
Abstract
In the ischemic brain, reperfusion with tissue plasminogen activator (tPA) sometimes causes catastrophic hemorrhagic transformation (HT); however, the mechanism remains elusive. Here, we show that the basement membrane, and not the endothelial cells, is vulnerable to ischemic/reperfusion injury with tPA treatment. We treated a spontaneously hypertensive rat model of middle cerebral artery occlusion (MCAO) with vehicle alone, tPA alone, or a free radical scavenger, edaravone, plus tPA. Light and electron microscopic analyses of each microvascular component revealed that the basement membrane disintegrated and became detached from the astrocyte endfeet in tPA-treated animals that showed HT. On the other hand, edaravone prevented the dissociation of the neurovascular unit, dramatically decreased the HT, and improved the neurologic score and survival rate of the tPA-treated rats. These results suggest that the basement membrane that underlies the endothelial cells is a key structure for maintaining the integrity of the neurovascular unit, and a free-radical scavenger can be a viable agent for inhibiting tPA-induced HT.
Keywords
Introduction
Ischemic brain damage can be ameliorated, if cerebral blood flow is restored by thrombolytic agents, for example, tissue plasminogen activator (tPA), within a short time (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995), but delayed reperfusion with tPA can cause hemorrhagic transformation (HT) (The NINDS t-PA Stroke Study Group, 1997). Cerebral microvascular integrity depends largely on three components, namely the vascular wall, formed by endothelial cells; the blood–brain barrier (BBB), provided by endothelial tight junctions; and the basal membrane, which lines the endothelial cells (del Zoppo, 2006). Free radicals, the fundamental mediators of reperfusion injury (Chan, 2001) and reperfusion-related HT (Maier et al, 2006), are generated soon after vessel occlusion, with explosive propagation after reperfusion. Various proinflammatory mediators (matrix metalloproteinases (MMPs), thrombin, vascular endothelial growth factor, and bradykinin) (Aschner et al, 1997; Kamiya et al, 1993; Rosenberg, 2002; Suarez and Ballmer-Hofer, 2001) increase in the ischemic brain, accompanied by brain edema, endothelial cell death (Maier et al, 2006), disruption of tight junctions, and basal membrane/extracellular matrix (collagenIV, laminin-1, and fibronectin) loss (del Zoppo and Mabuchi, 2003). Any of these changes could be involved in HT associated with tPA treatment. To elucidate the mechanism underlying tPA-associated HT, here we treated a spontaneously hypertensive rat model of middle cerebral artery occlusion (MCAO) with vehicle or tPA, and observed the neurovascular unit components, with and without a free radical scavenger, edaravone (Figure 1). We found that the basement membrane is the major structure disrupted in tPA-induced HT, and edaravone could prevent the HT-associated damage.
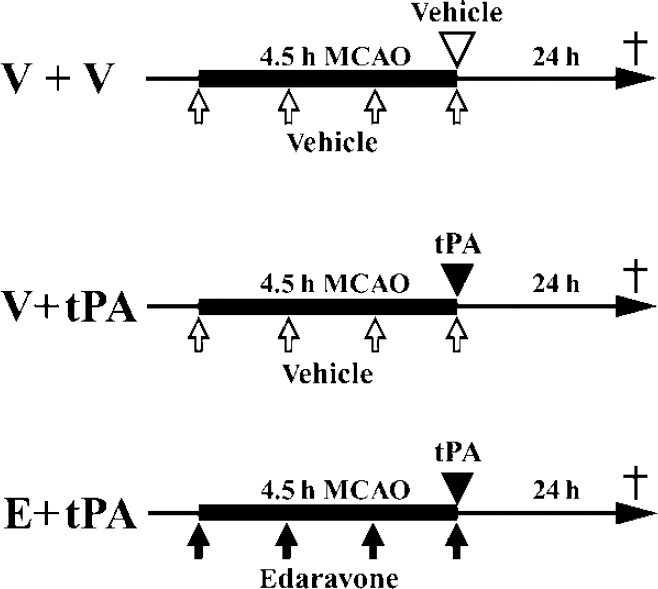
Experimental groups included the V+V group (
Materials and methods
Animals
Spontaneously hypertensive rats (male, 11 weeks old, 250 to 280 g, total
Experimental Groups and Drug Treatment
Three groups of rats were studied. The V+V control group (
Focal Cerebral Ischemia
To mimic a clinical situation reperfused by tPA, we designed the present experiment whereby tPA was administrated just before a reperfusion by pulling out a nylon thread from the origin of MCA, so that the tPA-associated damage could be assessed under similar conditions of reperfusion (Supplementary Table 1). However, in this model, it was not addressed whether edaravone could affect the clot bursting effect of tPA. In brief, the rats were anesthetized with a nitrous oxide/oxygen/isoflurane mixture (69/30/1%) administered through an inhalation mask. The left carotid bifurcation was exposed, and the external carotid artery was coagulated distal to the bifurcation. A silicone-coated 4–0 nylon thread was then inserted through the stump of the external cerebral artery and gently advanced (∼18mm) to occlude the origin of the MCA. After occlusion for 4.5 h, which could cause hemorrhagic infarction after tPA treatment (Zhang et al, 2004), the nylon was gently withdrawn to restore the blood flow in the MCA territory and the incision was closed. The caudal tail artery and left femoral vein were cannulated for continuous monitoring of the mean arterial blood pressure, analysis of arterial blood gases, blood glucose level, and serum cytokines/chemokines/MMPs, and for intravenous drug administration. During surgery, the rectal and left temporal muscle temperatures were maintained at 37.0°C±0.3°C by placing the animals on a heating bed (Model BWT-100; Bio Research Center, Nagoya, Japan). The cortical cerebral blood flow in the left MCA territory was measured during ischemia by laser-Doppler flowmetry using an MBF3D (Moor Instruments Ltd, Axminster, UK).
Physiologic parameters in the three experimental groups
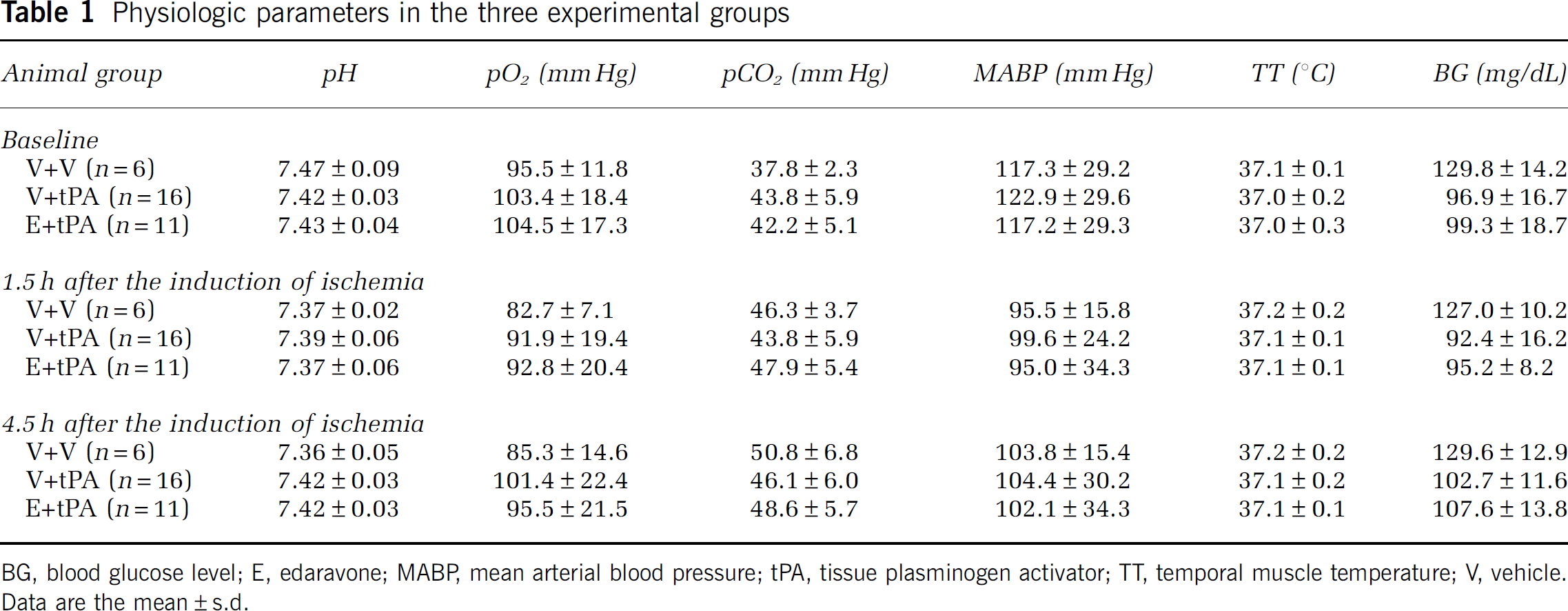
BG, blood glucose level; E, edaravone; MABP, mean arterial blood pressure; tPA, tissue plasminogen activator; TT, temporal muscle temperature; V, vehicle. Data are the mean±s.d.
Multiplex ELISA
We used an ELISA (enzyme-linked immunosorbent assay) micro-array kit (Pierce Endogen, Woburn, MA, USA) to assess the levels of secreted cytokines/chemokines/MMPs (IL-1β, IL-6, tumor necrosis factor-α, macrophage inflammatory protein-1α, Fractalkine, and MMP-9). Blood was drawn from the left femoral vein at 0 and 3 h after the induction of ischemia and at 24 h after reperfusion, and the serum level of each cytokine, chemokine, and MMP was measured according to the manufacturer's specifications, as described earlier in detail by Lee et al (2007).
Behavioral Analysis
At 24 h after reperfusion, the surviving rats were tested for behavioral changes and scored as described by Bederson et al (1986), with minor modifications, as follows: 0, no observable neurologic deficits; 1, failure to extend the right forepaw; 2, circling to the contralateral side; 3, falling to the right; and 4, unable to walk spontaneously.
Histochemistry
We focused on the changes at and around blood vessels, and analyzed the infarcted brain tissue mainly through histochemistry in this study. For light microscopy, the surviving rats (V +V group,
Electron Microscopic Analysis
For electron microscopy (V + tPA group,
Quantitative Analysis
For the semiquantitative evaluation of rat IgG staining, sections every 300 µm were captured through a microscope (Axioplan 2; Carl Zeiss, Jena, Germany). The staining intensity was measured with an image processing software (Scion Image, Scion Corporation, Frederick, MD, USA). For the semiquantitative evaluation of histochemical stainings, such as for iron, 4-HNE, HEL, MMP-9, NAGO, occludin, or collagenIV-positive blood vessels, the stained sections were selected from three levels of the caudate putamen (1.2, 0.7, and 0.2mm rostral to the bregma; Paxinos and Watson, 1982) of each animal, and three areas in the periinfarct cortex in each section were chosen randomly and captured at −200 magnification with a microscope (BX51; Olympus, Tokyo, Japan). We confirmed the border between the ischemic core and peri-infarct lesion through cresyl violet staining of adjacent sections as reported earlier (Omori et al, 2002). Blood vessels were identified by their morphology, and the staining intensity (iron, 4-HNE, HEL, MMP-9, NAGO, and collagenIV) was measured using the software (Scion Image). To quantify the degradation of occludin protein surrounding the vascular wall, we scored the occludin expression on each blood vessel (3, more than 75% of the vascular wall was positive for occludin; 2, 50% to 75%; 1, 25% to 49%; 0, > 25%). To assess the detachment of astrocyte endfeet from the basement membrane in the GFAP/collagenIV double-labeled sections, three levels of sections were selected as described above, and four areas in the ipsilateral peri-infarcted cortex in each section were chosen randomly and captured at −100 magnification with a confocal laser microscope (LSM510; Carl Zeiss). We measured the area between the astrocyte endfeet and basement membrane of each blood vessel, and the length of each blood vessel. We then calculated the ratio of the area to the length, which we called the ‘vascular dissociation index’ in this study. This index was very useful for analyzing the spatial dissociation of the vessels from the astrocyte endfeet.
Statistical Analysis
Values are expressed as means±s.d. The frequencies of death (survival rates) were compared by an
Results
We carefully monitored several physiologic parameters, including regional cerebral blood flow during the experiments, and found no significant differences among the three experimental groups (Table 1 and Supplementary Table 1). We then evaluated the survival rate, motor function, brain hemorrhage, and infarct volume in the three groups. Despite 4.5 h of MCAO, which caused severe brain infarction, no vehicle-treated (V+ V) rats had died 24 h after the reperfusion. However, 34% of the tPA-treated (V + tPA) rats had died, and all the dead animals showed obvious and massive intracerebral hemorrhage. In contrast, none of the rats treated with edaravone plus tPA (E + tPA) had died (Figure 2A). We also compared the rats' behavior using the protocol described by Bederson et al (1986), and found that motor function was significantly better in the E + tPA group than in the V+ tPA group (*
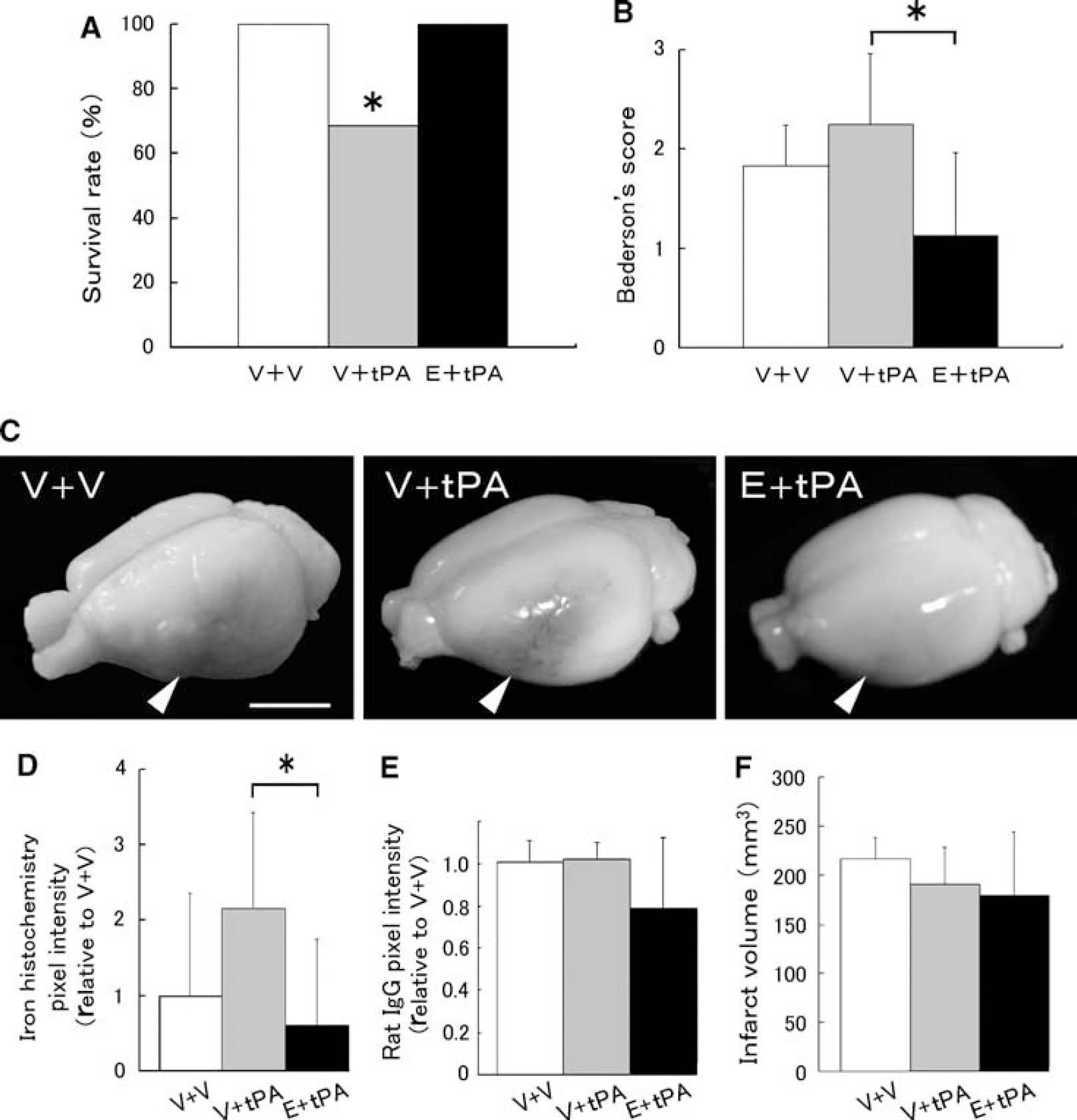
Tissue plasminogen activator administration into ischemic rat brain resulted in a lower survival rate, worsening motor function, and obvious intracerebral hemorrhage. Edaravone, a free radical scavenger, controlled these tPA-mediated exacerbations. (
We next evaluated the effects of the tPA and E + tPA treatments on the level of a spectrum of factors, including lipid peroxide, cytokines, chemokines, and MMPs. We frequently observed blood vessels positive for 4-HNE, an end product of lipid peroxidation, in the peri-infarct lesions of the V+ tPA group (Figure 3A, black arrowheads), consistent with the theory that reactive oxygen species (ROS) are mainly produced in the peri-infarct area (Peters et al, 1998). Intracerebral hemorrhage was also observed in the peri-infarct lesions (Figure 3A, white arrowheads), indicating the association of an activated radical system with intracerebral hemorrhage. We did not detect the expression of 4-HNE or HEL, a marker of early lipid peroxidation, in the contralateral side of the three experimental groups (data not shown). Tissue plasminogen activator treatment increased the 4-HNE expression of blood vessels in the peri-infarct lesions of the ipsilateral side (**
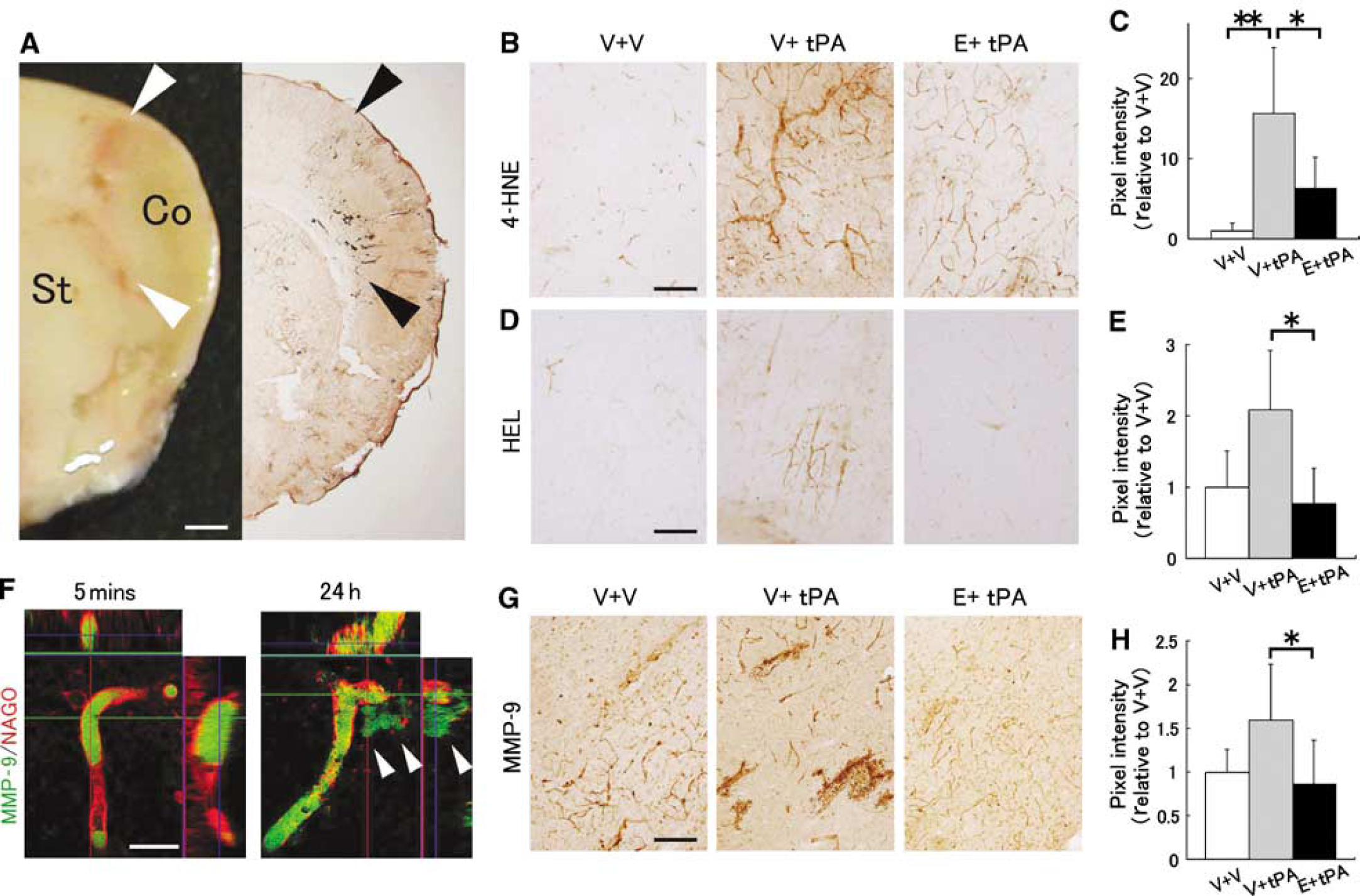
The free radical scavenger, edaravone, decreased lipid peroxidation and MMP-9 expression in the peri-infarct area. (
Multiplex cytokines/chemokines/MMPs array analysis in rat serum
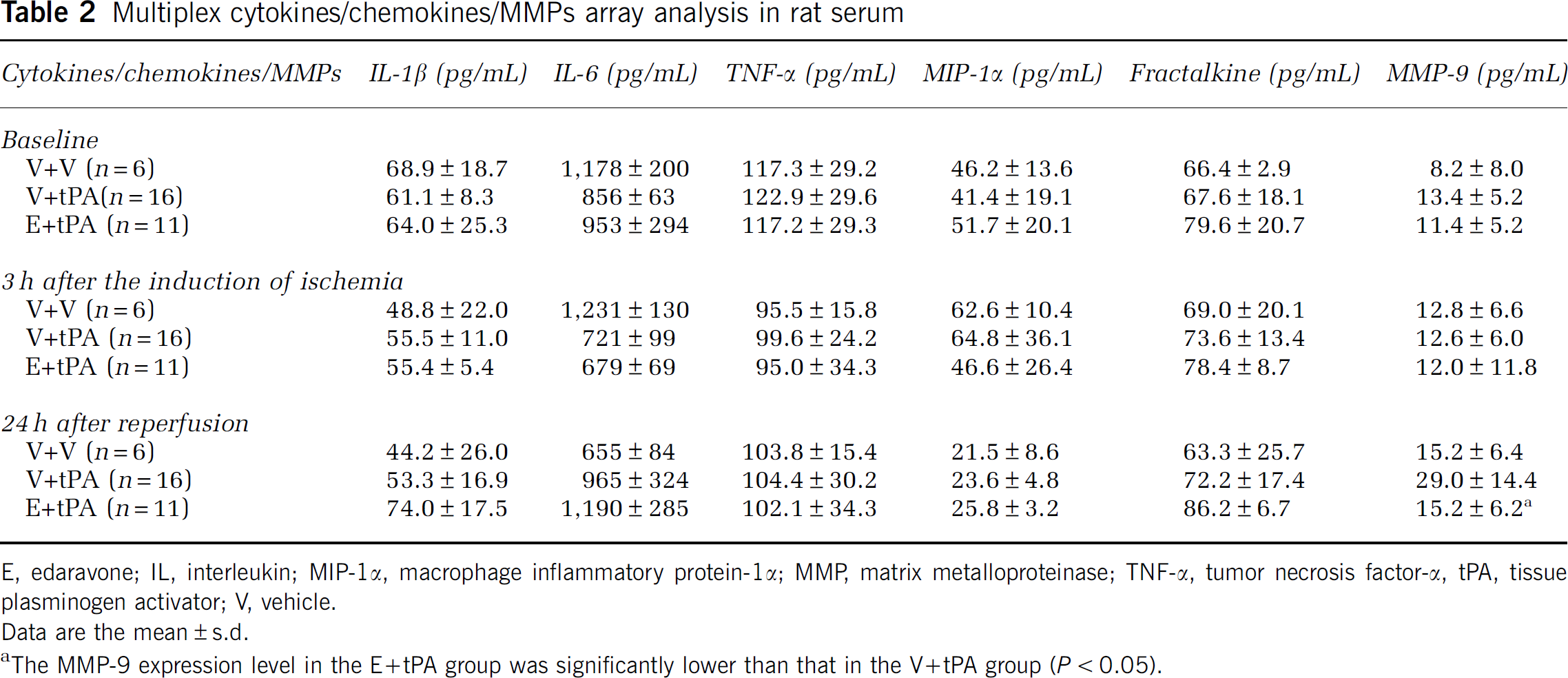
E, edaravone; IL, interleukin; MIP-1α, macrophage inflammatory protein-1α; MMP, matrix metalloproteinase; TNF-α, tumor necrosis factor-α, tPA, tissue plasminogen activator; V, vehicle.
Data are the mean±s.d.
The MMP-9 expression level in the E+tPA group was significantly lower than that in the V+tPA group (
Reperfusion-derived free radicals induce lipid peroxidation, especially in the membrane phospholipids of vascular endothelial cells (Abe et al, 1988; Phelan and Lange, 1991; Zhang et al, 2004). Moreover, lipid peroxide and MMP-9 are reported to cause the deterioration of tight junction proteins and the basement membrane (Usatyuk et al, 2006; Yang et al, 2007). To determine which component of the vascular unit was most disrupted in the three experimental models, we analyzed markers for vascular endothelial cells (NAGO), tight junctions (occludin), and basement membrane (collagenIV) located from the inner lumen side to the outer peri-vascular side of the cerebrovascular endothelium. To our surprise, there was no statistically significant difference in the expression of the endothelial cell marker, NAGO, in the three experimental models (Figures 4A and 4B). In contrast, edaravone treatment inhibited the disruption of the tight junction protein, occludin (*
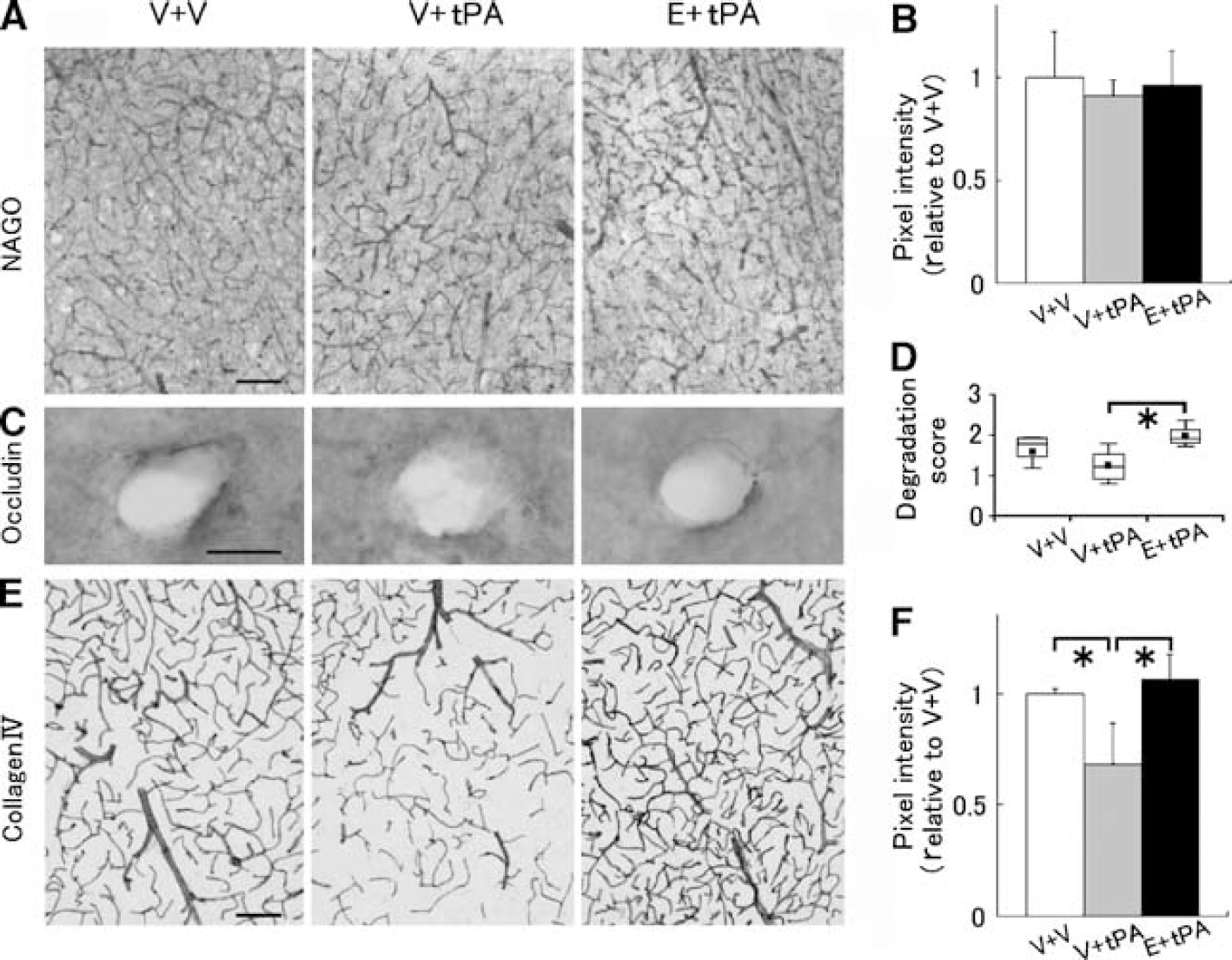
Tissue plasminogen activator injection disrupted the basement membrane around vessels, rather than the endothelial cells that are exposed to the blood flow. (
This disruption of the basement membrane led us to hypothesize that the tPA-induced dissociation of the neurovascular unit involves detachment of the astrocyte endfeet surrounding the vessel from the basement membrane. We therefore examined whether dissociation occurred between the collagenIV-positive basement membrane and the GFAP-positive astrocyte foot processes. In the tPA-treated rat, no dissociation of the neurovascular unit was found on the contralateral, nonischemic side (Figure 5A); however, marked dissociation was observed on the ipsilateral side (**
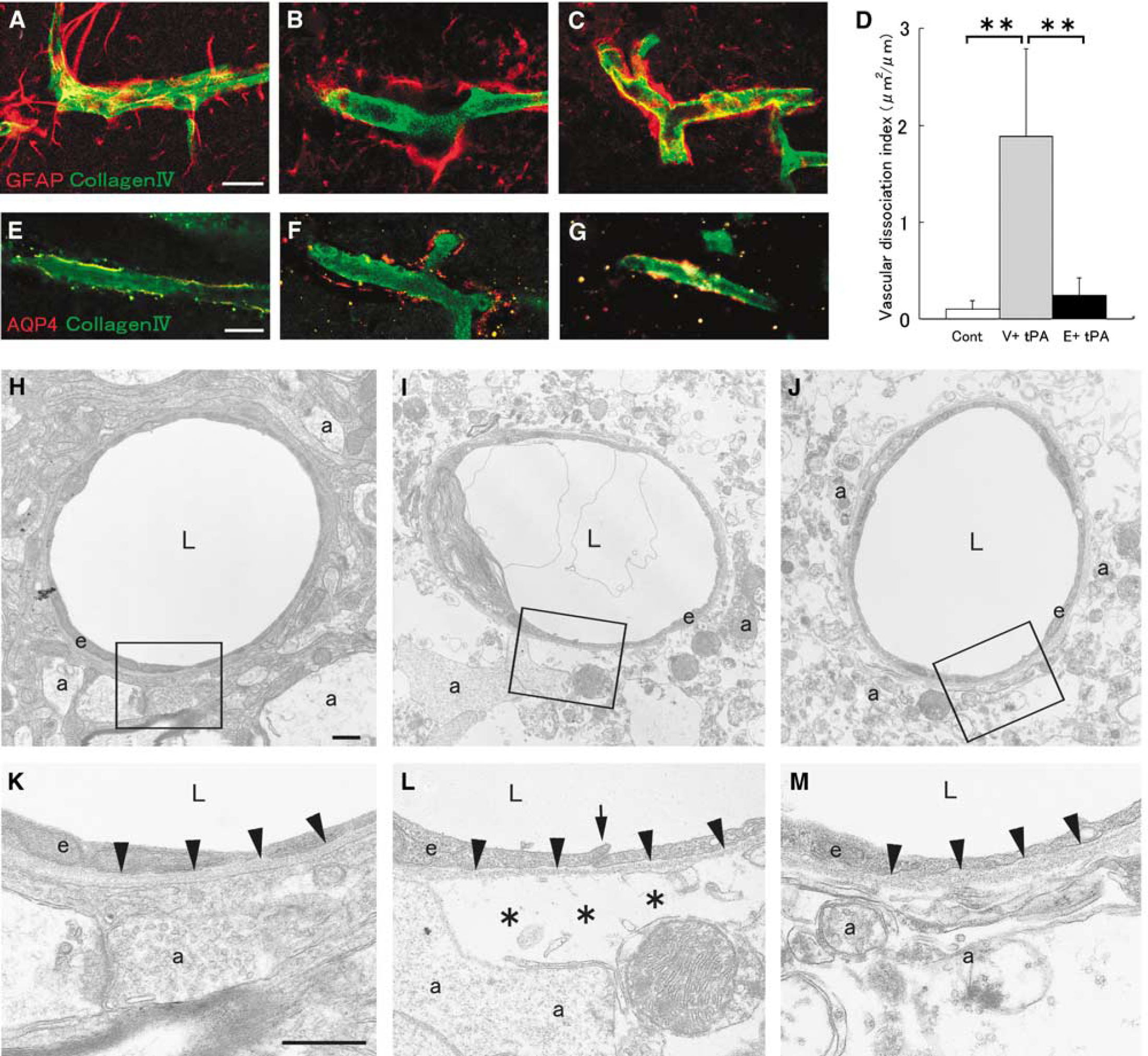
Reperfusion with tPA induced detachment of the astrocyte endfeet from the basement membrane. (
Discussion
Together, our findings indicated that it was not the inner lumen of cerebral endothelial cells, but the outer, peri-vascular side of the basement membrane that was severely degraded in the rat ischemic brain treated with tPA. At the peri-vascular extracellular space in an animal model of cerebral ischemia, large quantities of ROS are generated after the onset of ischemia (Kontos et al, 1992). In addition, after MCAO, SOD2-KO mice exhibit a significant increase in MMP-9 and a higher rate of brain hemorrhage (Maier et al, 2006), indicating that the excess ROS activate MMP-9, which can degrade the major basal membrane component, collagenIV. Several groups have reported the decreased expression of adhesion molecules, such as laminin (Hamann et al, 1996) and integrin α1β6 (Wagner et al, 1997), in the perivascular extracellular space in the ischemic animal brain. In contrast, vascular endothelial cells, which are constantly exposed to blood flow, rarely show dUTP incorporation into their DNA in the ischemic animal brain, indicating a low level of injury (Tagaya et al, 1997). In addition, an
The mechanism by which edaravone exerts its salutary effect on tPA-induced HT remains obscure, but several inferences can be drawn from this study. We observed that tPA treatment dramatically increased the 4-HNE expression of blood vessels in peri-infarct lesions (Figure 3B). Iron and thrombin, which can be released from blood clots after tPA treatment, have been reported to induce ROS (Nakamura et al, 2008). These results lead to speculation that exogenous tPA may, directly or indirectly, induce plenty of ROS in an infarcted brain. Ischemia/reperfusion injury also generates ROS, which can be associated with MMP-9 activation (Maier et al, 2006). We also showed that administration of edaravone resulted in the reduction of both 4-HNE and MMP-9 expressions on blood vessels of the peri-infarct area (Figures 3B and 3G). Therefore, edaravone may inhibit the activation of MMP-9, which can cause HT, by scavenging the tPA-induced ROS or by trapping the ROS derived from ischemia/reperfusion injury. However, other mechanisms are possible, and additional studies are needed to understand it in detail.
We reported earlier that edaravone ameliorates edema in the ischemic brain (Abe et al, 1988), and reduces the infarct size after 1.5 h of MCAO in rat brain reperfused with tPA, accompanied by reductions in the free radical end products of lipids, proteins, and DNA (Zhang et al, 2004). In a clinical trial, edaravone strongly attenuated the resulting disability in humans 90 days after acute ischemic stroke without serious adverse events (The Edaravone Acute Brain Infarction Study Group, 2003), and it has been used clinically in Japan as a neuroprotective agent for acute stroke patients since 2001. In a large clinical trial, another free radical scavenger, NXY-059, initially appeared to reduce disability after stroke (the first Stroke-Acute-Ischemic-NXY-Treatment trial, SAINT I) (Lees et al, 2006), but this effect could not be reproduced (SAINT II) (Shuaib et al, 2007). However, one mutual characteristic of NXY-059 was the potential to decrease symptomatic intracerebral hemorrhage after tPA treatment (Lees et al, 2006). NXY-059 is water soluble (octanol/water partition coefficients; cLog