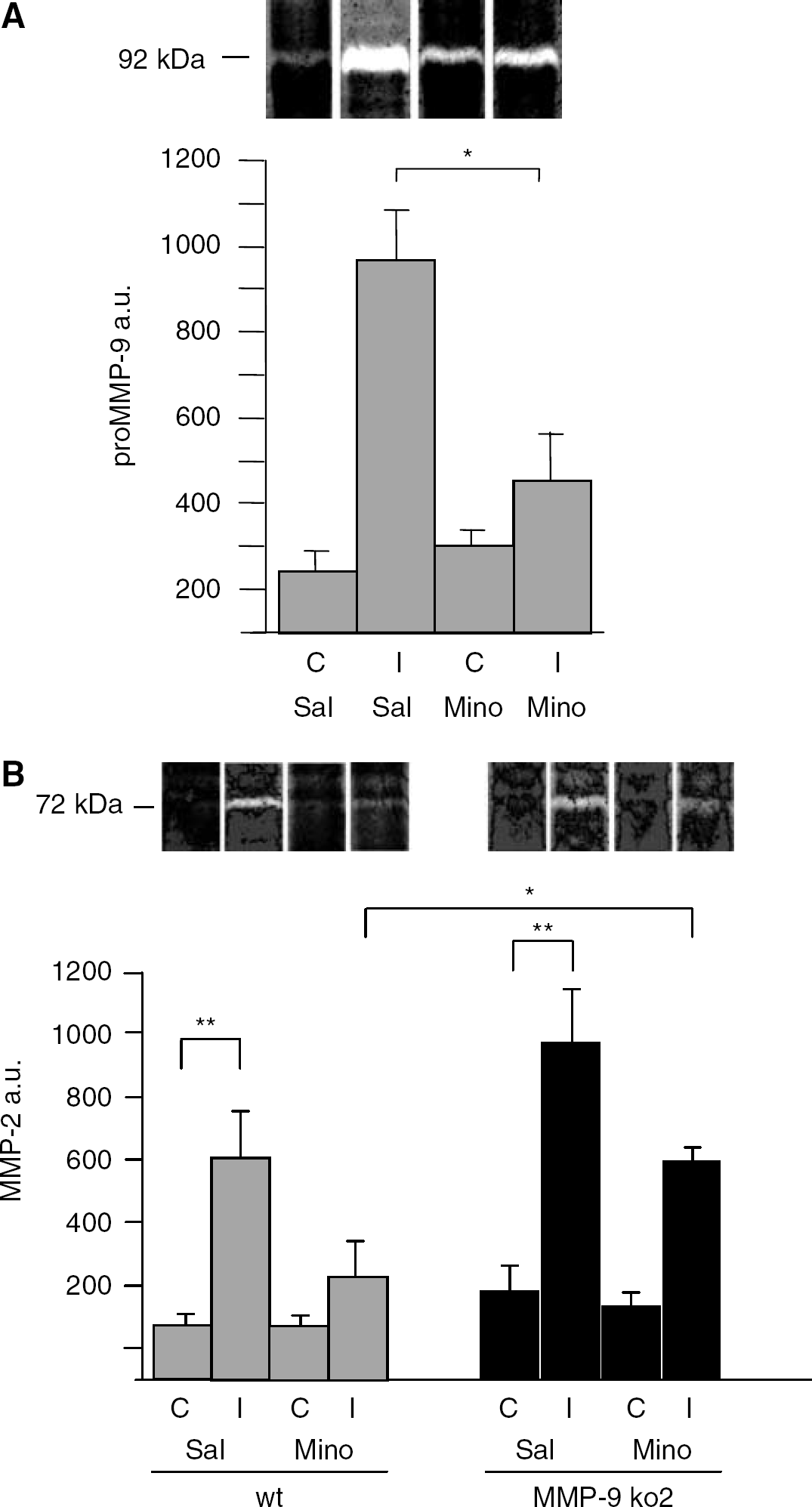
Zymography analysis of MMP-9 (
Discussion
Minocycline treatment has proven to be beneficial in animal models of transient brain ischemia (Wang et al, 2003; Xu et al, 2004; Yrjänheikki et al, 1998, 1999) and chronic neurodegenerative diseases (Brundula et al, 2002; Chen et al, 2000; Du et al, 2001; Tikka et al, 2001; Zhu et al, 2002). We now show that minocycline treatment is protective also in a mouse model of permanent cerebral ischemia, suggesting that minocycline is beneficial in cerebral ischemia by mechanisms that are not linked to the production of free radicals during reperfusion. Our model of permanent MCAO in mice results in consistent cortical infarcts and previously reported neuroprotection with an iNOS inhibitor, aminoguanidine (Zhang and Iadecola, 1998), is seen also in this model. Minocycline pretreatment (12 hours before onset of permanent MCAO) reduced the ischemic infarct size by 33% at 24 hours, and, depending on the mouse strain, by 26% to 50% at 72 hours after the onset of MCAO. However, when the treatment was started 2 hours after the onset of permanent MCAO, no significant protection was achieved. It is important to notice that another semisynthetic tetracycline, doxycycline, showed similar protective effect of permanent stroke in this model.
Tetracyclines, including minocycline, inhibit various MMPs, including MMP-9 and MMP-2 (Golub et al, 1998; Paemen et al, 1996; Sorsa et al, 1998; Van den Steen et al, 2002). We used two different gene ko mouse lines to study the role of MMP-9 in the protective effects of minocycline in permanent cerebral ischemia. In MMP-9 ko-1 mouse line, pro-MMP-9 is expressed as a truncated form in the brain, where its levels were not affected by ischemia or minocycline. In the nonleaky MMP-9 ko-2 mouse line, MMP-9 is not detectable in any organ (Opdenakker et al, 2003). Three important findings were made with these mice. First, the protective effect of minocycline was lost in MMP-9 ko mice. Second, MMP-9 deficiency did not provide protection in permanent MCAO model at 72 hours, and third, the ischemia-induced increase in pro-MMP-2 levels was inhibited by minocycline in wt mice but not substantially in MMP-9 ko mice. Previous studies have showed that both pro-MMP-2 and pro-MMP-9 are induced after cerebral ischemia (Asahi et al, 2000; Chang et al, 2003; Clark et al, 1997; Fujimura et al, 1999; Fukuda et al, 2004; Gasche et al, 1999; Romanic et al, 1998; Rosenberg et al, 1996). From these two MMPs, MMP-9 is more clearly linked to the pathogenesis of cerebral ischemia, because genetic (Asahi et al, 2000), immunologic (Romanic et al, 1998) or pharmacologic inhibition (Asahi et al, 2000; Romanic et al, 1998) of MMP-9 results in decreased size of ischemic lesions. However, MMP-9 deficiency was not associated with reduced infarcts in our ischemia model. Compared with the previous study, in which MMP-9 deficiency decreased the lesion size, the ischemia model, and especially the strain of MMP-9 ko mice used by us, were different (Asahi et al, 2000). Even though the ischemia model is a determinant of ischemic lesion, both the strain (Majid et al, 2003) and the postischemia follow-up time might be more important factors that could explain the controversy. Even though MMPs may contribute to the opening of the blood–brain barrier and development of edema and secondary inflammation (Barone and Feuerstein, 1999; del Zoppo and Mabuchi, 2003; Gasche et al, 1999; Rosenberg et al, 1998; Van den Steen et al, 2002), MMPs also play a role in physiologic remodeling, neurite outgrowth and release of growth factors (Del Zoppo and Mabuchi, 2003; Van den Steen et al, 2002), all of which might be beneficial shortly after the acute injury. Because the infarct volumes were quantified at 24 hours by Asahi et al (2000), and at 72 hours in the current study, the diverted functions of MMP-9 after the first 24 hours could explain why the final outcome of permanent MCAO in MMP-9 ko mice is not beneficial. Alternatively, compensatory changes in MMP-2 levels may occur when MMP-9 expression is silenced, as has been reported in myocardial infarction model with another MMP-9 ko line (Ducharme et al, 2000). In all our four different treatment groups (saline-treated shams, minocycline-treated shams, saline-treated MCAO, minocycline-treated MCAO), the pro-MMP-2 levels were slightly higher in MMP-9 ko2 mice compared with wt mice, but the difference was significant only in minocycline-treated ischemic mice. Even though MMP-2 gene knock out in one study (Asahi et al, 2001) had no effect on brain injury 24 hours after transient focal ischemia, several studies suggest that MMP-2 may contribute to ischemic injury (Clark et al, 1997; Chang et al, 2003; Fujimura et al, 1999; Fukuda et al, 2004).
The idea that MMP-2 may play a role in permanent focal ischemia is also supported by our finding that minocycline provides protection only when it is able to downregulate pro-MMP-2 near to control level. High levels of pro-MMP-2 after minocycline treatment in MMP-9 ko2 mice may also explain why MMP-9 deficiency was not sufficient to result in reduced brain infarcts. It is of interest that minocycline has been reported to inhibit p38 MAPK (Du et al, 2001; Tikka et al, 2001), which in some models regulates MMP-2 expression (Park et al, 2002; Vayalil and Katiyar, 2004; Vitale et al, 2004) and contributes to infarction after permanent MCAO (Koistinaho et al, 2002). Finally, upregulation of other extracellular proteases occurs in cardiac ischemia of another MMP-9 ko mouse line (Ducharme et al, 2000), suggesting that the lack of MMP-9 might be compensated by other proteases in ischemic tissues. Such compensation could also explain the different effects of minocycline and MMP-9 deficiency on infarct volumes. However, the present and previous experiments with our MMP-9 ko1 or ko2 mice have not revealed compensatory changes in various proteases, yet the possibility that other, unknown factors compensate the MMP-9 deficiency cannot be excluded.
In conclusion, our results support the hypothesis that minocycline provides protection in cerebral ischemia by interfering with MMPs. In agreement with the recent studies on the role of MMP-2 in stroke (Chang et al, 2003; Fukuda et al, 2004), our study points to the possibility that MMP-2 may play a destructive role in acute ischemic injury in the brain.