
Abstract
Delayed complications of subarachnoid hemorrhage (SAH) such as angiographic vasospasm, cortical spreading ischemia, microcirculatory dysfunction, and microthrombosis are reported in both patients and animal models of SAH. We demonstrated previously that SAH is associated with increased oxidative stress in the brain parenchyma, and that this correlates with dysfunction of endothelial nitric oxide synthase (eNOS) (homodimeric uncoupling). Uncoupling of eNOS exacerbated oxidative stress and enhanced nitric oxide (NO) depletion, and was associated with multiple secondary complications such as microthrombosis, neuronal apoptosis, and release of reactive oxygen species. Thus, we hypothesized that genetic abbrogation of eNOS would confer a beneficial effect on the brain after SAH. Using a prechiasmatic injection model of SAH, we show here that eNOS knockout (KO) significantly alleviates vasospasm of the middle cerebral artery and reduces superoxide production. Endothelial nitric oxide synthase KO also affected other nitric oxide synthase isoforms. It significantly increases neuron nitric oxide synthase expression but has no effect on inducible nitric oxide synthase. Endothelial nitric oxide synthase KO decreases Zn2+ release after SAH, reduces microthrombi formation, and prevent neuronal degeneration. This work is consistent with our findings where, after SAH, increased oxidative stress can uncouple eNOS via Zn2+ thiolate oxidation, or theoretically by depletion or oxidation of tetrahydrobiopterin, resulting in a paradoxical release of superoxide anion radical, further exacerbating oxidative stress and microvascular damage.
INTRODUCTION
Delayed complications of subarachnoid hemorrhage (SAH) such as angiographic vasospasm, cortical spreading ischemia, microcirculatory dysfunction, and microthrombosis are reported in some animal models of SAH.1-9 They are postulated to contribute to delayed cerebral ischemia in humans and thus, contribute to death and disability in patients with SAH. 3
One theory behind how these secondary complications cause cerebral ischemia and infarction revolves around the depletion of or excess generation of the potent vasodilator nitric oxide (NO).3,6,10 Nitric oxide is synthesized enzymatically by three main nitric oxide synthase (NOS) isoforms, endothelial, neuronal, and inducible NOS, or in a nonenzymatic fashion via a series of nitrate-nitrite reduction-oxidation reactions. 11 Nitric oxide produced under physiologic conditions confers a number of beneficial effects such as vasodilation of the microcirculation and maintenance of normal vascular tone, antithrombotic effects, prevention of excess platelet adhesion and aggregation, inhibition of endothelial apoptosis, and vascular smooth muscle cell hyperplasia. 12 We demonstrated previously that SAH in mice is associated with oxidative stress in the brain parenchyma, and that this correlates with dysfunction of eNOS (homodimeric uncoupling). Uncoupling of eNOS exacerbated oxidative stress and enhanced NO depletion, and was associated with multiple secondary complications such as microthrombosis, neuronal apoptosis, and release of reactive oxygen species (ROS).4,10
Therefore, in the current study, we hypothesized that genetic abrogation of eNOS would confer a beneficial effect on the brain after SAH. Additionally, we investigated the mechanisms behind eNOS dysfunction, the potential role of other isoforms, and their potential contributions to secondary complications after SAH, if any. We show here that eNOS knockout (KO) significantly alleviates vasospasm of the middle cerebral artery (MCA) and reduces superoxide production, increases nitric oxide synthase (nNOS) expression but has no effect on inducible nitric oxide synthase (iNOS). Endothelial nitric oxide synthase KO decreases Zn2+ release after SAH, reduces microthrombi formation, and prevents neuronal degeneration.
MATERIALS AND METHODS
Animals and Subarachnoid Hemorrhage Model
Experiments were approved by the Animal Care Committee of St Michael's Hospital, University of Toronto and complied with regulations of the Canadian Council on Animal Care and guildlines of ARRIVE (http://www.nc3rs.org/ARRIVE). The eNOS KO mice were from Jackson Laboratory (Stock #007073, Bar Harbor, ME, USA). The KO mice were created by targeted mutation with a 1.2-kb neomycin cassette replaced with 129 bp of exon 12 of the gene. This replacement disrupts the calmodulin-binding site essential to eNOS function and introduces a premature translation stop codon into the eNOS transcripts. 13 Mice homozygous for the eNOS-targeted mutation were viable and fertile. All animals were bred in our animal care facility.
Subarachnoid hemorrhage was created (by MS) by injection of autologous blood into the prechiasmatic cistern as previously reported. 14 The head was fixed in a stereotactic frame equipped with a mouse adaptor (Stoelting Company, Wood Dale, IL, USA). Relative cerebral blood flow (CBF) was measured using a laser Doppler flow probe (BLT21, Transonics systems, New York, NY, USA). Body temperature was maintained at 37 ± 0.5 °C with a homeothermic heating pad (Harvard apparatus, Holliston, MA, USA) and monitored with a rectal probe. A 0.9-mm hole was drilled in the midline of the skull 4.5mm anterior to the bregma. The drill was angled 40° caudally, and a 27-gauge spinal needle was advanced at the same angle through the burr hole to the base of the skull. For the SAH group, 80-00 μl autologous blood (nonheparinized) was withdrawn from the ventral tail artery using a 25-gauge needle and 60 μl was injected through the spinal needle over 15 seconds. For the control group, the same volume of saline was injected. Animals in the sham groups underwent insertion of the needle but no injection. Allocation to groups was sequential with an SAH and a control animal operated each day. Mice were killed 48 hours after surgery. For histologic studies, mice were perfused through the left cardiac ventricle with NaCl, 0.9%, 10 mL, followed by 150 mL, 4% paraformaldehyde in phosphate-buffered saline. Brains were removed and postfixed in 4% paraformaldehyde for 48 hours. Sections of brain were processed and embedded in paraffin, and 7-μm sections were cut using a microtome (Leica, Wetzlar, Germany). For western blot and NO/O2− detection, brains were isolated without perfusion. They were used fresh or immediately frozen at -80 °C for later analysis.
Hematoxylin and Eosin Staining and Vasospasm Measurement
Paraffin sections were incubated in xylene for deparaffinization and rehydrated through a decreasing gradient of ethanol solutions. Slides were stained with hematoxylin and eosin. After dehydration through an increasing gradient of ethanol solutions and three changes of xylene, slides were coverslipped with xylene-based mounting medium (Permount, Sigma, Oakville, Ontario, Canada) and viewed under a light microscope. The lumen perimeter of the MCA was quantified by a masked observer (by EL) at x 200 magnification using Image J (National Institutes of Health (NIH), Bethesda, MD, USA).
Zinc Assay
Free zinc (Zn2+) content in brain homogenates was measured using 4-(2-pyridylazo)-resorcinol disodium salt (PAR) assay. 15 Absorbance at 500 nm was measured using a spectrofluorometer (SpectraMAX-Gemini, Molecular Devices, Sunnyvale, CA, USA). PAR in the absence of Zn2+ is associated with low basal absorbance. However, when it is bound with Zn2+, the PAR2Zn2+ complex has absorbance at 500nm. The PAR assay specificity was tested by adding 5 μl of samples into a cuvette containing 150 μM PAR in Chelex buffer prepared with Chelex 100-treated 50 mM Tris, 100 mM NaCl, pH 7.8. Chelex was added to eliminate background cations and to test for the specificity of PAR assay, and was not utilized in subsequent buffers for brain homogenates. Assays were run at 23°C in rapidly stirred cuvettes, and the total assay volume was 1.5 mL. Toward the end of the assay, 1mM N,N-bis(carboxymethyl) glycine (NTA) was added. Under these assay conditions, NTA selectively chelates Zn2+ from the PAR2Zn2+, resulting in a gradual reduction in absorbance at 500 nm, allowing for the calculation of the amount of Zn2 + released from the protein.
NO Superoxide Anion Radical Detection
Superoxide anion radical (O−2) and NO were detected in homogenized fresh or frozen brain tissue using spectrophotometric methods. The cell-permeable fluorophore 4,5-diaminofluorescein-2-diacetate (DAF-2DA, Alexis Biochemicals, Gruenberg, Germany) was used to detect NO, and a chemiluminescence probe, 2-methyl-6-(
Western Blots for Endothelial Nitric Oxide Synthase, Neuron Nitric Oxide Synthase and Inducible Nitric Oxide Synthase
Brain tissue was homogenized in 300 μl 1% RIPA buffer with 0.1% protease inhibitor, and centrifuged at 13,000 r.p.m. for 12 minutes at 4 °C. Protein was quantified using the Bradford method, with RIPA buffer used as the blank standard. Thirty micrograms protein was loaded and separated by electrophoresis on 8% sodium dodecyl sulfate—polyacrylamide gels and transferred onto nitrocellulose membrane. We used Ponceau S and Gel Code to stain the membrane and gel, respectively. Blots were incubated with 5% milk for 60 minutes, followed by incubation with primary monoclonal antibodies (1:1000 dilution) against phosphorylated S1177-eNOS (BD Biosciences, San Jose, CA, USA), nNOS (1:500 Abcam, Cambridge, MA, USA) and iNOS (1:500 Abcam). After washing in PBS, membranes were incubated in horseradish peroxidase-conjugated antigoat polyclonal antibody (Abcam) at a dilution of 1:1000 for 50 minutes at room temperature. Reactions were developed with ECL reagent mix (Amersham Biosciences, Amersham, UK). Protein intensities were quantified by densitometric analysis utilizing Image J software (NIH). Values are expressed as relative unit or arbitrary unit after normalization to beta-tubulin controls.
For eNOS monomer and dimer detection, we used low-temperature sodium dodecyl sulfate—polyacrylamide.16,17 Samples were subjected to sodium dodecyl sulfate—polyacrylamide on 8% gels that were kept in an ice bath at 4 °C. The gels were then blotted onto nitrocellulose membranes and blocked. The membranes were incubated with primary antibodies against eNOS (Cell Signalling, Danvers, MA, USA) and the remainder of the procedure was identical to the standard western blot steps.
Immunohistological Staining of Fibrinogen for Microthrombi
Coronal brain sections were dereparaffinized and rehydrated. Antigen was retrieved by heating the sections for 20 minutes in 0.01 mmol/L sodium citrate (pH 6.0) at 96 °C. We permeabilized sections with 0.3% Triton X-100 for 10 minutes and then incubated them with 10% normal goat serum, which was diluted in 1% bovine serum albumin and 0.1% sodium azide, for 60 minutes. Slides were incubated with primary antibody (rabbit polyclonal antifibrinogen 1:200, Abcam) and secondary antibody (Alexa Fluor 568-conjugated antirabbit for fibrinogen (1:1000, Invitrogen, Burlington, Ontario, Canada). Slides were washed and coverslipped with antifading mounting medium and sealed with nail polish. Slides were viewed in a confocal microscope and images were captured using consistent parameters (pinhole size, exposure time, and laser intensity).
Fluoro-Jade Staining
Fluoro-jade B (Histo-Chem, Jefferson, AR, USA) staining was performed according to a previously published protocol. 14 After deparaffinization and rehydration, the slides were incubated in 0.06% potassium permanganate (VWR International, Strasbourg, France) for 15 minutes. Slides were then rinsed in deionized water and immersed in 0.001% Fluoro-jade B in 0.1% acetic acid for 30 minutes. They were washed and dried at 60 °C for 15 minutes. Sections were cleared in xylene and coverslipped with a nonaqueous, low-fluorescence, styrene-based mounting medium (Sigma). Slides were viewed under a confocal microscope and images taken using constant parameters (laser power, exposure time, and pinhole size).
Statistical Analysis and Data Quantification
All data are presented as means ± standard deviation (SD), and were compared between groups by analysis of variance (ANOVA). If significant variance was found, a
RESULTS
All assessment was done 48 hours after creation of SAH or control surgery.
Endothelial Nitric Oxide Synthase Knockout Does Not Affect Cerebral Blood Flow Acutely after Subarachnoid Hemorrhage The CBF changes are similar to that in our previous reports.4,6,14 In sham-operated mice, there were no changes in CBF after the insertion of the needle (Figure 1A). Subarachnoid hemorrhage animals showed a steep drop in CBF seconds after the injection. Raw CBF data of tissue perfusion units were standardized to baseline, and expressed as percent change from an averaged baseline. At the time of injection or needle insertion, the average CBF decrease was to 6.5% ± 2.2% for wild-type SAH, 5.7% ± 3.4% for eNOS KO mice, 55% ± 4.4% for saline-injected, and 95.2% ± 1.9% for sham controls (
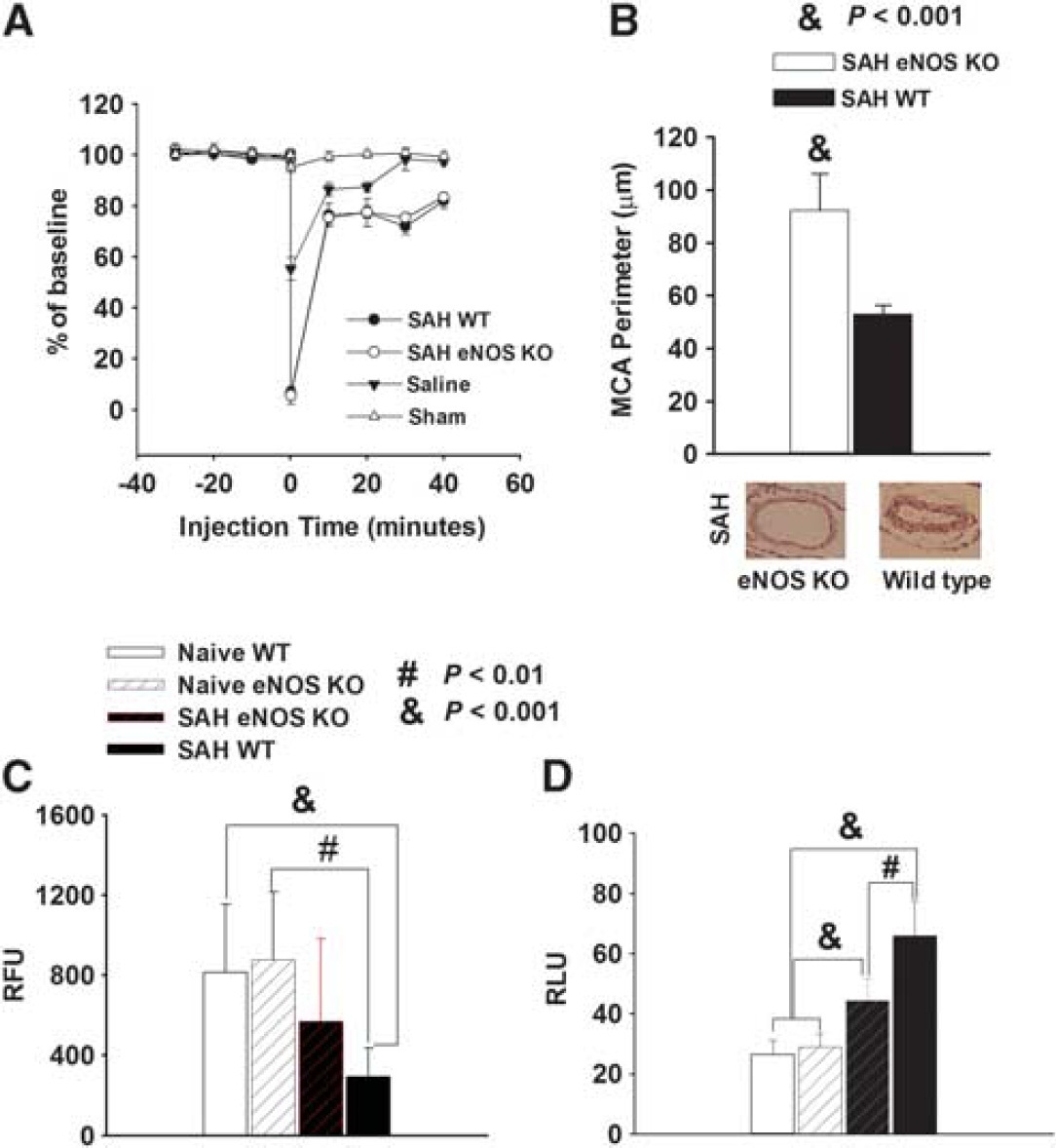
Endothelial nitric oxide synthase (eNOS) knockout (KO) reduces vasospasm, nitric oxide (NO) depletion, and O2− production with no effect on cerebral blood flow (CBF). (
Endothelial Nitric Oxide Synthase Knockout Decreases Vasospasm The eNOS KO mice exhibited less vasospasm after SAH compared with wild-type mice (Figure 1B). The perimeter of the MCA was 93 ± 14 μm for eNOS KO SAH mice and 53 ± 4 μm for wild-type SAH mice (mean ± s.d.,
Endothelial Nitric Oxide Synthase Knockout Prevents Nitric Oxide Reduction after Subarachnoid Hemorrhage We previously reported that SAH is associated with decreased NO production.
4
To determine whether reduction of vasospasm by eNOS KO is related to NO production, we measured NO in brains from mice with or without SAH (Figure 1C). Nitric oxide value is expressed as relative fluorescence units (RFU). The eNOS KO mice without SAH induction had the same NO production as wild-type mice (NO was 875 ± 340 RFU for naive eNOS KOas compared with 813 ± 339 RFU for wild-type naive animals,
Endothelial Nitric Oxide Synthase Knockout Reduces Superoxide Anion Radical after Subarachnoid Hemorrhage
We previously showed that SAH resulted in upregulated but uncoupled eNOS, which produced O2− instead of NO (O2− values are expressed as relative luminescence unit (RLU)).
4
Similarly, in this study, SAH produced a significantly higher amount of O2− than naive controls in the wild-type mice (66 ± 12 RLU for SAH, 27 ± 5 RLU for naive wild-type mice,
Endothelial Nitric Oxide Synthase Knockout Increases Neuron Nitric Oxide Synthase Expression but has no Effect on Inducible Nitric Oxide Synthase
In wild-type animals, there was significantly more eNOS monomer than dimer after SAH as compared with that in eNOS KO mice, where both eNOS monomer and dimer were expressed at a similar, almost undetectable level (monomer/dimer ratio for wild type is13.6 ± 0.9, for KO is 1.4 ± 0.03,
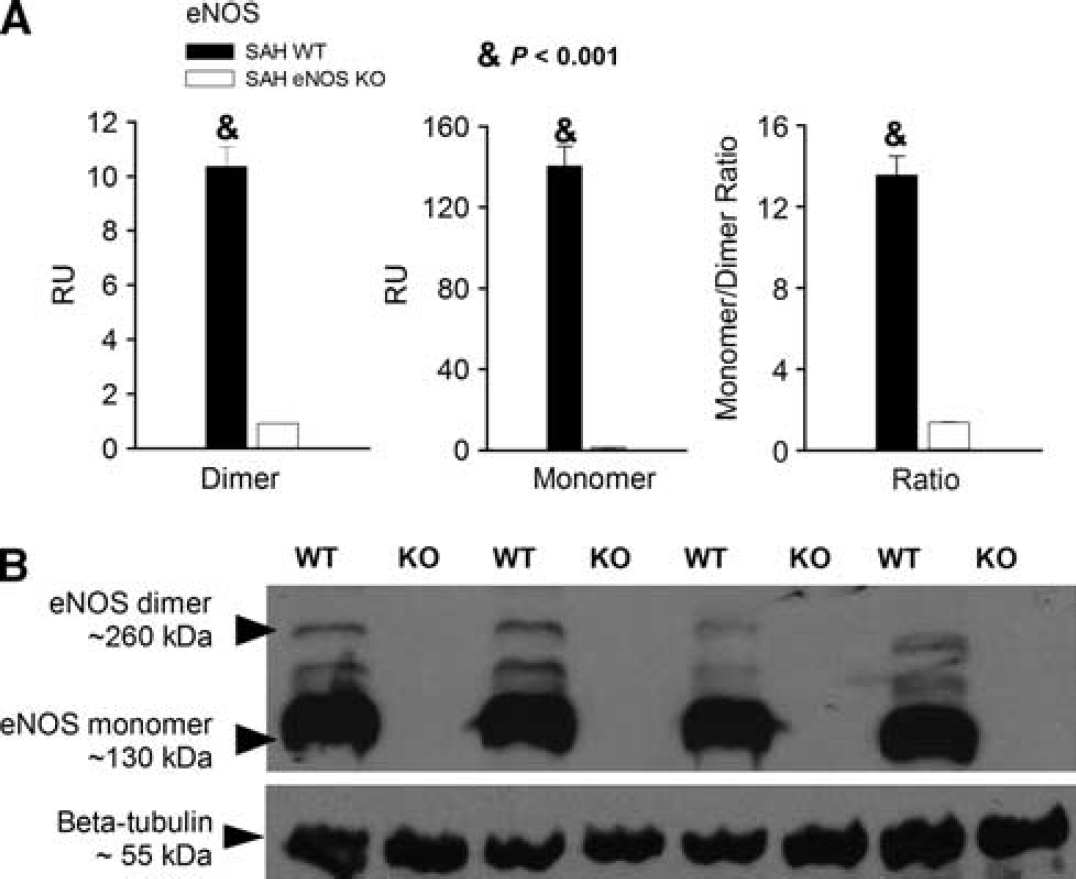
Endothelial nitric oxide synthase (eNOS) expression. (
There was a significant increase of nNOS expression in eNOS KO mice after SAH as compared with wild-type mice (nNOS expression was 167 ±13 AU for eNOS KO mice and 129 ± 4AU for wild-type mice,
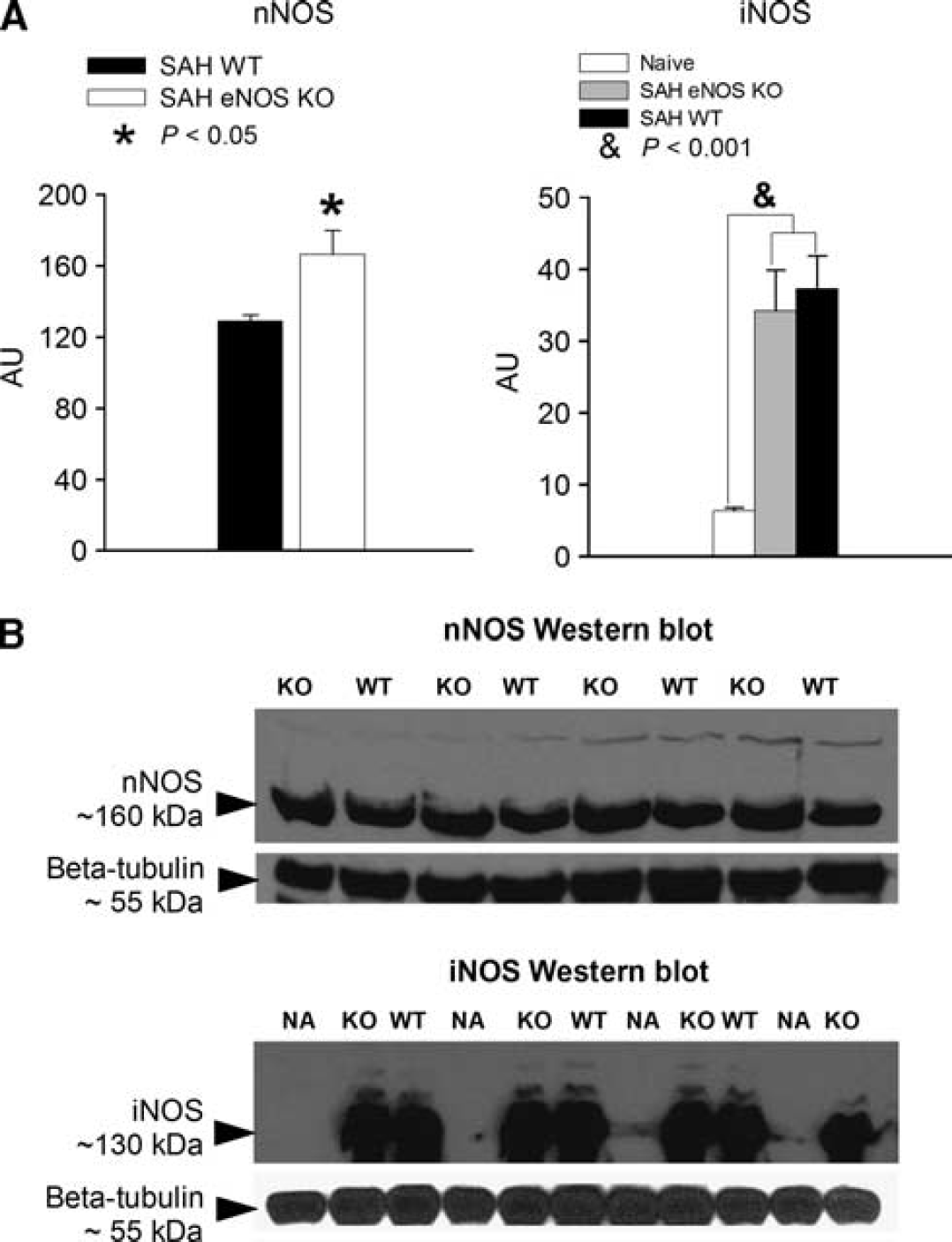
Neuron nitric oxide synthase (nNOS) and inducible nitric oxide synthase (iNOS) expression. (
Endothelial Nitric Oxide Synthase Knockout Decreases Zinc Release after Subarachnoid Hemorrhage
A hallmark of eNOS uncoupling is Zn2+ release from its thiolate complex. To investigate further the effect of SAH and eNOS KO on NOS uncoupling, we measured Zn2+ content in brain homogenates from different groups of animals. Zn2+ value is expressed as relative absorbance (RA) at 500 nm. Figure 4 shows that SAH-induced Zn2+ release was significantly decreased in eNOS KO animals as compared with wild-type animals (90 ± 10 RA for eNOS KO, 161 ± 25RA for wild type, P<0.001). In contrast, in sham-operated or saline-injected mice, eNOS KO animals had a significant increase in Zn2+ release compared with wild-type animals. In sham animals, Zn2+ release was 51 ± 7 RA for eNOS KO mice and 34±6RA for wild-type mice. In saline-injected animals, Zn2 + release was 49 ± 10 RA for eNOS KOs and 36 ± 7 RA for wild-type mice (
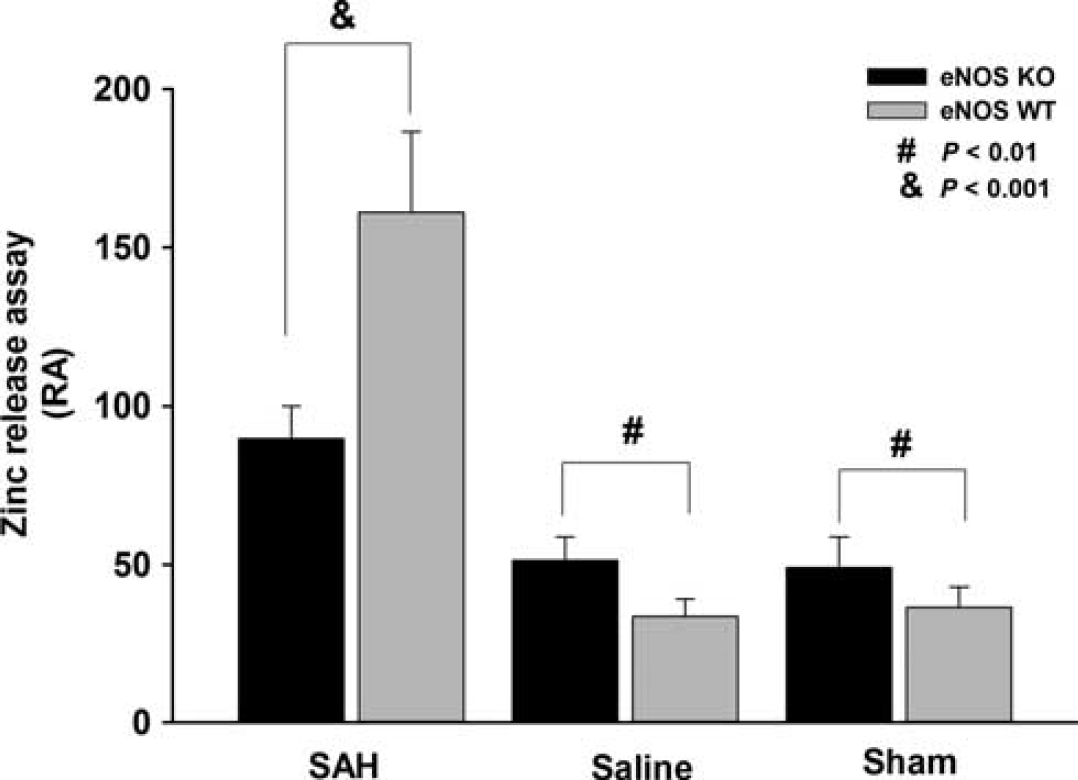
Zn2+ release determined by 4-(2-pyridylazo)-resorcinol disodium salt assay. Bar graph shows that subarachnoid hemorrahge-induced Zn2+ release was significantly decreased in endothelial nitric oxide synthase (eNOS) knockout (KO) animals as compared with wild-type animals (WT). In contrast, in sham-operated or saline-injected control groups, eNOS KO animals have a significant increase of Zn2+ release than that in WT animals. Values are means ± s.d.,
Endothelial nitric oxide synthase Knockout Reduces Microthrombi and Neuronal Degeneration
Having observed that mice with no eNOS had less vasospasm, a trend towards less NO production and a significantly higher O2− as compared with wild-type SAH mice, we investigated whether these changes were accompanied by prevention of microthrombi that have been postulated to contribute to brain injury after SAH.2,3 Antifibrinogen staining of cortical and hippocampal sections of saline-injected and SAH mice showed that mice with SAH had many fibrinogen-positive stained microvessels scattered throughout multiple layers of the cerebral cortex and hippocampus (Figure 5B). In comparison, sections from eNOS KO mice showed fewer fibrinogen-stained microvessels in the brain (Figure 5B). The number of microthrombi in wild-type SAH animals was significantly increased in both cortical and hippocampal sections in comparison to that in eNOS KO SAH animals (Figure 5A). In hippocampus, the counts are 79 ± 16 for wild-type SAH and 29 ± 16 for eNOS KO SAH, (
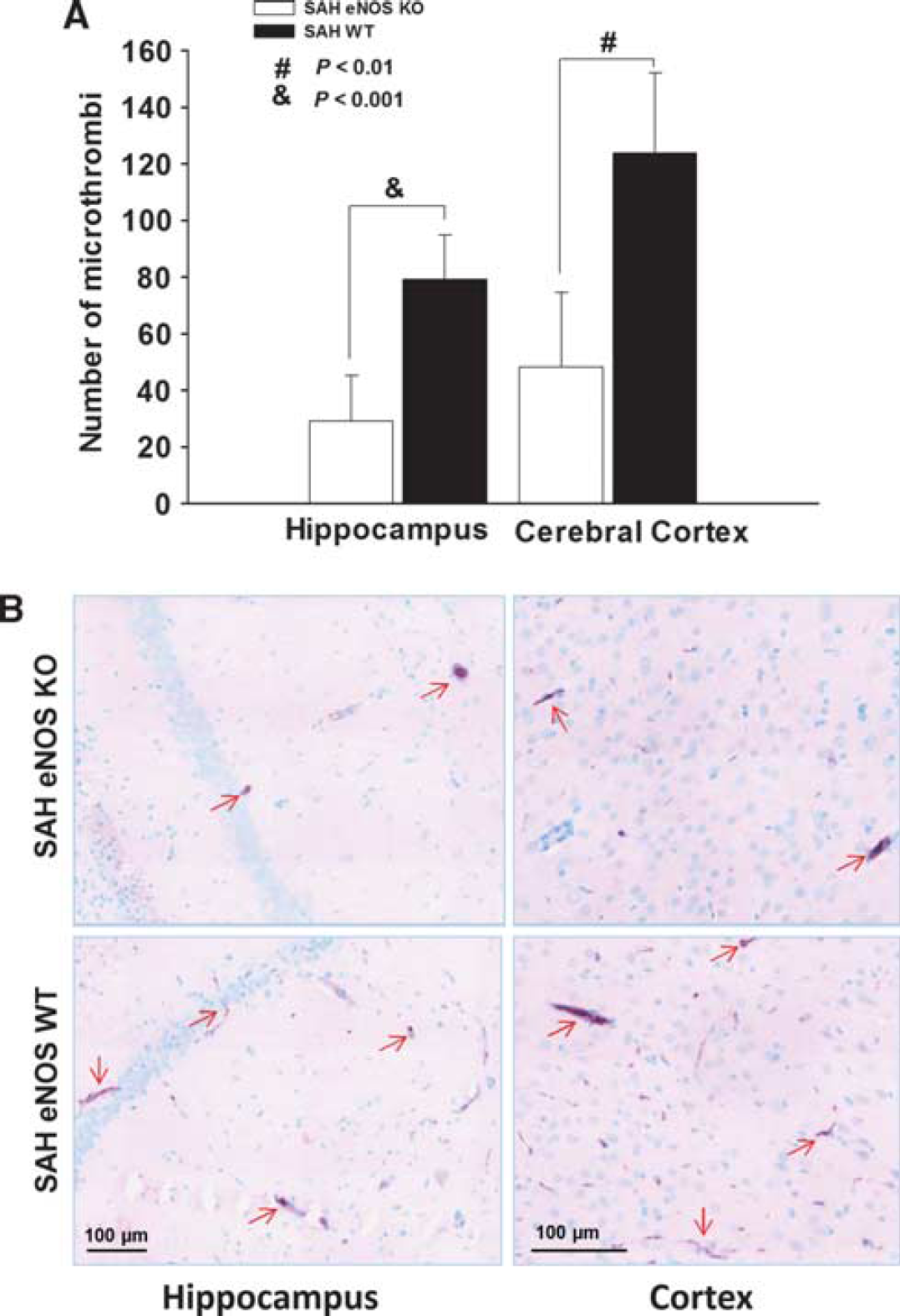
Quantification of microthrombi. (
Fluoro-jade staining to assess neuronal degeneration gave similar results to those of microthrombi counts (Figure 6). Subarachnoid hemorrhage was associated with degenerating neuronal cells in cerebral cortex and hippocampus in the wild-type mice (Figure 6B). In comparison, eNOS KO reduced the number of degenerating neurons in both regions. In hippocampus, the counts of degenerating cells were 86 ± 19 for wild-type SAH and 19 ± 8 for eNOS KO SAH (
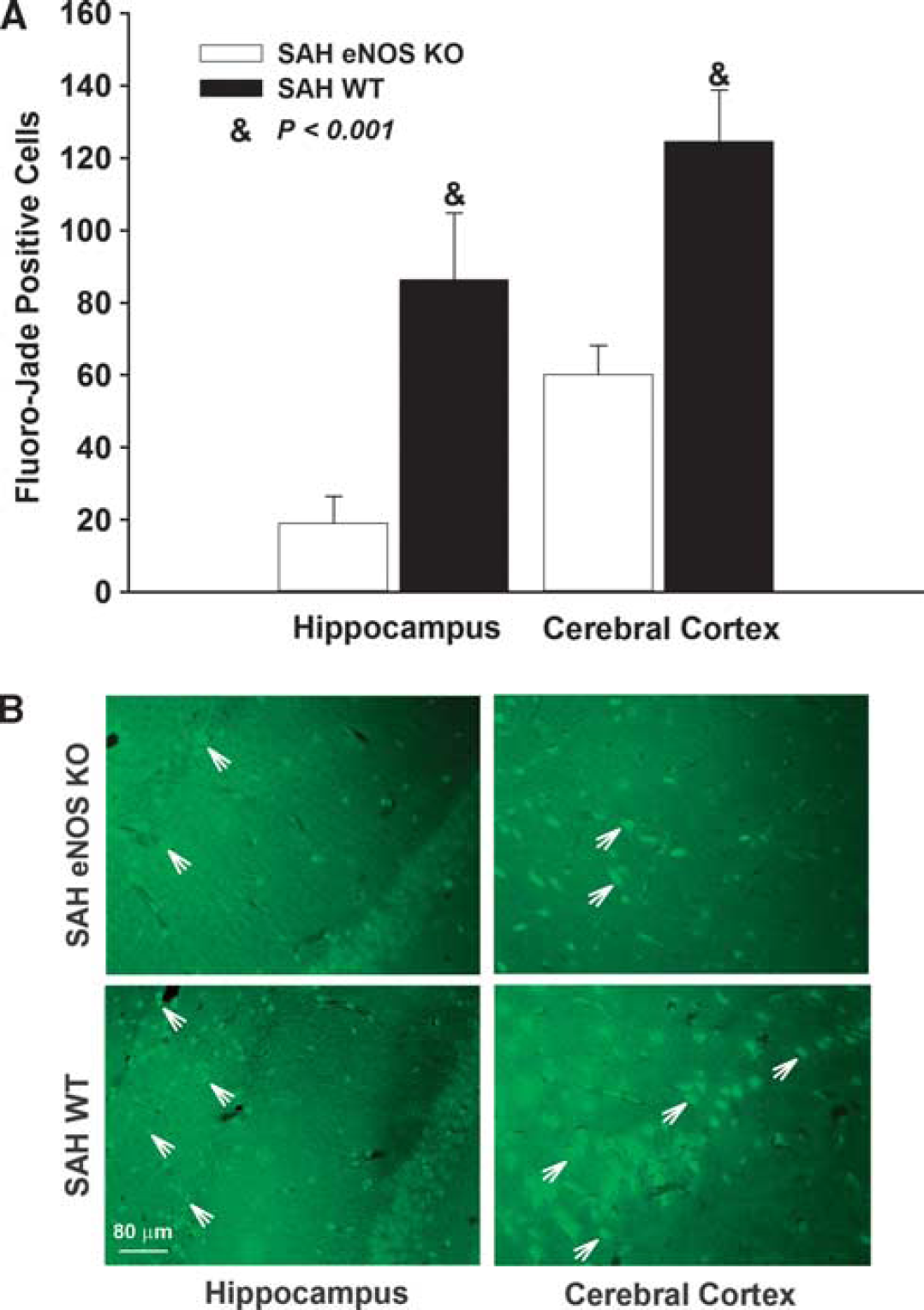
Quantification of degenerating neuronal cells. (
DISCUSSION
Subarachnoid hemorrhage in various animal models and in humans was associated with oxidative stress, likely due to clot-derived ROS.19-23 We demonstrated that this oxidative stress was associated with eNOS uncoupling in the brain parenchyma and that this contributed to nitrosative stress, resulting in neuronal apoptosis, microvascular dysfunction, and large artery vasospasm.10,24 The eNOS uncoupling could be reversed by drugs like simvastatin, which has antioxidant and other effects that may preserve eNOS function.4,24 However, simvastatin has multiple potential mechanisms of action that preclude definition of the exact mechanisms of its beneficial effects. A recent randomized, double-blind, placebo-controlled trial of simvastatin does not support a beneficial effect of simvastatin in patients with SAH, which suggests that translation from bench-to-bedside could be difficult. 25 A larger phase-3 trial (SimvasTatin in Aneurysmal Subarachnoid Hemorrhage (STASH)) is in progress (http://urlm.co/www.stashtrial.com).
In the current experiments, eNOS KO mice were used to overcome these limitations and gain more insight into the role of eNOS in brain injury after SAH. It should be emphasized that at baseline, naive eNOS KO mice are similar on all the endpoint measurements as compared with wild-type controls. They differed, however, in the response to SAH. The eNOS KO mice exhibited less vasospasm, microthrombi, and brain injury after SAH. Furthermore, they had reduced oxidative stress as measured by reduced O2− content. Another new finding of this study is that other isoforms of NOS changed differently in eNOS KO animals in comparison to wild-type mice. Subarachnoid hemorrhage increased iNOS in both wild-type and eNOS KO mice, which is consistent with other studies in wild-type animals.26,27 Increased iNOS activity could produce more ROS and aggravate brain injury and large artery vasospasm.28-30 Yatsushige
Neuronal NOS has been less studied after SAH. After SAH in humans, nNOS messenger ribonucleic acid was unchanged in brain tissue except in patients in poor clinical condition in whom it was increased. 32 We found that nNOS was significantly elevated after SAH in eNOS KO but not wild-type mice, suggesting that this is a compensatory response to the lack of eNOS. Prior studies have suggested that nNOS decreases in brain 33 and cerebral arteries 34 after SAH, which is similar to the current findings.
The hypothesis supported by the current data is that eNOS is uncoupled and causes detrimental effects after SAH. 12 In the current study, eNOS monomers were increased after SAH in wild-type mice, and we further demonstrate that SAH in wild-type mice was associated with Zn2+ release in comparison to animals injected with saline or in sham-operated mice. The eNOS KO mice with SAH also demonstrated a similar increase in Zn2+, however, there was a much smaller increase in comparison to wild-type mice. These data are consistent with previously published work showing peroxynitrite-induced oxidation of Zn2+ from the Zn2+ thiolate cluster of eNOS, which results in the formation of disulfide bonds between the monomers. This could explain why the dimer can be separated in SDS-reducing gel conditions. 35 It has been documented that oxidative stress and increased peroxynitrite target the thiolate complex in eNOS rather than free thiols. 35 Experiments demonstrated that peroxide-oxidized thiols had no added effect on eNOS dimer or Zn2+ content. Recent studies demonstrated that Zn2+ homeostasis is very important for normal function of neurons and glia in the central nervous system. Zn2+ accumulation may contribute to selective and delayed degeneration of hippocampal pyramidal neurons in the CA1 subregion after focal and transient global ischemia. 36 Intramitochondrial Zn2+ accumulation leads to a loss of mitochondrial membrane potential and increased generation of ROS that exacerbate oxidative stress and eventually lead to cell death. 36 This suggests that Zn2+ accumulation from eNOS uncoupling after SAH may represent a novel mechanism and potential target for therapy of observed neuronal cell death in the disease.
Interestingly, even in the absence of eNOS, there was some Zn2+ release after SAH, possibly due to release from other NOS isoforms (nNOS and iNOS). Neuronal NOS can also uncouple and release Zn2+ under some conditions.35,37–39
There are multiple possible mechanisms underlying the uncoupling of eNOS after SAH, such as depletion of tetrahydro-biopterin (BH4) or oxidization to dihydrobiopterin (BH2), processes that have been demonstrated to result in eNOS dysfunction in a number of studies. For example, Santhanam,
DISCLOSURE/CONFLICT OF INTEREST
RL Macdonald is a consultant for Actelion Pharmaceuticals and Chief Scientific Officer of Edge Therapeutics. The remaining authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
RL Macdonald receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Stroke Network, Canadian Institutes of Health Research and the Heart and Stroke Foundation of Ontario.