
Abstract
Erythropoietin (EPO) plays a prominent role in the regulation of the hematopoietic system, but the potential function of this trophic factor as a cytoprotectant in the cerebral vascular system is not known. The authors examined the ability of EPO to modulate a series of death-related cellular pathways during free radical–induced injury in cerebral microvascular endothelial cells (ECs). Endothelial cell injury was evaluated by trypan blue, DNA fragmentation, membrane phosphatidylserine exposure, apoptotic protease–activating factor-1 (Apaf-1), and Bcl-xL expression, mitochondrial membrane potential, cytochrome c release, and cysteine protease activity. They show that constitutive EPO is present in ECs but is insufficient to prevent cellular injury. Signaling through the EPO receptor, however, remains biologically responsive to exogenous EPO administration to offer significant protection against nitric oxide–induced injury. Exogenous EPO maintains both genomic DNA integrity and cellular membrane asymmetry through parallel pathways that prevent the induction of Apaf-1 and preserve mitochondrial membrane potential in conjunction with enhanced Bcl-xL expression. Consistent with the modulation of Apaf-1 and the release of cytochrome c, EPO also inhibits the activation of caspase-9 and caspase-3–like activities. Identification of novel cytoprotective pathways used by EPO may serve as therapeutic targets for cerebral vascular disease.
Keywords
Erythropoietin (EPO) is well known as a mediator of erythroid maturation in the hematopoietic system, but new work has identified this trophic factor as a potential cytoprotectant in the nervous system. Initial studies showed the existence of EPO in cerebral endothelial cells (Bernaudin et al., 1999) and in the brain (Marti et al., 1996), with subsequent work illustrating the expression of EPO and the EPO receptor (EPOR) in both neurons and astrocytes (Nagai et al., 2001). In neuronal injury paradigms, EPO has been shown to provide protection against toxic insults, such as ischemia and free radical injury (Bernaudin et al., 1999; Digicaylioglu and Lipton, 2001; Wen et al., 2002; Chong et al., 2003). Recent work that is both inclusive and exclusive of the nervous system also has suggested a potential role for EPO to avert vascular injury (Carlini et al., 1999; Chong et al., 2002a).
To initiate the development of EPO as a potential vascular protectant for cerebral endothelial cells (ECs), it is necessary to define the cellular pathways that determine injury in ECs and are susceptible to modulation by EPO. Injury in ECs can lead to the active destruction of the endothelium and precipitate both acute and chronic vascular degeneration that destroys cortical function (Zhang et al., 2000). In this regard, oxygen free radicals, such as nitric oxide (NO), have been established as significant precipitants of vascular disorders, such as Alzheimer disease and cerebral ischemia (Anderson et al., 2001; Chong et al., 2002b). Nitric oxide can trigger the induction of apoptosis, also referred to as programmed cell death, which involves both genomic DNA fragmentation and membrane phosphatidylserine (PS) exposure (Maiese and Vincent, 2000; Lin and Maiese, 2001). DNA fragmentation, which is considered to be a late component of apoptosis, can be a committed event that results in EC destruction (Jessel et al., 2002). Exposure of membrane PS residues in ECs, however, may play a more significant role that involves the precipitation of a procoagulant environment (Bombeli et al., 1997) and cellular inflammation (Dombroski et al., 2000).
Critical to the prevention of EC injury are the preservation of mitochondrial membrane integrity and the modulation of cysteine protease activity. Cellular release of NO can directly lead to mitochondrial membrane depolarization and the opening of mitochondrial permeability transition pores (Boyd and Cadenas, 2002; Chong et al., 2002c). As a result, cytochrome c is released from mitochondria and eventually leads to the activation of a family of executioner cysteine proteases (caspases). These pathways can be modulated by the Bcl-2 family member Bcl-xL to prevent cytochrome c release and cellular apoptosis (Yamaguchi and Wang, 2002). Once cytochrome c is released into the cytoplasm, however, it can then bind to the apoptotic protease–activating factor-1 (Apaf-1). Apaf-1 consists of three different domains that include a caspase recruitment domain, repeats of tryptophan and aspartate residues (WD-40 repeats), and a nucleotide-binding domain CED-4. Binding of cytochrome c to Apaf-1 results in the removal of the WD-40 domain, masking the CED-4 and caspase recruitment domain domains, and leads to the oligomerization of Apaf-1 under the assistance of dATP/ATP (Hu et al., 1999).
After the oligomerization of Apaf-1, caspase-9 is activated and proteolytically cleaves and activates procaspase-3 to begin to elicit genomic DNA destruction (Li et al., 1997). Caspase-9 is activated through a process that requires the cytochrome c–Apaf-1 complex together with dATP to form a cell death machinery apoptosome. Activated caspase-9 cleaves downstream procaspases, which include procaspase-3. Active caspase-3 is subsequently associated with cleavage of DNA protein kinase, actin, and fodrin that results in both DNA fragmentation and membrane PS exposure (Lin and Maiese, 2001; Mandal et al., 2002). Given the potential ability of EPO to offer protection against vascular disease, we examined the cellular mechanisms modulated by EPO that may foster cerebral function and plasticity during EC injury.
MATERIALS AND METHODS
Cerebral microvascular endothelial cell cultures
Vascular ECs were isolated from Sprague-Dawley adult rat brain cerebra by using a modified collagenase–dispase-based digestion protocol (Lin and Maiese, 2001; Chong et al., 2002c). Briefly, ECs were cultured in endothelial growth media consisting of M199E with 20% heat-inactivated fetal bovine serum, 2 mmol/L l-glutamine, 90 μg/mL heparin, and 20 μg/mL EC growth supplement (ICN Biomedicals, Aurora, OH, U.S.A.). Cells from the third passage were identified by positive direct immunocytochemistry for factor VIII–related antigen (>98% purity) (Antonov et al., 1986) and were negative for glial fibrillary acidic protein immunocytochemistry.
Experimental treatments
Nitric oxide administration was performed by replacing the culture media with media containing sodium nitroprusside (SNP; 1.00 mmol/L; Sigma, St. Louis, MO, U.S.A.) or 6-(2-hydroxy-1-methyl-2-nitrosohydrazino) -N-methyl-1-hexanamine (NOC-9; 1.00 mmol/L; Calbiochem, San Diego, CA, U.S.A.) per the experimental paradigm (Maiese and Vincent, 2000). More than one NO generator was used as a control to show that the ECs were responding to NO rather than to other byproducts of these agents. During both pre- and post-paradigm applications, EPO or the EPO antibody (R&D Systems, Minneapolis, MN, U.S.A.) application was continuous.
Assessment of cell survival
Endothelial cell injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 h after NO exposure per our previous protocols (Lin and Maiese, 2001). The mean survival was determined in 24-well plates with an initial cell count of 1.5 × 105 cells/mm2 (70% to 80% confluence) by counting 8 randomly selected nonoverlapping fields with each containing approximately 20 cells (viable + nonviable). Each experiment was replicated six times independently with different cultures.
Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick-end labeling (TUNEL) assay (Lin and Maiese, 2001; Chong et al., 2002c). Briefly, ECs were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI, U.S.A.) followed by streptavidin–peroxidase and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, U.S.A.). The mean number of cells positive for TUNEL was determined by counting 8 randomly selected nonoverlapping fields with each containing approximately 20 cells [TUNEL (+) + TUNEL (−)]. Each experiment was replicated six times independently with different cultures.
Assessment of membrane phosphatidylserine residue externalization
Per our prior protocols (Maiese and Vincent, 2000; Lin and Maiese, 2001; Chong et al., 2002c), a 30 μg/mL stock solution of annexin V conjugated to phycoerythrin (R&D Systems) was diluted to 3 μg/mL in warmed calcium containing binding buffer (10 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). Plates were incubated with 500 μL of diluted annexin V for 10 minutes. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL, U.S.A.) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single-excitation light at 490 nm and detected emission at 585 nm. The mean number of cells positive for membrane PS exposure was determined by counting 8 randomly selected nonoverlapping fields with each containing approximately 20 cells [PS (+) + PS (−)]. Each experiment was replicated six times independently with different cultures.
Immunocytochemical detection for erythropoietin and the erythropoietin receptor
Endothelial cells were fixed in 4% paraformaldehyde, blocked with horse serum, and then incubated with primary rabbit polyclonal antibody against EPO (1:1,000) or EPOR (1:1,000; Santa Cruz Biotechnologies, Santa Cruz, CA, U.S.A.) overnight at 4°C. Biotinylated horse anti-rabbit antibody was used as a secondary antibody (1:100). ABC reagent was applied to detect EPO and EPOR with 3,3′-diaminobenzidine (Vector Laboratories). Absence of the primary antibody was used as an initial negative control. The specificity of the antibodies was previously confirmed by prior studies (Acs et al., 2001; Siren et al., 2001). In addition, the specificity of the immunoreactivities for the antibodies was evaluated with antibody absorption tests. The primary antibody against EPO was preincubated with EPO (10:1, peptide-to-antibody ratio; R&D Systems), or the primary antibody against EPOR was preincubated with blocking peptide for EPOR (Santa Cruz Biotechnologies). This resulted in the complete abolition of immunocytochemical staining for EPO and the EPOR.
Assessment of mitochondrial membrane potential
The fluorescent probe JC-1 (Molecular Probes, Eugene, OR, U.S.A.) was used to assess the mitochondrial membrane potential. Endothelial cells in 35-mm plates were incubated with 2 μg/mL JC-1 in growth medium for 30 minutes. After washing, ECs were analyzed immediately under a Leitz DMIRB microscope (Leica) with a dual-emission fluorescence filter with 515 to 545 nm for green fluorescence and emission at 585 to 615 nm for red fluorescence. The relative ratio of red-to-green fluorescent intensity of mitochondrial staining was measured in 4 independent experiments by counting 8 randomly selected nonoverlapping fields with each containing approximately 20 cells.
Assessment of caspase activity
The activities of caspase-9 and caspase-3 were assessed by determination of the cleavage of their colorimetric substrates (Ac-LEHD-pNA, caspase-9; Ac-DEVD-pNA, caspase-3; Calbiochem) as previously described (Maiese and Vincent, 2000; Lin and Maiese, 2001; Chong et al., 2002c).
Modulation of caspase activity
Modulation of cysteine protease activity in ECs was performed by using the irreversible and cell-permeable caspase inhibitors Z-LEHD-FMK for caspase-9 (LEHD) and Z-DEVD-FMK (DEVD) for caspase-3, obtained from Pharmingen Inc. (Livermore, CA, U.S.A.). Inhibitors were added directly to the cultures at a concentration of 50 μmol/L 1 h before NO application.
Western blot analysis for erythropoietin, erythropoietin receptor, apoptotic protease–activating factor-1, Bcl-xL, and cytochrome c
Cells were homogenized and after protein determination, each sample (50 μg/lane) was then subjected to 7.5% (Apaf-1, EPO, EPOR) or 12.5% (cytochrome c, Bcl-xL) sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The membranes were incubated with a rabbit polyclonal antibody against EPO (1:1,000), EPOR (1:1,000; Santa Cruz Biotechnologies), Apaf-1 (1:2,000; PharMingen, San Diego, CA, U.S.A.), Bcl-xL (1:100; Santa Cruz Biotechnologies), and mouse polyclonal antibody against cytochrome c (1:2,000; PharMingen, San Diego). After washing, the membranes were incubated with a horseradish peroxidase–conjugated secondary antibody (goat anti–rabbit immunoglobulin G [IgG], 1:15,000 [Apaf-1, EPO, EPOR, Bcl-xL]; goat anti–mouse IgG, 1:2,000 [cytochrome c]; Pierce, Rockford, IL, U.S.A.). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.).
Statistical analysis
For each assessment involving EC survival, DNA fragmentation, membrane PS exposure, and caspase activity, the mean and standard deviation were determined from four to six replicate experiments. Statistical differences between groups were assessed by means of analysis of variance and with the post hoc Student's t-test. Results are expressed as the mean ± the standard deviation. Statistical significance was considered at P < 0.05.
RESULTS
Erythropoietin and the erythropoietin receptor are constitutively expressed in endothelial cells without change in expression during nitric oxide exposure
Endothelial cells were exposed to the NO donors NOC-9 (1.00 mmol/L) or SNP (1.00 mmol/L), and expression of EPO and the EPOR was assessed 24 h later with immunocytochemistry and Western blot analysis. In untreated control ECs not exposed to a NO donor, both endogenous EPO and the EPOR were present in all EC populations examined (Figs. 1B and 1F). Application of NO did not significantly alter the expression of EPO or the EPOR when examined at 24 h after NO application (Figs. 1C and 1G). Western blot analysis also confirmed that the constitutive expression of EPO and the EPOR was not altered in response to NO exposure (Fig. 1D and 1H).

Erythropoietin (EPO) and the EPO receptor (EPOR) are constitutively expressed in endothelial cells (ECs) without alteration during nitric oxide (NO) exposure. EC cultures were subjected to immunocytochemical detection and Western blot detection for EPO and the EPOR by using a rabbit primary polyclonal anti-EPO and anti-EPOR antibodies. For EPO detection,
Erythropoietin provides efficacious cytoprotection during pre- and posttreatment regimens that is concentration and time dependent
Increasing concentrations of EPO (0.01 ng/mL to 1,000 ng/mL) were administered directly to cultures, and cell survival was evaluated by a trypan blue dye exclusion method 24 h later to examine the possible cytotoxicity of EPO in ECs (Fig. 2A). As compared with EC survival in untreated control cultures (96 ± 4%), no significant toxicity over a 24-h period was present in the cultures exposed to EPO in the concentrations of 0.01 ng/mL to 1,000 ng/mL.
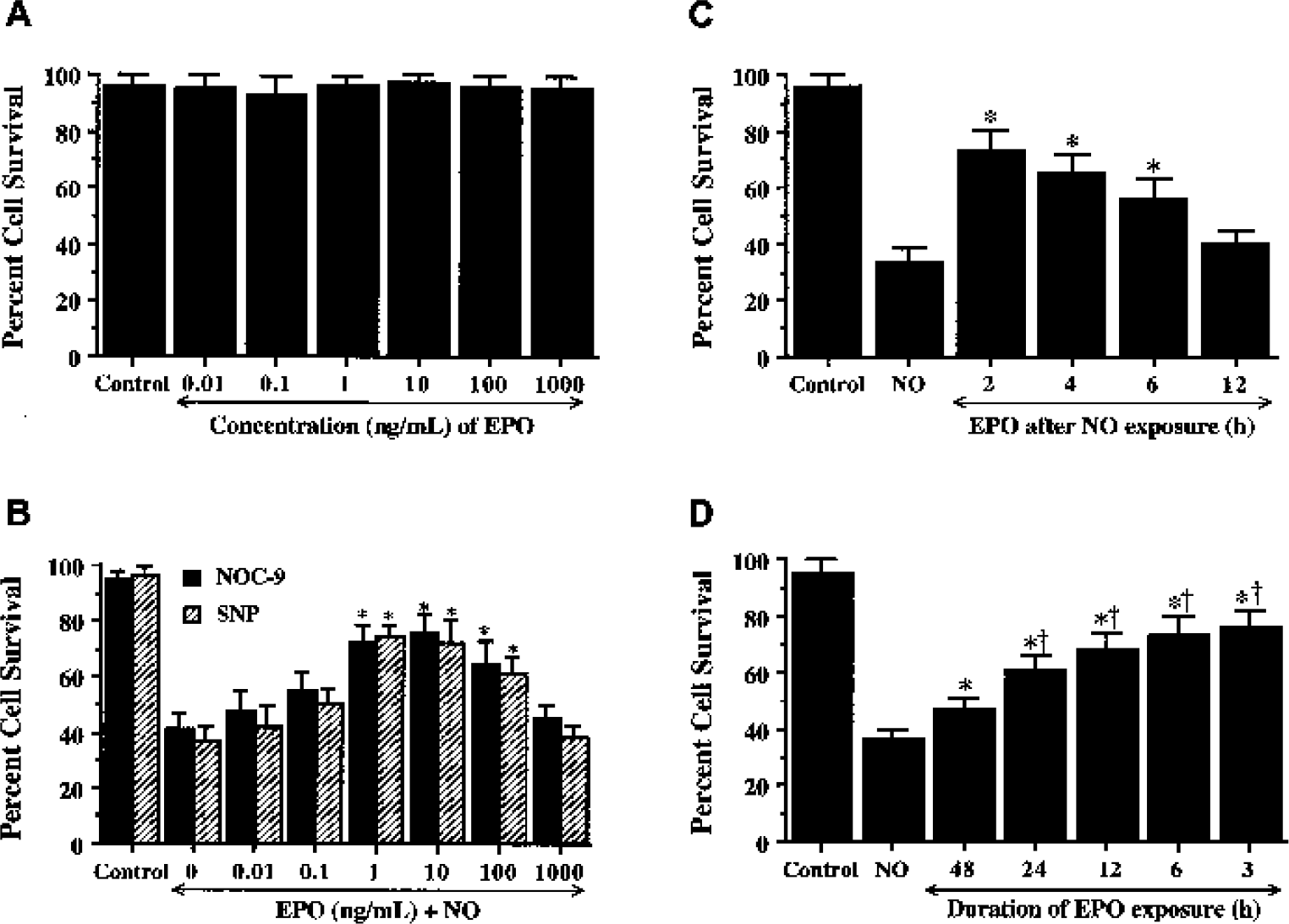
Protection by erythropoietin (EPO) is concentration and time dependent.
Endothelial cell survival was significantly reduced to 41 ± 6% after exposure to NOC-9 (1.00 mmol/L) and to 37 ± 5% after exposure to SNP (1.00 mmol/L) when compared with untreated control cultures (95 ± 3%, P < 0.01) (Fig. 2B). In contrast, application of EPO with the concentrations of 1 ng/mL to 100 ng/mL significantly increased EC survival. A concentration of EPO of 10 ng/mL achieved the maximum EC survival (76 ± 6%, NOC-9; 72 ± 8%, SNP), but concentrations lower than 1 ng/mL or higher than 100 ng/mL did not improve EC survival during NO exposure.
We next assessed whether changes in the temporal administration of EPO altered its protective ability during free radical exposure. As shown in Fig. 2C, EPO applied at 2, 4, and 6 h after NO exposure significantly increased EC survival from 34 ± 5% (NO alone) to 73 ± 7% (P < 0.01), 66 ± 6% (P < 0.01), and to 56 ± 7% (P < 0.01), respectively. In contrast, posttreatment with EPO at 12 h after NO exposure did not increase EC survival. The time of pretreatment of EPO also altered EC survival. Erythropoietin applied at time points closest to the application of NO yielded the greatest protection (Fig. 2D). For example, administration of EPO at 3 h before NO exposure yielded the highest level of EC survival (76 ± 6%, P < 0.01). EPO administered at 48 h (47 ± 4%, P < 0.01), however, achieved the least improvement of EC survival when compared with ECs exposed to NO only (36 ± 4%). In Figs. 2C and 2D, data for the two NO donors were combined because no significant differences in cell injury were present between the two agents (Fig. 2B).
Erythropoietin is necessary and sufficient for endothelial cell protection during nitric oxide exposure
Administration of EPO antibody in a series of concentrations of 0.01 to 1.00 μg/mL did not significantly alter EC survival when compared with untreated control cultures (Fig. 3A). In the presence of NO, application of the EPO antibody (0.01 to 1.00 μg/mL) also did not alter EC survival when compared with cultures treated with NO alone (Fig. 3B), suggesting that constitutive EPO in ECs is insufficient to prevent cellular injury. In the presence of the EPO antibody, the concentrations of EPO antibody of 0.10,0.50, and 1.00 μg/mL significantly decreased the protective capacity of EPO, yielding EC survivals of 44 ± 6% (P < 0.01), 38 ± 6% (P < 0.01), and 39 ± 4% (P < 0.01), respectively (Fig. 3C).
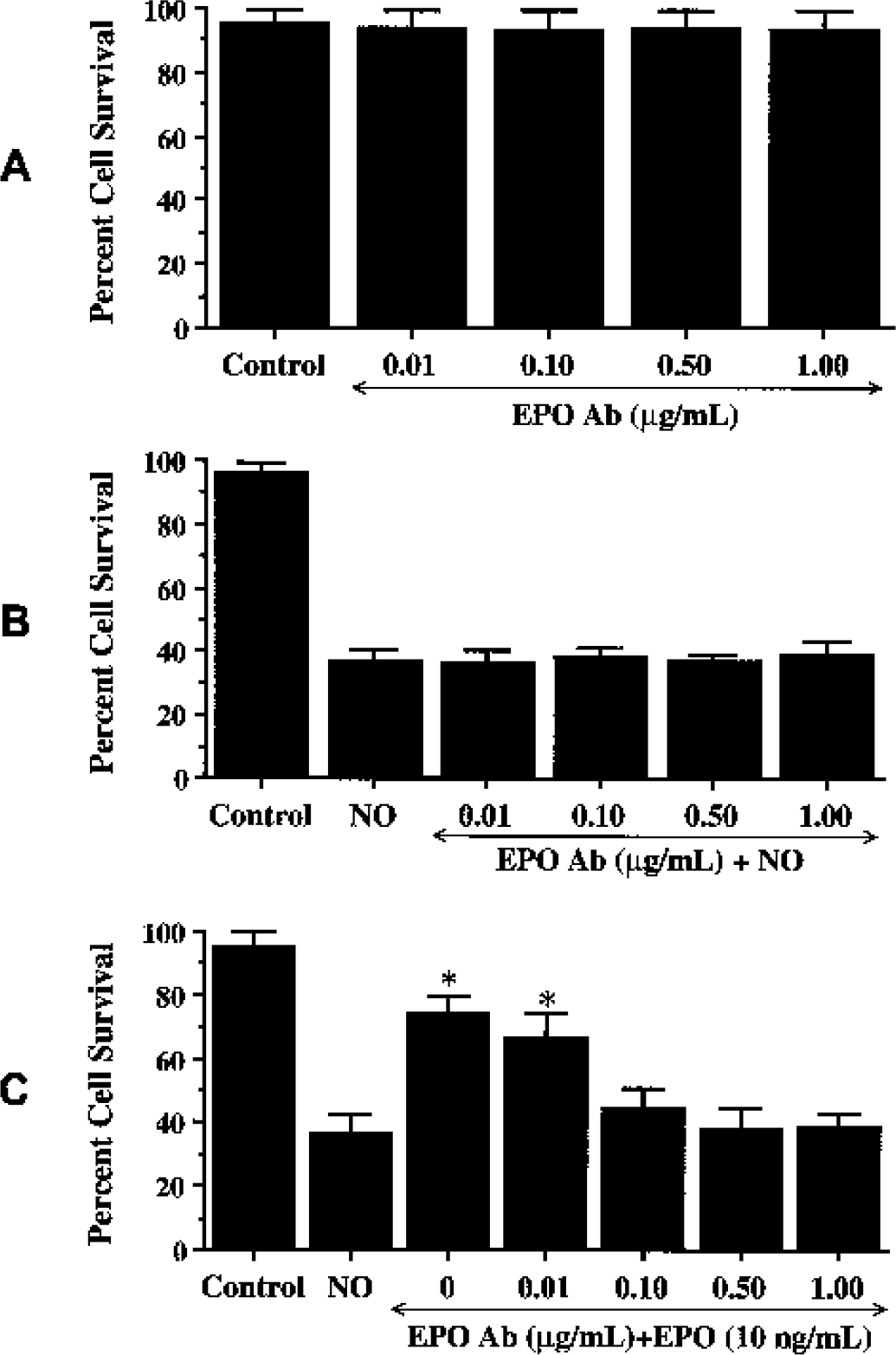
Erythropoietin (EPO) is necessary and sufficient for endothelial cell (EC) protection during NO exposure.
Erythropoietin prevents DNA fragmentation and membrane phosphatidylserine exposure in endothelial cells
Twenty-four hours after NO exposure (NOC-9 or SNP, 1.00 mmol/L), nuclear chromatin condensation was observed in ECs (Fig. 4A). In contrast, ECs pretreated with EPO (10 ng/mL) 1 h before NO exposure were without nuclear fragmentation. As shown in Fig. 4Aa, NO resulted in a significant increase in percent DNA fragmentation (64 ± 8%) when compared with untreated control cultures (2 ± 1%). DNA fragmentation was reduced to 24 ± 7% in EC cultures during the application of EPO over a 24-h period.
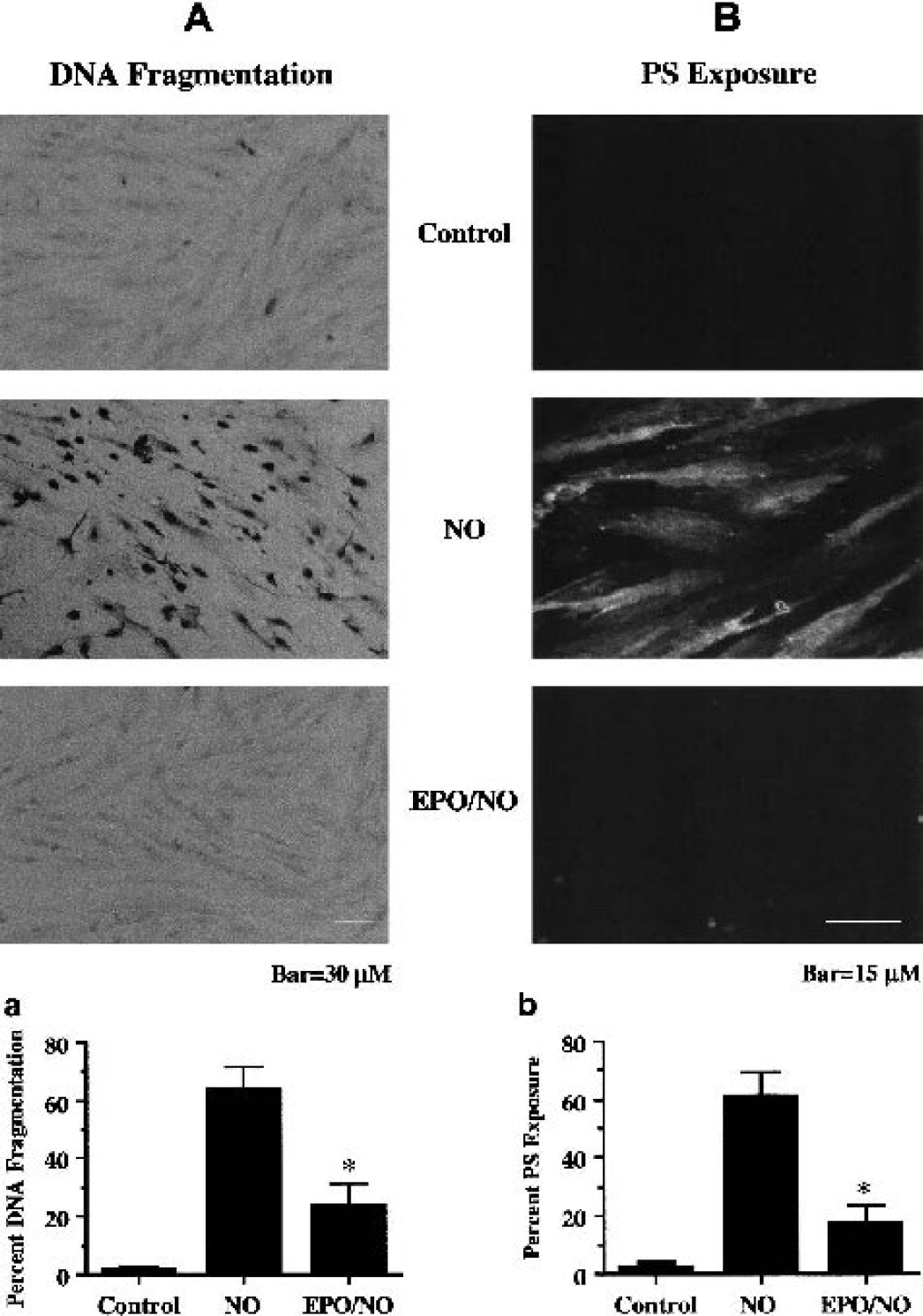
Erythropoietin (EPO) prevents DNA fragmentation and externalization of membrane phosphatidylserine (PS) residues in endothelial cells (ECs).
In Fig. 4B, NO exposure resulted in the marked induction of membrane PS exposure that is present throughout the membrane of ECs. Administration of EPO (10 ng/mL) 1 h before NO prevented the externalization of membrane PS residues in ECs. In Fig. 4Bb, a significant increase in membrane PS residue exposure was observed in EC cultures at 24 h after NO (61 ± 8%) when compared with untreated control cultures (3 ± 2%). Application of EPO (10 ng/mL) 1 h before NO significantly inhibited externalization of membrane PS residues to 18 ± 6%.
Erythropoietin protects endothelial cells through Bcl-xL expression and the prevention of mitochondrial membrane depolarization and the release of cytochrome c
Western blot assay was performed for Bcl-xL at 24 h after NO (NOC-9 or SNP, 1.00 mmol/L) application. In Fig. 5A, expression of Bcl-xL was decreased by NO. In contrast, administration of EPO (10 ng/mL) 1 h before NO exposure significantly increased the expression of Bcl-xL within 24 h after NO exposure. Given the ability of EPO to enhance Bcl-xL in ECs, we next assessed whether EPO could modulate mitochondrial membrane depolarization after NO exposure. Exposure to NO (NOC-9 or SNP, 1.00 mmol/L) produced a significant decrease in the red–green fluorescence intensity ratio using a cationic membrane potential indicator JC-1 within 3 h when compared with untreated control cultures (Figs. 5B and 5C), suggesting that NO results in mitochondrial membrane depolarization. Application of EPO (10 ng/mL) 1 h before NO exposure significantly increased the red–green fluorescence intensity of the ECs, indicating that mitochondrial permeability transition pore membrane potential was restored to baseline (Figs. 5B and 5C). Administration of EPO (10 ng/mL) 1 h before NO maintained mitochondrial permeability transition pore function and prevented mitochondrial cytochrome c release as shown by Western blot analysis (Fig. 5D).
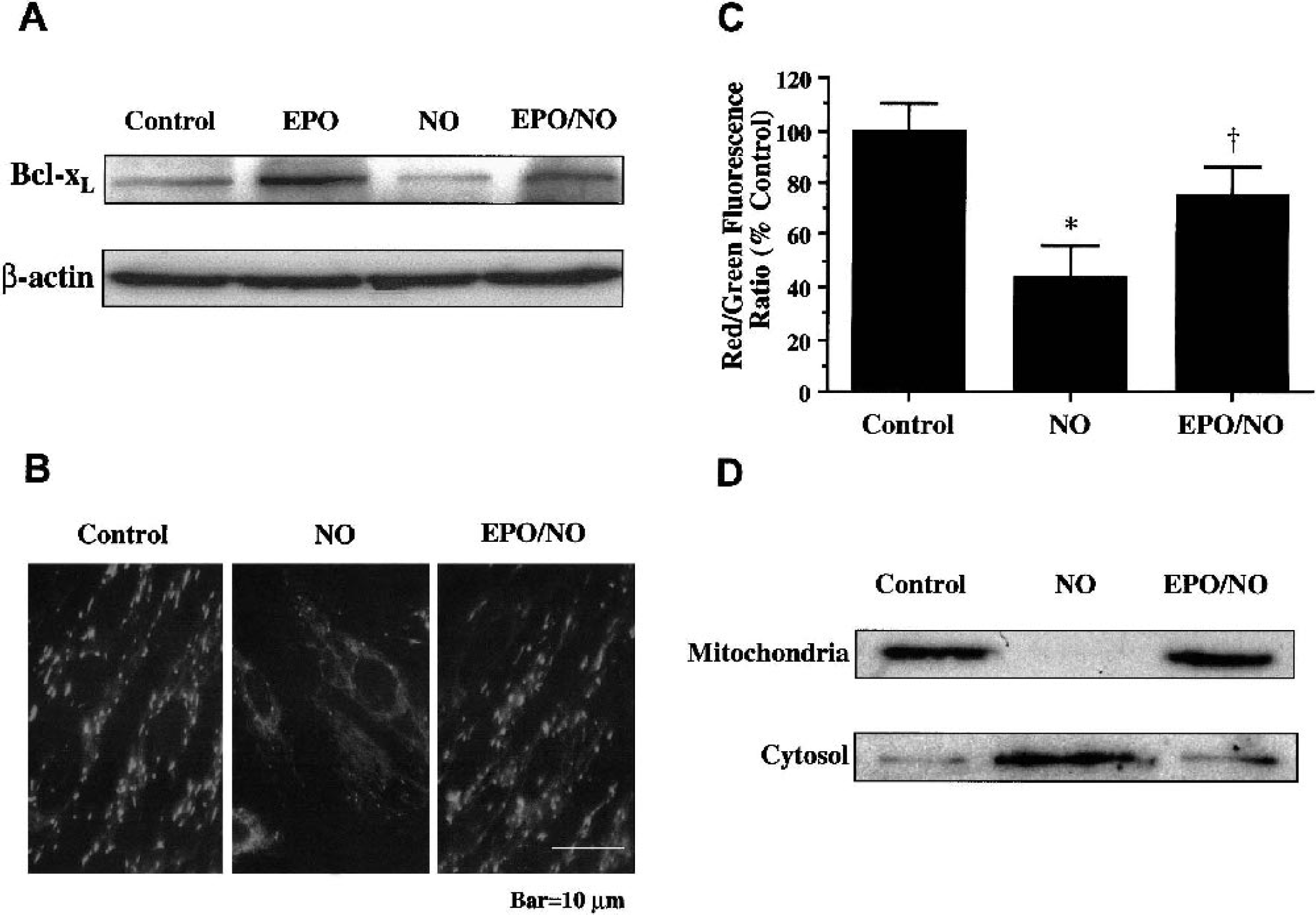
Erythropoietin (EPO) increases Bcl-xL expression and prevents mitochondrial membrane depolarization and cytochrome c release.
Protection by erythropoietin is associated with inhibition of Apaf-1 expression
Western blot assay was performed for Apaf-1 at 6 and 12 h after NO (NOC-9 or SNP, 1.00 mmol/L) application. In Fig. 6A, NO independently increased the expression of Apaf-1 within 6 h after the initial insult. Expression of Apaf-1 remained elevated over a 12-h period. Application of EPO (10 ng/mL) 1 h before NO exposure significantly decreased the expression of Apaf-1 within 6 h after NO exposure. This inhibition in Apaf-1 expression by EPO was maintained throughout a 12-h period (Fig. 6A).
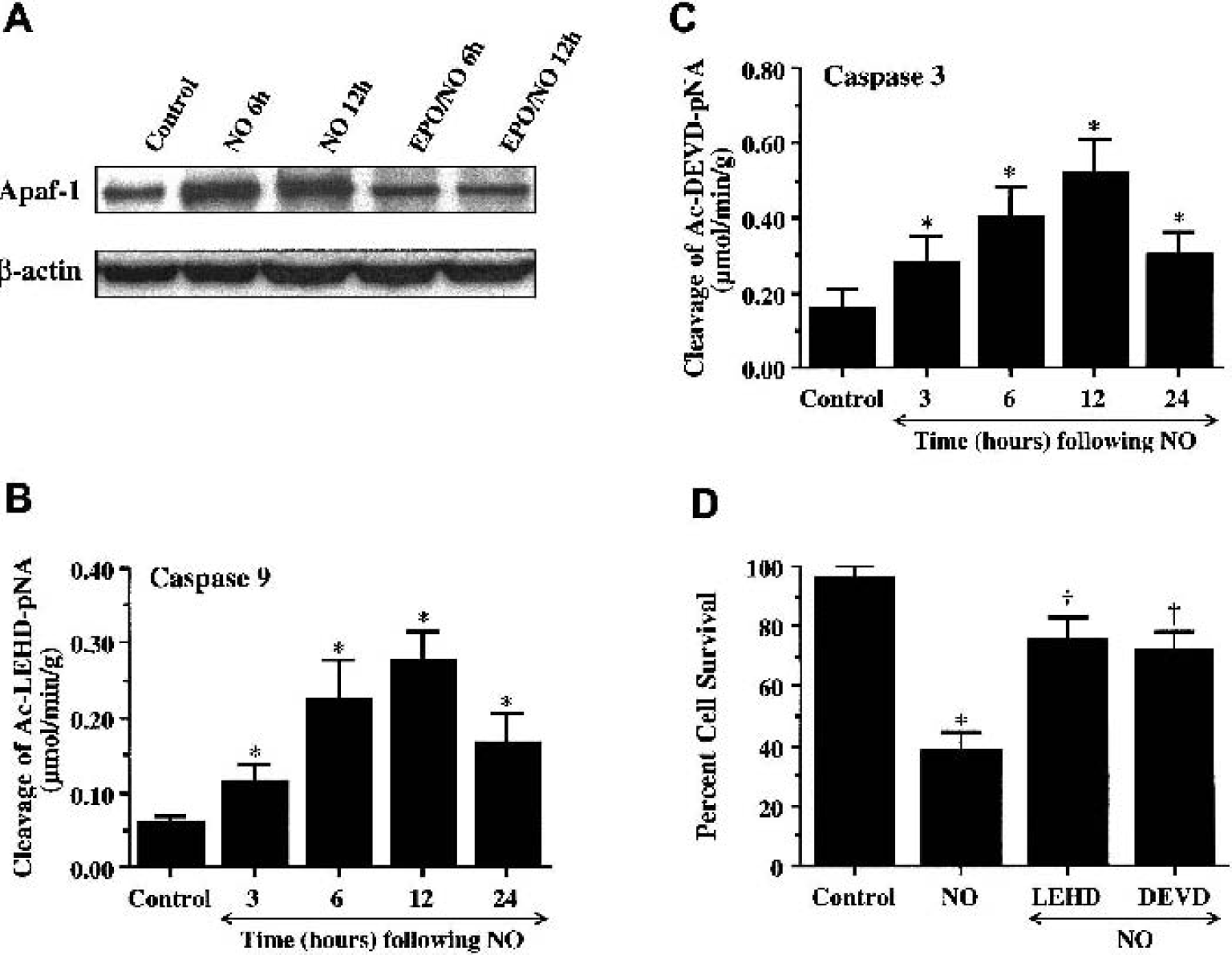
Nitric oxide (NO) exposure leads to enhanced expression of apoptotic protease–activating factor-1 (Apaf-1) and induction of caspase-9 and caspase-3–like activities.
Erythropoietin decreases caspase-9 and caspase-3–like activities after nitric oxide exposure
In Figs. 7A and 7B, EPO (10 ng/mL) was applied to EC cultures 1 h before NO application (NOC-9 or SNP, 1.00 mmol/L), and data for caspase-9 and caspase-3 activities were obtained 12 h after NO because this period represented the peak activities for these cysteine proteases (Lin and Maiese, 2001; Chong et al., 2002c). Administration of EPO significantly decreased caspase-9-like activity to 0.14 ± 0.04 μmol · min−1 · g−1 (P < 0.01) (Fig. 7A). Similarly, EPO pretreatment significantly reduced the activity of caspase-3–like activity (0.26 ± 0.08 μmol · min−1 · g−1) when compared with cultures treated with NO alone (0.52 ± 0.09 μmol·min−1 · g−1) (Fig. 7B).
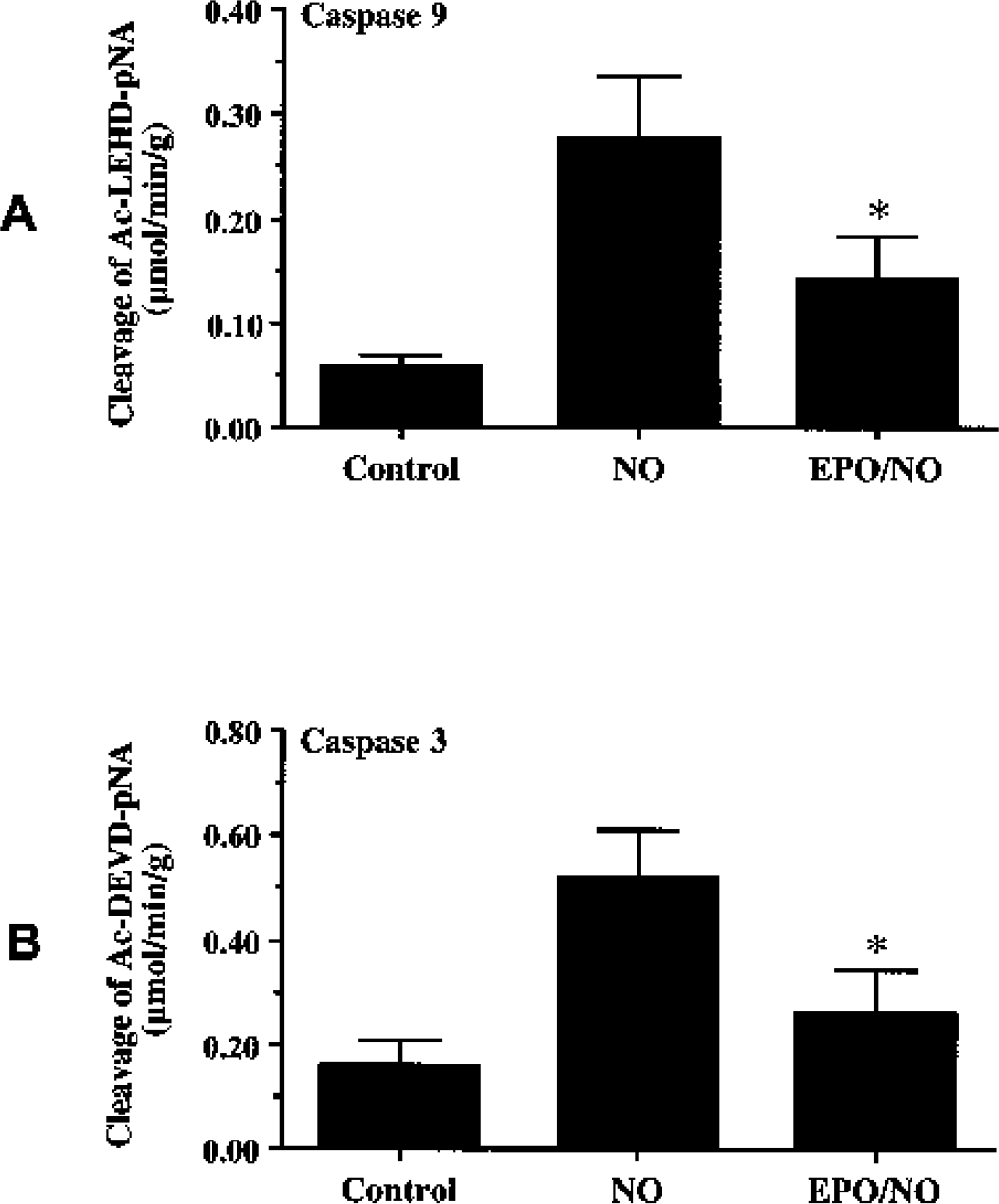
Erythropoietin (EPO) protects endothelial cells (ECs) from injury through the modulation of caspase-9 and caspase-3–like activities. ECs were exposed to a nitric oxide (NO) donor (NOC-9 or SNP, 1.00 mmol/L), and caspase-9
DISCUSSION
Given that cerebral microvascular endothelial cells are a critical component for the maintenance and function of the CNS, the use EPO as a cytoprotectant may offer a novel avenue for the treatment of vascular disease in the CNS. It is the identification of the underlying cellular mechanisms used by EPO, however, that will serve to lay the foundation for future drug development against vascular degenerative disorders. In this respect, we identified some of the downstream cellular pathways modulated by EPO that were critical for protection against EC injury and apoptosis.
Acute ischemic cortical injury in humans can enhance the expression of EPO in the vascular endothelium, suggesting a cytoprotective role for EPO (Siren et al., 2001). We show that EPO and the EPOR are constitutively expressed in cerebral ECs and that the expression of EPO or the EPOR is not altered during free radical exposure. As a result, the ability of EPO to offer EC protection after a toxic insult may be solely dependent on the presence of an intact EPOR for each EC. It therefore would be predicted that the efficacy of EPO vascular protection would be strongly linked to the concentration of exogenous EPO administration, because endogenous EPO is insufficient in providing protection against toxic insults.
Interestingly, protection with EPO is achieved only in a limited concentration range that is dependent on the temporal exposure of EPO. Concentrations of EPO less than 1 ng/mL or greater than 100 ng/mL did not enhance EC survival during free radical exposure. This concentration range for protection with EPO is similar to other injury paradigms in both in vitro (Wen et al., 2002; Chong et al., 2003) and in vivo models (Bernaudin et al., 1999; Grasso et al., 2002) that illustrate a tight therapeutic concentration range for EPO.
Posttreatment strategies with EPO illustrate that EC injury is reversible in nature but resides in a fixed time frame within a 6-h period after the onset of a toxic exposure. This “window of opportunity” for protection by EPO most likely coincides with the progressive induction of secondary cellular pathways during this 6-h time span, such as cytochrome c release (Fig. 5) and cysteine protease induction in cerebral ECs (Fig. 6) (Kwon et al., 2001; Lin and Maiese, 2001; Chong et al., 2002a). Furthermore, prolonged administration of EPO during acute injury paradigms also reduces the ability of EPO to provide cytoprotection. We show that, with preadministration of EPO, the greatest cell survival was achieved with administration periods closest to the application of NO exposure, such as at 3, 6, and 12 h before NO administration. Several factors may determine both the concentration and temporal parameters that regulate the protective ability of EPO. During chronic administration, EPO can result in the formation of antierythropoietin antibodies (Casadevall et al., 2002) and decrease the expression of the EPOR on the cell surface (Verdier et al., 2000). As a result, biologic function of EPO can be blocked at any concentration level.
Administration of exogenous EPO is both necessary and sufficient to protect ECs during NO exposure. Application of the EPO antibody, which can bind to EPO and block its biologic activities in cells (Koshimura et al., 1999), did not alter EC survival when compared with untreated control cultures. Administration of exogenous EPO, however, offers robust protection against NO exposure that is prevented only with coapplication of the EPO antibody. These results illustrate that EPO provides necessary and sufficient protection against EC injury.
Erythropoietin maintains EC survival during free radical exposure through the prevention of DNA fragmentation and the maintenance of cellular membrane asymmetry. Similar to the ability of EPO to protect against lipopolysaccharide-induced apoptosis in pulmonary artery ECs (Carlini et al., 1999), immediate protection by EPO in cerebral ECs is also afforded through the maintenance of intact genomic DNA. Long-term protection results through the inhibition of membrane PS residue exposure, because membrane PS externalization marks cells for phagocytic elimination (Verhoven et al., 1999; Maiese and Vincent, 2000), leads to the propagation of a procoagulant surface (Bombeli et al., 1997; Dombroski et al., 2000), and can promote cellular inflammation (Dombroski et al., 2000). Prior work has shown that administration of EPO in patients undergoing chronic hemodialysis reduces the extent of leukocyte activation and tissue inflammation (Bombeli et al., 1997; Dombroski et al., 2000). Our present work provides further insight into the cellular mechanisms used by EPO to protect ECs from inflammatory injury and delayed phagocytic removal.
A series of cellular pathways seem to be responsible for cytoprotection by EPO. One particular pathway of interest is the modulation of mitochondrial membrane potential. Mitochondrial-mediated apoptosis has been shown to be initiated by free radical injury and result in the cytoplasmic release of cytochrome c (Kwon et al., 2001; Chong et al., 2002c). In our present studies, we show that the free radical NO leads to the depolarization of the mitochondrial membrane in ECs with the subsequent release of cytochrome c. Consistent with earlier clinical studies in patients with chronic renal failure that associated EPO administration with preserved mitochondrial function (Miro et al., 2002), our present studies illustrate that EPO maintains mitochondrial membrane potential and prevents the release of cytochrome c in ECs. In cerebral ECs, EPO may modulate the release of cytochrome c directly or through the regulation of the Bcl-2 family member Bcl-xL. In both erythroid cells and neurons, Bcl-xL has been shown to be strongly expressed during EPO administration (Gregoli and Bondurant, 1997; Wen et al., 2002). In addition, EPO may require Bcl-xL expression for cytoprotection, because without EPO, Bcl-xL is not expressed and apoptotic cell death results in hematopoietic cells (Silva et al., 1996). We now illustrate that upregulation of Bcl-xL by EPO is conserved in ECs and may be necessary for the prevention of apoptotic EC loss by blocking the release of cytochrome c (Yamaguchi and Wang, 2002).
Erythropoietin may foster EC protection through an additional pathway, namely, Apaf-1, which is parallel, but possibly independent of the loss of mitochondrial membrane potential and cytochrome c release during EC injury (Marsden et al., 2002). Central to the enhancement of caspase activity is the expression of Apaf-1 and its oligomerization with cytochrome c to activate caspase-9. We show that NO exposure directly increases Apaf-1 expression within 6 h after the initial exposure. This upregulation of Apaf-1 expression is maintained over a minimum period of 12 h. Application of EPO prevents the induction of Apaf-1 activity over a 12-h course after NO exposure. Thus, EPO independently decreases Apaf-1 expression in ECs and offers a “two-pronged” approach to protect ECs against free radical injury. One pathway involves the prevention of Apaf-1 expression. The second pathway consists of the ability of EPO to maintain mitochondrial membrane potential and limit cytochrome c release through Bcl-xL expression. As a result, oligomerization of Apaf-1 with cytochrome c is prevented and induction of caspase-9 activity is blocked (Fig. 8).
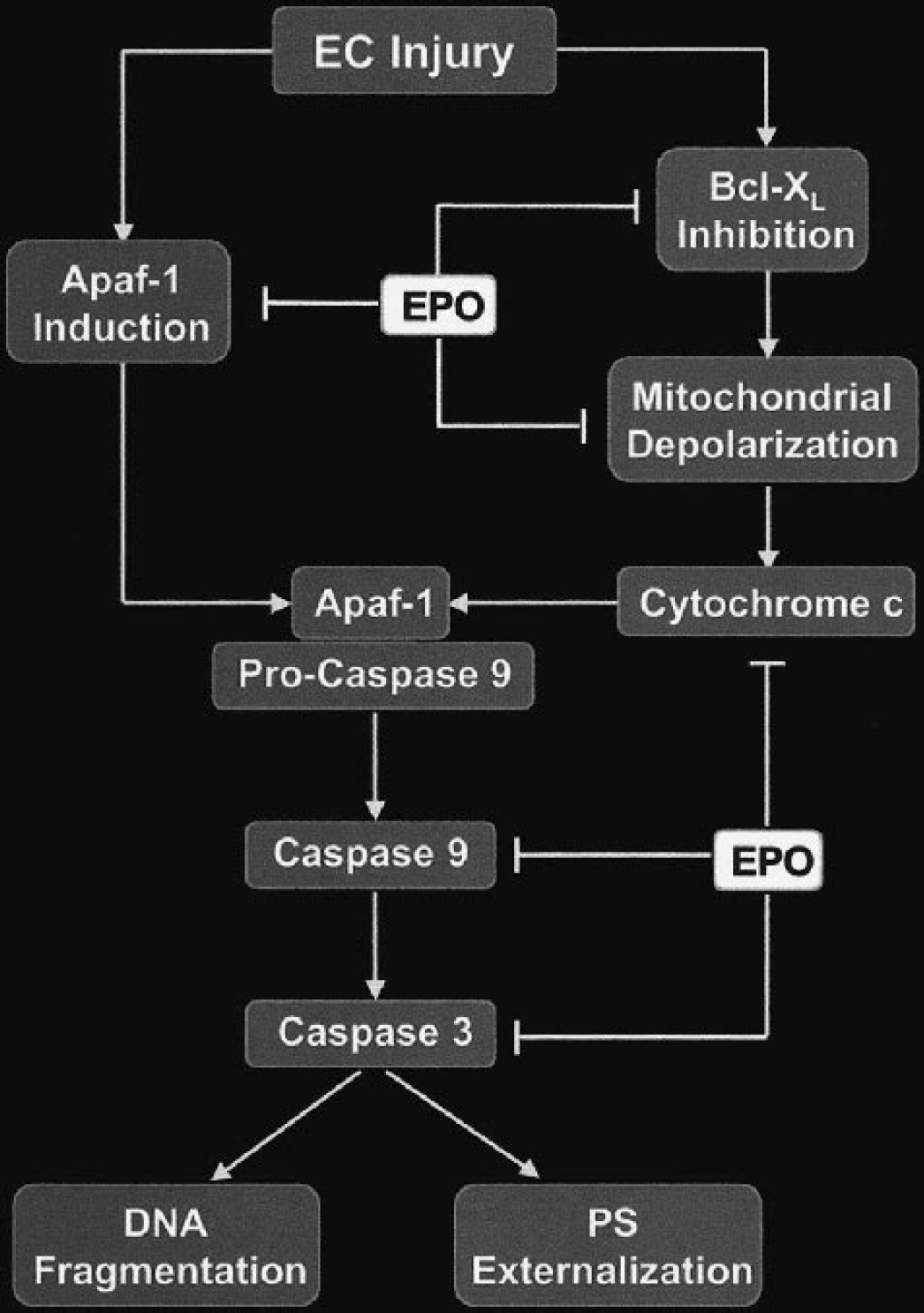
Erythropoietin (EPO) prevents endothelial cell (EC) injury through parallel pathways at the level of apoptotic protease–activating factor-1 (Apaf-1) and the mitochondria with Bcl-xL expression. After a free radical insult to ECs, such as with NO, Apaf-1 expression increases and the mitochondrial membrane becomes depolarized, resulting in the release of cytochrome c. Subsequently, cytochrome c can bind to Apaf-1, oligomerize with cytochrome c, and activate caspase-9. Active caspase-9 can then lead to the induction of caspase-3 to lead to EC apoptosis manifested by DNA fragmentation and membrane phosphatidylserine (PS) externalization. The prevention of EC apoptosis by EPO during NO exposure can occur through parallel pathways that involve Apaf-1 and the maintenance of mitochondrial membrane stability with the upregulation of Bcl-xL. Alternatively, EPO also may act directly on cytochrome c, caspase-9, or caspase-3 to ensure EC survival during toxic insults.
Free radical exposure in ECs can stimulate caspase-3–like activity (Kwon et al., 2001; Lin and Maiese, 2001; Chong et al., 2002a). This activation of cysteine proteases by NO is maintained in our present studies but also includes the activation of caspase-9. As previously described, caspase-9 is activated through a process that involves the cytochrome c–Apaf-1 complex and has been associated with oxidized low-density lipoprotein programmed cell death in systemic ECs (Zhang et al., 2001). In addition, caspase-9 activity is intimately linked to genomic DNA degradation and membrane PS exposure through the activation of caspase-3 (Li et al., 1997; Chong et al., 2002b). The present work shows that EPO significantly prevents the induction of caspase-9 and caspase-3–like activities during NO exposure. The ability of EPO to prevent cysteine protease activity may occur directly at the level of caspase induction or require the prevention of mitochondrial membrane depolarization, the release of cytochrome c, and the generation of Apaf-1 (Fig. 8). This inhibition of caspase-9 and caspase-3–like activities by EPO also is biologically relevant because reduction in the activity of these cysteine proteases prevents EC destruction during free radical injury.