
Abstract
The authors developed a novel diketopiperazine that shows neuroprotective activity in a variety of in vitro models, as well as in a clinically relevant experimental model of traumatic brain injury (TBI) in rats. Treatment with 1-ARA-35b (35b), a cyclized dipeptide derived from a modified thyrotropin-releasing hormone (TRH) analog, significantly reduced cell death associated with necrosis (maitotoxin), apoptosis (staurosporine), or mechanical injury in neuronal–glial cocultures. Rats subjected to lateral fluid percussion–induced TBI and then treated with 1 mg/kg intravenous 35b thirty minutes after trauma showed significantly improved motor recovery and spatial learning compared with vehicle-treated controls. Treatment also significantly reduced lesion volumes as shown by magnetic resonance imaging, and decreased the number of TUNEL-positive neurons observed in ipsilateral hippocampus. Unlike TRH or traditional TRH analogs, 35b treatment did not change mean arterial pressure, body temperature, or thyroid-stimulating hormone release, and did not have analeptic activity. Moreover, in contrast to TRH or typical TRH analogs, 35b administration after TBI did not alter free-magnesium concentration or cellular bioenergetic state. Receptor-binding studies showed that 35b did not act with high affinity at 50 classical receptors, channels, or transporters. Thus, 35b shows none of the typical physiologic actions associated with TRH, but possesses neuroprotective actions in vivo and in vitro, and appears to attenuate both necrotic and apoptotic cell death.
Traumatic brain injury (TBI) remains a leading cause of neurologic disability and causes approximately 75,000 deaths and 350,000 hospitalizations in the United States annually (Krause et al., 1996). Experimental head injury models have been developed in rodents to study pathophysiologic mechanisms that may relate to clinical head injury as well as to examine potential pharmacological therapies. The motor and cognitive impairments that are found in these models parallel those observed clinically (Dixon et al., 1991; Fox et al., 1998; Hamm et al., 1992; McIntosh et al., 1989; Povlishock, 1996; Smith et al., 1995). Such deficits result, in part, from delayed secondary tissue damage resulting from altered metabolic and biochemical processes that are induced by injury (Faden, 1996b; Raghupathi et al., 2000; Vink et al., 1987; Yakovlev et al., 1997). Proposed injury factors include changes in ionic homeostasis, excitatory amino acids, free radicals, proteases, and inflammatory–immune responses, among others; together, they form an interactive cascade that may lead to a reduction in local blood flow, decrease in metabolism, damage to cell membranes, and/or the initiation apoptosis (Faden, 1996b; Raghupathi et al., 2000; Yakovlev et al., 1997; Yamakami et al., 1995).
Many pharmacological treatment strategies have been proposed that are intended to interrupt or inhibit a particular component of this secondary injury cascade. Although several experimental studies have reported improved functional outcome in treated animals (for review, see Faden, 1996b; McIntosh et al., 1998), neuroprotective treatment after clinical TBI has been disappointing (Faden, 2002; Maas, 2001). This failure may reflect the highly multifactorial nature of tissue damage after trauma, which includes varying combinations of ischemia or hypoxia, hematoma, contusion, diffuse axonal injury, and edema (Bullock et al., 1999), as well as issues relating to injury severity, sample size, or lack of appropriate stratification (Faden, 2002; Maas, 2001). Therefore, effective clinical treatment may require either combination drug strategies or the use of single drugs that modulate multiple injury factors (Faden, 2001, 2002).
Thyrotropin-releasing hormone (TRH) and certain TRH analogs have shown neuroprotective effects across species in various CNS injury models, through mechanisms that are believed to be multifactorial (Faden, 1996b). For example, TRH can modulate the effects of endogenous opioids, leukotrienes, platelet-activating factor, glutamate, and sodium channel activity, as well as improve cerebral blood flow and cellular bioenergetic state after CNS trauma (Faden, 1996b). Moreover, TRH has a large therapeutic window (Faden et al., 1984) and is well tolerated in humans (Pitts et al., 1995). Cognitive-enhancing properties have also been demonstrated for TRH and certain TRH analogs (Olson et al., 1995).
However, TRH or conventional TRH analogs have other physiologic effects that may limit their potential utility for the treatment of TBI (Faden et al., 1993; Metcalf, 1982). For example, pressor actions may serve to exacerbate posttraumatic hemorrhage. Temperature regulating effects may limit the potential use of hypothermia as a treatment. Analeptic actions may preclude the implementation of pharmacological coma or inhibit anesthesia required for surgery. In addition, endocrine effects could limit chronic treatment, as may be desired for its possible nootropic actions.
Structure-activity studies have suggested that the neuroprotective actions of TRH do not relate to effects at its classical high-affinity receptor (Faden et al., 1988, 1993, 1999) and may be separable from its other major physiologic actions. Thyrotropin-releasing hormone is rapidly metabolized through the action of endopeptidases (Bassiri and Utiger, 1973; Metcalf, 1982). Its metabolic product, the cyclic dipeptide cyclo(his-pro) (CHP) or histidyl-proline diketopiperazine, has considerable physiologic activity, as do other naturally occurring diketopiperazines (Prasad, 1995). We have developed a series of diketopiperazines related to derivatives of TRH analogs and have examined them for neuroprotective effects as well as for other pharmacological actions. One such compound, 35b, shows considerable neuroprotective activity in vivo and in vitro, but is devoid of many of the other physiologic actions of TRH.
In the present studies, we used both in vivo and in vitro injury models to examine the effects of 35b treatment. Neuronal–glial cocultures subjected to necrotic (maitotoxin), apoptotic (staurosporine), or traumatic insults were used to evaluate the potential neuroprotective actions of the diketopiperazine. Potential autonomic, endocrine, and analeptic effects were examined in uninjured animals and compared with a classical TRH analog (YM-14673) with established neuroprotective properties. Using a lateral fluid percussion TBI model, effects of 35b treatment or vehicle on motor and cognitive recovery and lesion volumes were compared using T2-weighted magnetic resonance imaging (MRI). Numbers of TUNEL-positive neurons were also compared in ipsilateral hippocampus between treated/injured animals and controls. 31P magnetic resonance spectroscopy (MRS) was used to evaluate possible actions of 35b on posttraumatic bioenergetics. Finally, receptor-binding profiles were performed with regard to classical neurotransmitters and transporters.
MATERIALS AND METHODS
Synthetic chemistry
1-(t-Butoxycarbonylamino)-1-cyclohexanecarboxylic acid: 1-amino-1-cyclohexanecarboxylic acid (1.0 g, 6.98 mmol) was dissolved in a mixture of dioxane (15 mL) and NaOH (0.5 mol/L, 15 mL). To this solution was added di-tert-butyl dicarbonate (1.6 g, 7.33 mmol) and the resulting mixture was stirred at room temperature for 15 hours. The reaction mixture was concentrated under reduced pressure and diluted with ethyl acetate (EtOAc; 50 mL). The aqueous solution was extracted with EtOAc (3 × 50 mL), and the collected organic phase was washed with brine (50 mL), dried over sodium sulfate, and concentrated under reduced pressure to yield the title compound 2 (0.98 g, 60%) as a white solid; melting point (mp) 175°C to 177°C; 1H NMR (CD3OD) δ 1.08 to 1.19 (multiplet [m], 2H), 1.40 (singlet [s], 9H), 1.42 to 1.58 (m, 4H), 1.59 to 1.66 (m, 2H), 1.80 to 1.98 (broad singlet, 2H), 6.88 (s, 1H), 12.08 (s, 1H) (Fig. 1).
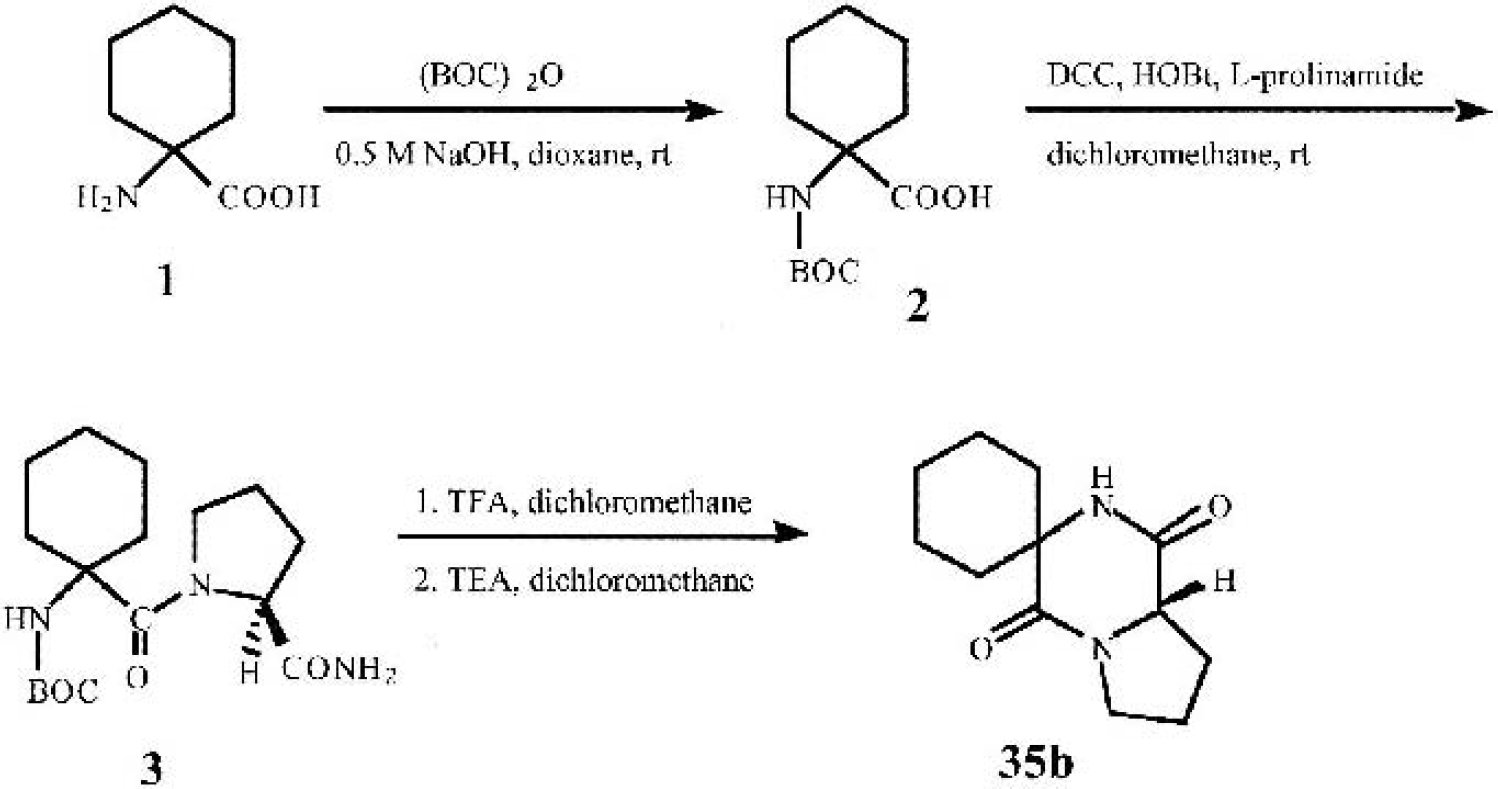
Synthetic chemistry. 1-Amino-1-cyclohexanecarboxylic acid
N[1-(t-Butoxycarbonylamino)-1-cyclohexanecarbonyl]-l-prolinamide in dry dichloromethane (20 mL) was dissolved at 0°C 1-(t-butoxycarbonylamino)-1-cyclohexanecarboxylic acid (0.60 g, 2.47 mmol), l-prolinamide (0.28 g, 2.47 mmol), and dicyclohexylcarbodimide (0.51 g, 2.47 mmol). The resulting mixture was stirred at 0°C for 10 minutes, and HOBt (4.93 mL of 0.5-mol/L solution in DMF, 2.47 mmol) was added and the mixture stirred overnight at room temperature. Insoluble matters were filtered off, and the filtrate was concentrated under reduced pressure. The crude residue was washed with hexane/EtOAc (9:1, 20 mL) to yield the title compound 3 (0.50 g, 60%) as a white solid; mp: 220°C; 1H NMR (CD3OD) δ 1.02 to 1.90 (m, 12H), 1.42 (s, 9H), 1.91 to 2.10 (m, 2H), 3.36 to 3.41 (m, 1H), 3.72 to 3.82 (m, 1H), 4.08 to 4.14 (m, 1H), 6.85 (doublet, 2H), 7.50 (s, 1H) (Fig. 1).
Fluid percussion–induced traumatic brain injury
Fluid percussion–induced TBI was induced as previously described (McIntosh et al., 1989; Yakovlev et al., 1997). Male Sprague-Dawley rats (350 ± 50 g) were anesthetized with sodium pentobarbital (70 mg/kg, intraperitoneally), intubated, and implanted with femoral venous and arterial catheters (studies 1 and 3); in studies 2 and 4, the tail artery was cannulated and the tail vein was used for injection of drug or vehicle. Brain temperature was assessed indirectly through a thermistor in the temporalis muscle and body temperature was maintained at normal levels through a feedback-controlled heating blanket. Blood pressure was continuously monitored, and arterial blood gases were analyzed periodically. After the animal was placed in a stereotaxic frame, the scalp and temporal muscle were reflected; a small craniotomy (5 mm), located midway between the lambda and bregma sutures over the left parietal cortex, allowed insertion of a Leur-Loc that was cemented in place. The fluid-percussion head injury device, manufactured by the Medical College of Virginia (Richmond, VA, U.S.A.), consists of a plexiglas cylindrical reservoir filled with isotonic saline; one end includes a transducer that is mounted and connected to a 5-mm tube that attaches through a male Leur-Loc fitting to the female Leur-Loc cemented at the time of surgery. A pendulum strikes a piston at the opposite end of the device, producing a ∼22-millisecond pressure pulse, leading to the deformation of underlying brain. The degree of injury is related to the pressure pulse expressed in atmospheres (atm): 2.6 or 2.7 atm in our laboratory produces a moderately severe injury with regard to neurologic and histologic deficits. Sham (control) animals were subjected to anesthesia and surgery without fluid percussion brain injury.
Motor scores
Standardized motor scoring was performed at 1, 7, and 14 days after TBI, by persons unaware of treatment (Yakovlev et al., 1997). Motor function was evaluated using three separate tests, each of which is scored via an ordinal scale ranging from 0 (severely impaired) to 5 (normal function). Tests measure the ability to maintain position on an inclined plane in a vertical and two horizontal positions for 5 seconds, forelimb flexion (suspension by the tail), and forced lateral pulsion. Each of seven individual scores (vertical angle, right and left horizontal angle, right and left forelimb flexion, right and left lateral pulsion) was added to yield a composite neurologic score ranging from 0 to 35. This scoring method, which has been used in our laboratory extensively during the last 10 years, shows high interrater reliability (>93%) and is very sensitive to pharmacological manipulations (McIntosh et al., 1989).
Cognitive assessment: Morris water maze
Learning and memory deficits in laboratory models of brain injury can be detected using a water maze paradigm similar to that originally described by Morris (Morris, 1984), as previously described by us for experimental TBI (Faden et al., 1999; Fox et al., 1998). Spatial learning was assessed by training the animals to locate a hidden, submerged platform using extramaze visual cues. The apparatus used consisted of a large, white circular pool (165-cm diameter, water temperature 24°C + 1°C) with a plexiglas platform (110 mm2) painted white and submerged (20 mm) below the surface of water that was rendered opaque with the addition of white, nontoxic paint. During training, the platform was hidden in one quadrant, and the animal was gently placed into the water facing the wall at one of four randomly chosen locations separated by 90 degrees. The latency to find the hidden platform within a 90-second period was recorded by a blinded observer. On the first trial, rats failing to find the platform within 90 seconds were assisted to the platform. Animals were allowed to remain on the platform for 15 seconds on the first trial and 10 seconds on all subsequent trials. There was an intertrial interval of 30 minutes, during which time the animals were towel-dried and placed under a heat lamp. A series of 16 training trials administered in blocks of 4 were conducted on days 14 to 17 after injury. To control for visual discriminative ability or motor impairment, the same animals were required to locate a clearly visible black platform (placed in a different location) raised 5 mm above the water surface at least 2 hours after the last training trial.
Study 1 was performed in association with motor testing. Because lesion volumes were not evaluated in this study, a second study was performed using a more recently acquired computerized analytical system. In study 2, a PC-controlled video system (AccuScan Instruments, Inc., Columbus, OH, U.S.A.) was used to examine swim speed, distance, latency necessary to find a submerged resting platform, and time spent in the periphery versus the central zone adjacent to the platform. The swimming pool was monitored by a video camera (Sony CCD-TR517) equipped with a wide-angle objective. The signal was digitized (50 half-frame/s) and data were buffered in the computer's (Pentium-IV) main memory. The software (EZ-Video version 2.50, Automated Tracking System with Accu-Trak version 2.40, AccuPlot version 2.40, SerSwit 1.00, and ZoneMapper version 2.30) acquired, compressed, stored, and analyzed real-time pictures of animal movement. Swim speed was calculated by dividing path length by latency for each trial for each rat, and expressed as centimeters per second. The periphery was defined as a circular zone (zone 1) with a diameter of 20 cm (of 165 cm total). The remaining inner circle was divided into four equal parts (zones 2, 3, 4, and 5), with the platform located in the central zone 5 (Fig. 2). The mean latency and time periods spent in periphery and central zone adjacent to the platform were expressed in seconds. The effect of 35b on cognitive outcome was analyzed by comparison among groups (sham-operated controls, injured group without therapy, and injured group with 35b).
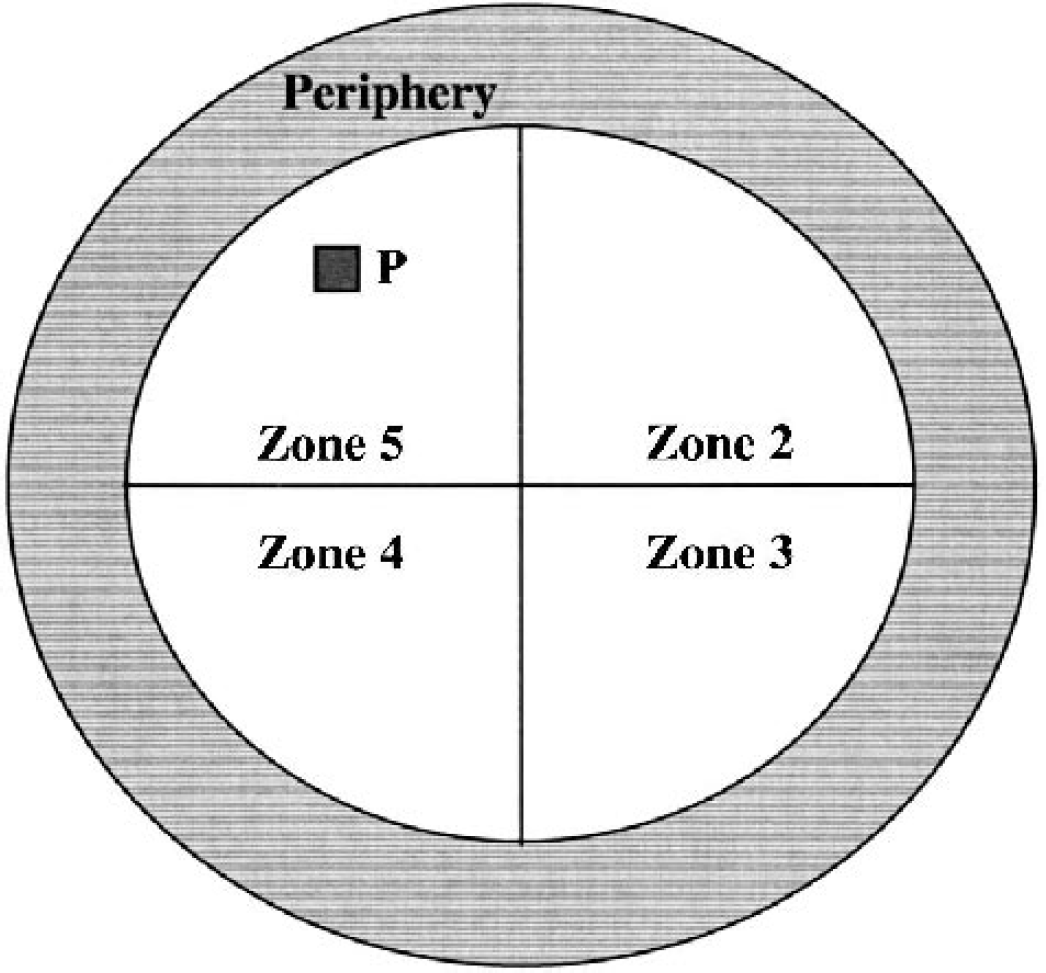
Diagrammatic representation of the map generated by a software used to analyze animal performance during the Morris water maze test. The periphery is defined as a circular zone (zone 1) with a diameter of 20 cm (of 165 cm total), whereas the remaining inner circle is divided into four equal parts. The hidden, submerged platform (P) is located in zone 5 (central zone).
Analeptic and autonomic assessment
Additional groups of uninjured rats were tested for autonomic and analeptic responses immediately before and up to 60 minutes after drug administration. For the analeptic study, rats were first anesthetized with 40 mg/kg sodium pentobarbital intraperitoneally and placed onto an unheated pad on the laboratory bench top at room temperature (22°C + 2°C). A thermistor probe was placed in the rectum to measure core body temperature. After a 10-minute period, vehicle or drug was administered via the tail vein. Time to recovery of the righting reflex was subsequently determined while temperature was recorded at 5-minute intervals for all animals.
To assess mean arterial pressure responses to the novel TRH analogs, a separate group of rats was anesthetized with 4% isoflurane (1.5 L/min). Catheters were then placed into the right carotid artery and internal jugular vein and exteriorized through the back of the neck. Rats were separated in individual cages and allowed to recover from anesthesia. Mean arterial blood pressure was continuously recorded via a transducer connected directly to the arterial catheter for the duration of the study. One hour after catheter placement, vehicle or drug was administered via the venous catheter.
Endocrine assessment
A radioimmunoassay for rat (r) TSH was used to assess endocrine function. Saline or 35b was administered intravenously via a chronically implanted catheter in anesthetized rats (70 mg/kg sodium pentobarbital; n = 6/group) at time 0, and 5 mL whole blood was collected 30 minutes after injection. Blood plasma was subsequently obtained, frozen at −70° C, and shipped to Covance Laboratories (Vienna, VA, U.S.A.) for appropriate analyses. The rTSH assay is a specific double-antibody radioimmunoassay using antiserum prepared in the rabbit against purified rTSH antigens (Kieffer et al., 1974). The assay is standardized against purified rTSH.
Drugs and design
Four separate TBI studies were performed. In all four studies, rats were injected with 1.0 mg/kg 35b or vehicle 30 minutes after injury. In study 1, cognitive and motor functions were assessed in animals treated with vehicle (n = 11) and 35b (n = 12), and all injections were made through the femoral vein. In study 2, vehicle-treated (n = 11) and 35b-treated (n = 11) animals were subjected to cognitive testing as assessed by multiple outcomes (i.e., distance swum, search strategy) and lesion volume measurements via MRI, and all injections were made via the tail vein. In study 3, vehicle-treated (n = 5) and 35b-treated (n = 5) animals were used for MRS analysis of bioenergetic status after injury, and all injections were made via the femoral vein. In study 4, vehicle-treated (n = 6) and 35b-treated (n = 5) animals were killed 24 hours after injury and their brains removed for TUNEL analysis. The numbers of animals given are those that survived the intended experimental period. The level of mortality in all studies was less than 20% and was not affected by treatment. Investigators were blinded as to treatment.
For autonomic, analeptic, or endocrine studies, rats were given normal saline (n = 6), YM-14673 (n = 6), or 35b (n = 6) at the times indicated previously. YM-14673 was used for comparison purposes because previous work showed that the TRH analog significantly improves outcome after CNS injury (Faden, 1989, 1993). Different anesthetic protocols were used in the various studies, as required by the different models and durations of measurement, but in all cases, nontreated vehicle controls were used for comparisons.
TUNEL studies
Rats were anesthetized with sodium pentobarbital (100 mg/kg) 24 hours after TBI and then killed by decapitation. Their brains were quickly removed and submersed in OCT (OCT Tissue-Tek; Andwin Scientific, Warner Center, CA, U.S.A.) media and frozen in cooled isopentane (−70°C). Later, 16-μmol/L sections were taken from −3.2 to −3.7 relative to bregma as described previously (Knoblach et al., 1999). Every tenth section was saved, and four of these sections (each at least 500 μm apart) were selected for analysis. Tissue was stained using the ApopTag Fluorescein-based indirect immunofluorescence staining kit (Serologicals Corporation, Norcross, GA, U.S.A.) with minor modifications. Some sections were double-labeled with anti-NeuN neuronal-specific marker antibody as detailed previously (Knoblach et al., 1999). Sections were viewed with a Nikon TE300 microscope, and all TUNEL-positive cells within the dentate granule cell layer (upper and lower blades) and CA3 region were counted at high magnification. Cell counts for all slides from each animal were averaged, and averages from each animal were combined and represented as the mean ± SEM for the group.
Magnetic resonance imaging
Three weeks after injury, animals included in study 2 were reanesthetized with sodium pentobarbital (70 mg/kg, intraperitoneally) and an MRI examination using a Bruker 7.0-T magnetic resonance spectrometer/imager was performed as described elsewhere (Albensi et al., 2000). Animals (n = 11/group) were placed in a plexiglas holder with a heating pad warmed to 37°C, and a respiratory motion detector was fastened over the thorax to facilitate respiratory gating. The plexiglas holder was then slid into proton-tuned birdcage coil, located in the center of the magnet bore. Field homogeneity across the brain was optimized, and sagittal and coronal scout images were collected to position the transverse slices throughout the brain region of interest. Multislice, multiecho T2-weighted images were acquired to obtain eight contiguous slices of cerebrum (field of vision, 4 × 4 cm; slice thickness, 2 mm; resolution, 256 × 256; repetition time to echo time, 2,000/20 milliseconds; four echo images and two averages). Lesion volumes were estimated from the summation of areas of hyperintensity in cerebrum on each slice, multiplied by slice thickness, for both ipsilateral and contralateral hemispheres, using Bruker Paravision 2.1 imaging software. Average lesion volume for each group (± SEM in μL) was calculated. A comparison was made between injured animals treated with 35b and those receiving vehicle after injury.
Magnetic resonance spectroscopy
Magnetic resonance spectra were obtained as detailed elsewhere (Vink et al., 1990). Briefly, animals were placed in a specially constructed, temperature-controlled plexiglas holder and a 5 × 9-mm surface coil was placed centrally around the craniotomy site. Skin and muscle were retracted away from the coil to prevent contributions from these tissues. The animals were inserted into the center of a 7.0-T magnet interfaced with a Bruker spectrometer, and field homogeneity was optimized on the water signal before acquisition of phosphorus spectra. Phosphorus spectra were obtained in 30-minute blocks before and for 4 hours after induction of fluid percussion–induced TBI using a 90° pulse calibrated for a 2-mm cortical depth, a 700-millisecond delay time, and a 4,000-Hz spectral width containing 2,048 data points. Rectal temperature and respiration were monitored at all times. Anesthesia was maintained via intravenous sodium pentobarbital (15 mg·kg−1·h−1). At the conclusion of the 4-hour acquisition period, animals were removed from the magnet and returned to their cages after their wounds were closed.
Phosphorus magnetic resonance spectra were analyzed using a Bruker computer software program. Following convolution difference (400/20 Hz), chemical shifts and integrals of the individual peaks were determined after line fitting. Intracellular pH, brain free-magnesium concentration, and cytosolic phosphorylation potential were then determined as described in detail elsewhere (Vink et al., 1990). Briefly, intracellular pH was determined from the chemical shift of the inorganic phosphate peak δPi) relative to phosphocreatine in the magnetic resonance spectra using the following equation:
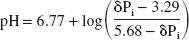
Similarly, free-magnesium concentration was determined from the chemical shift difference between the α and β peaks of ATP (Gupta et al., 1978) using the following equation:
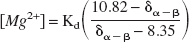
where X is the chemical shift difference between the α and β peaks of ATP. The Kd for MgATP was initially assumed to be 50 μmol/L at a pH 7.2 and an ionic strength of 0.15 mol/L, and was corrected for pH according to the method described by Bock et al. (1987).
Cytosolic phosphorylation potential (PP) was determined according to the following equation:

where Σ represents all the ionic forms of the free species. The concentration of ADP was calculated from the creatine kinase equilibrium equation after correcting the equilibrium constant for pH and free-magnesium concentration as described in detail elsewhere (Vink et al., 1994). Concentrations of the other metabolites were determined from the integrated peak areas of the respective MRS peaks, assuming that the normal, preinjury values for phosphocreatine and ATP were 4.72 and 2.59 μmol/g, respectively, and that the total creatine pool remained constant at 10.83 μmol/g (Siesjo, 1981; Veech et al., 1979). Brain water content was assumed to be 80%, with the intracellular compartment accounting for 78% of the total water (Siesjo, 1981).
In vitro studies
RESULTS
Neurologic outcome after traumatic brain injury
Study 1 examined the effects of 35b administration on motor and cognitive deficits induced by lateral fluid percussion injury. Significantly improved composite neurologic scores were observed 7 and 14 days after trauma in 35b-treated animals compared with vehicle-treated controls (Fig. 3). For spatial learning, the latency to find the submerged platform was significantly reduced on all testing days after the initial training day (Fig. 4).
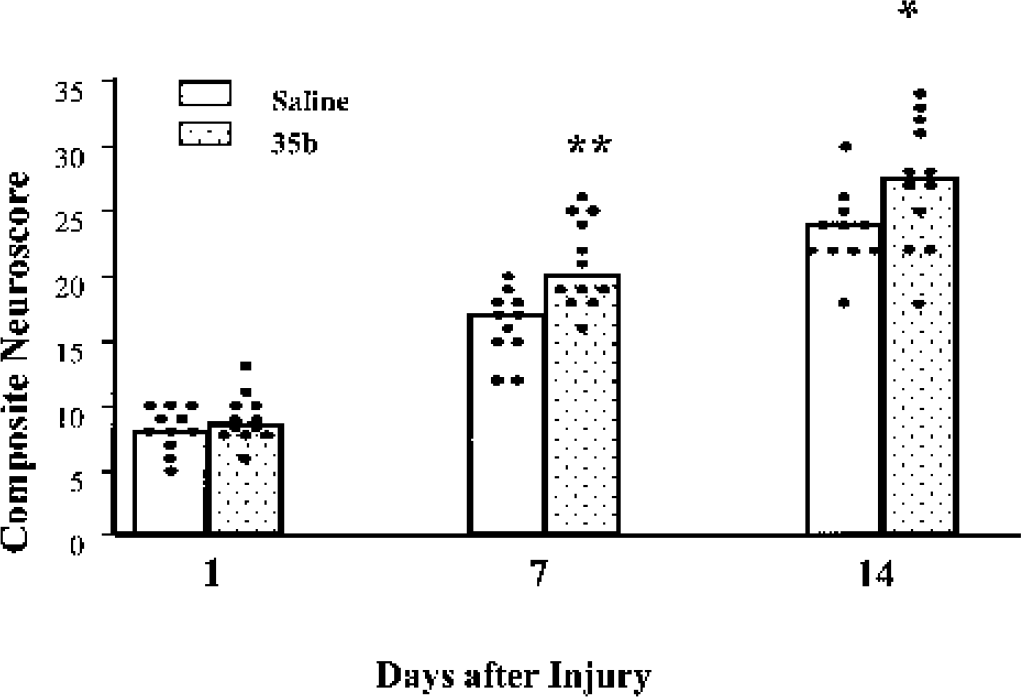
Effect of 35b on motor recovery after moderately severe (2.6 to 2.7 atmospheres) lateral fluid percussion injury in the rat. 35b (1 mg/kg, intravenously, n = 12) or saline vehicle (n = 11) was administered 30 minutes after injury. Columns represent median values for each treatment group, whereas each dot represents an individual animal score. *P < 0.05, **P < 0.01 versus saline group.
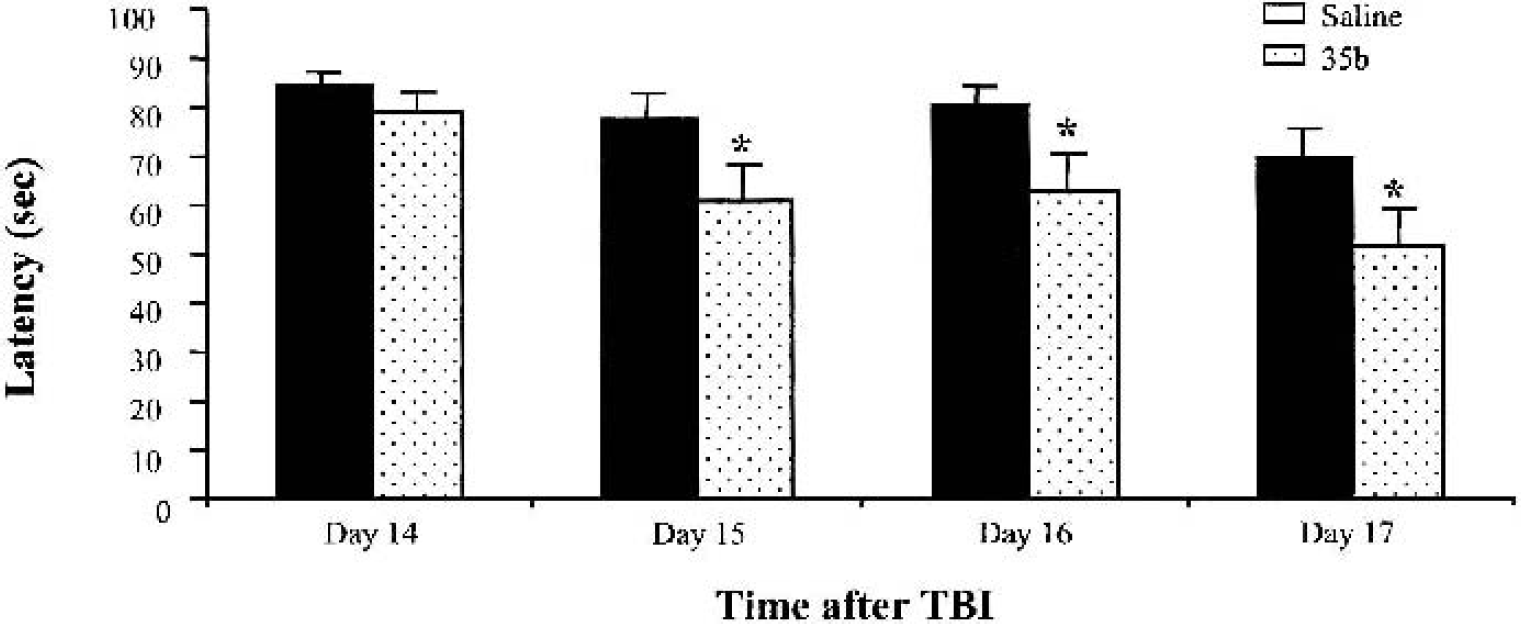
Latency to find the hidden platform in a version of the Morris water maze (study 1). Results are expressed as daily mean ± SEM for each group over four trials. *P < 0.05 versus saline group. TBI, traumatic brain injury.
Study 2 examined the effects of 35b injection on trauma-induced cognitive deficits, measured and analyzed in detail using the aforementioned PC-controlled video system for tracking and analyzing animal behavior. Parameters such as latency required to find a submerged resting platform, swim speed and distance, and time spent in periphery and in the central zone adjacent to the platform were evaluated and compared in 35b-treated injured, vehicle-treated injured, and sham-control animals (Fig. 5). The swim speed was similar in both 35b- and vehicle-treated injured groups (27 ± 6 vs. 25 ± 5 cm/s; data not shown). The latency necessary to find the platform in sham-operated controls was 38 ± 5 seconds. Administration of 35b significantly (P < 0.05) reduced mean latency on the fourth test day (day 17 after trauma) compared with the vehicle-treated injured group (71 ± 7 vs. 89 ± 8 seconds; Fig. 5A). Furthermore, 35b-treated animals covered less distance to the platform (1,948 ± 21 and 1,927 ± 18 cm vs. 2,407 ± 17 and 2,214 ± 15 cm, respectively; P < 0.05) than the nontreated animals on postinjury days 16 and 17, respectively (Fig. 5B). At the same time points, injured animals receiving 35b spent less time in periphery compared with the traumatized vehicle-treated group (57 ± 6 and 57 ± 8 seconds vs. 76 ± 8 and 79 ± 7 seconds; P < 0.05; Fig. 5C). Additionally, the 35b group spent more time in the central zone than the vehicle-treated group on the fourth day of testing (4.30 ± 0.30 vs. 2.83 ±0.19 seconds; P < 0.05) (Fig. 5D).
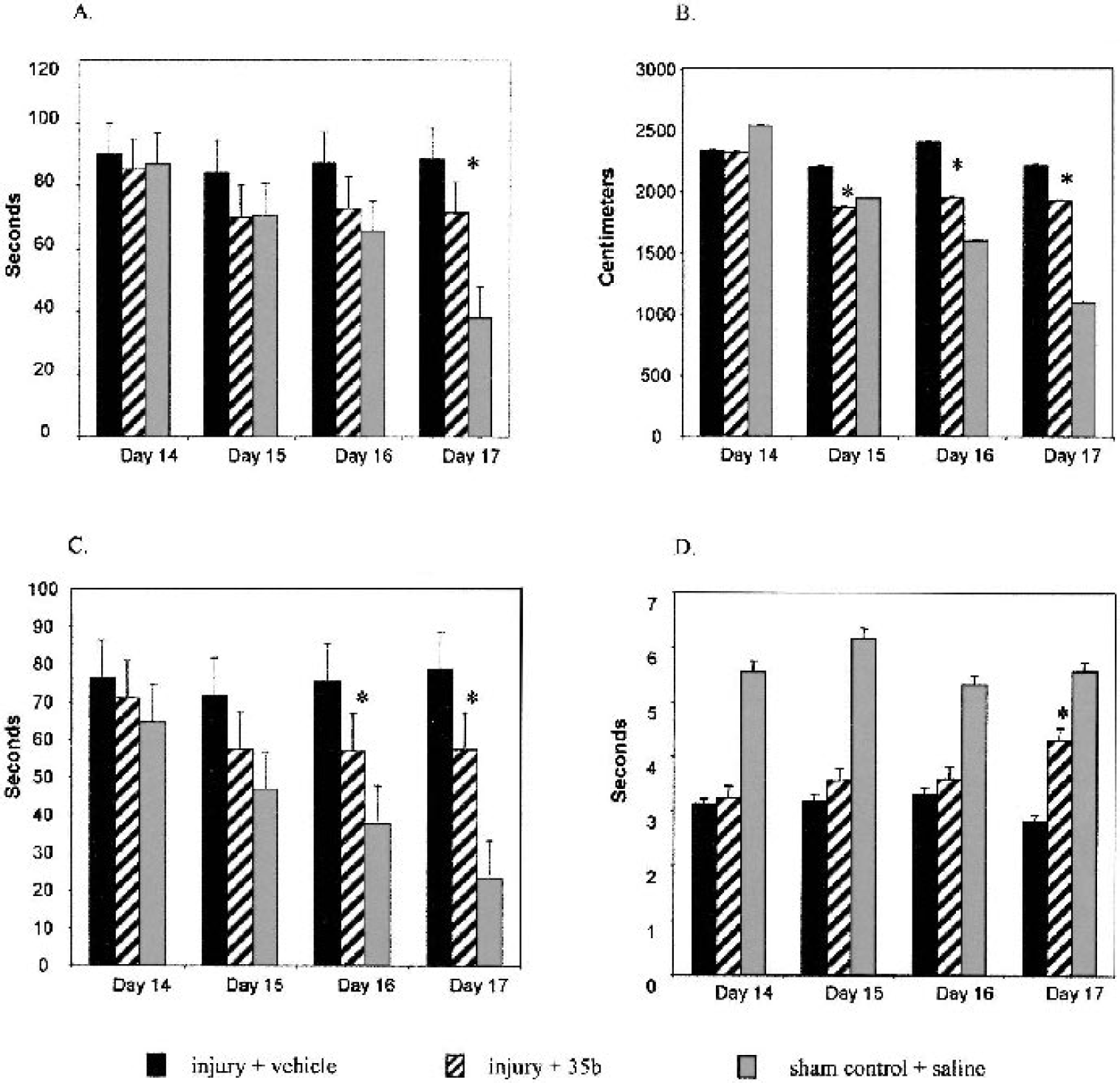
The effects of 35b on Morris water maze performance after traumatic brain injury as assessed using a computerized video system to acquire data on multiple outcomes. Bars represent the mean ± SEM averaged from the four trials. *P < 0.05 versus fluid percussion injury (injury + vehicle) from the same day.
Magnetic resonance imaging lesion volume measurement
T2-weighted images were used to assess lesion volume by MRI at 21 days after injury. Changes in magnetic resonance signal in injured animals were evident as increases in pixel intensity. Regions of hyperintensity were observed principally in the ipsilateral hemisphere, encompassing not only cortex and hippocampus but also the subcortical white matter tracts such as corpus callosum. Administration of 35b significantly (P < 0.05) reduced lesion volumes compared with the injured vehicle-treated animals (10.77 ± 1.39 vs. 21.75 ± 3.56 μL; Fig. 6).
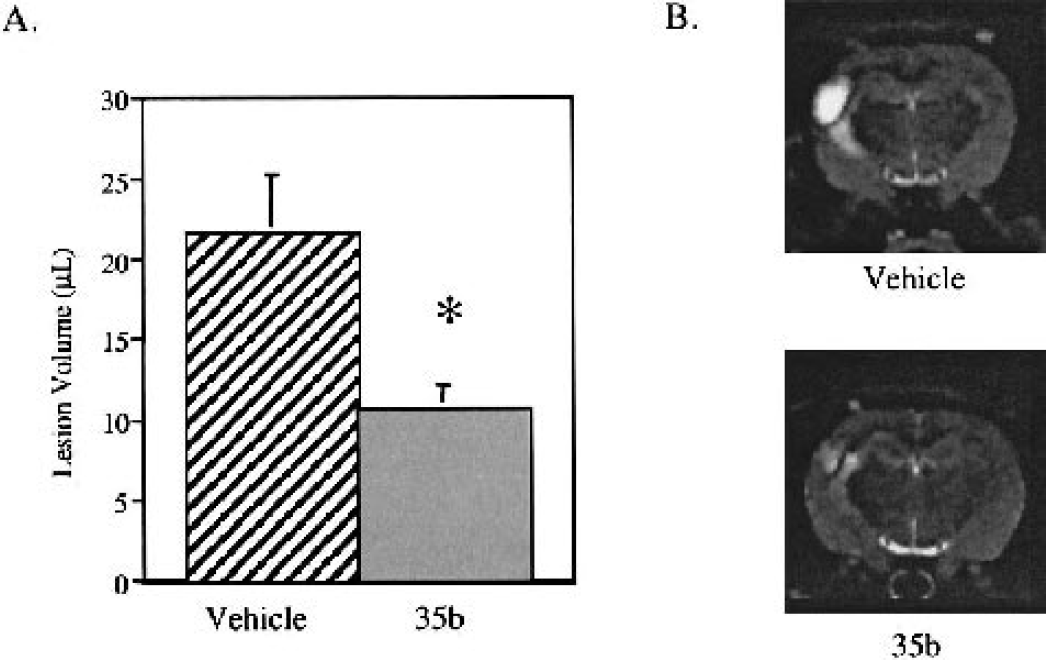
Effect of 35b on lateral fluid percussion–induced lesion volume 21 days after injury in the rat. Data are from rats treated 1 hour after injury with 35b or saline vehicle.
TUNEL staining after traumatic brain injury
The TUNEL-positive cells were distributed throughout the ipsilateral cortex of all animals 24 hours after TBI. In addition, TUNEL-positive cells were observed in the CA3, CA4/hilus and granule cell layer of the hippocampus. However, the number of TUNEL-positive cells in the granule cell layer of the hippocampus was significantly lower in 35b-treated rats compared with vehicle-treated controls (P < 0.05 for treated vs. control; Fig. 7).
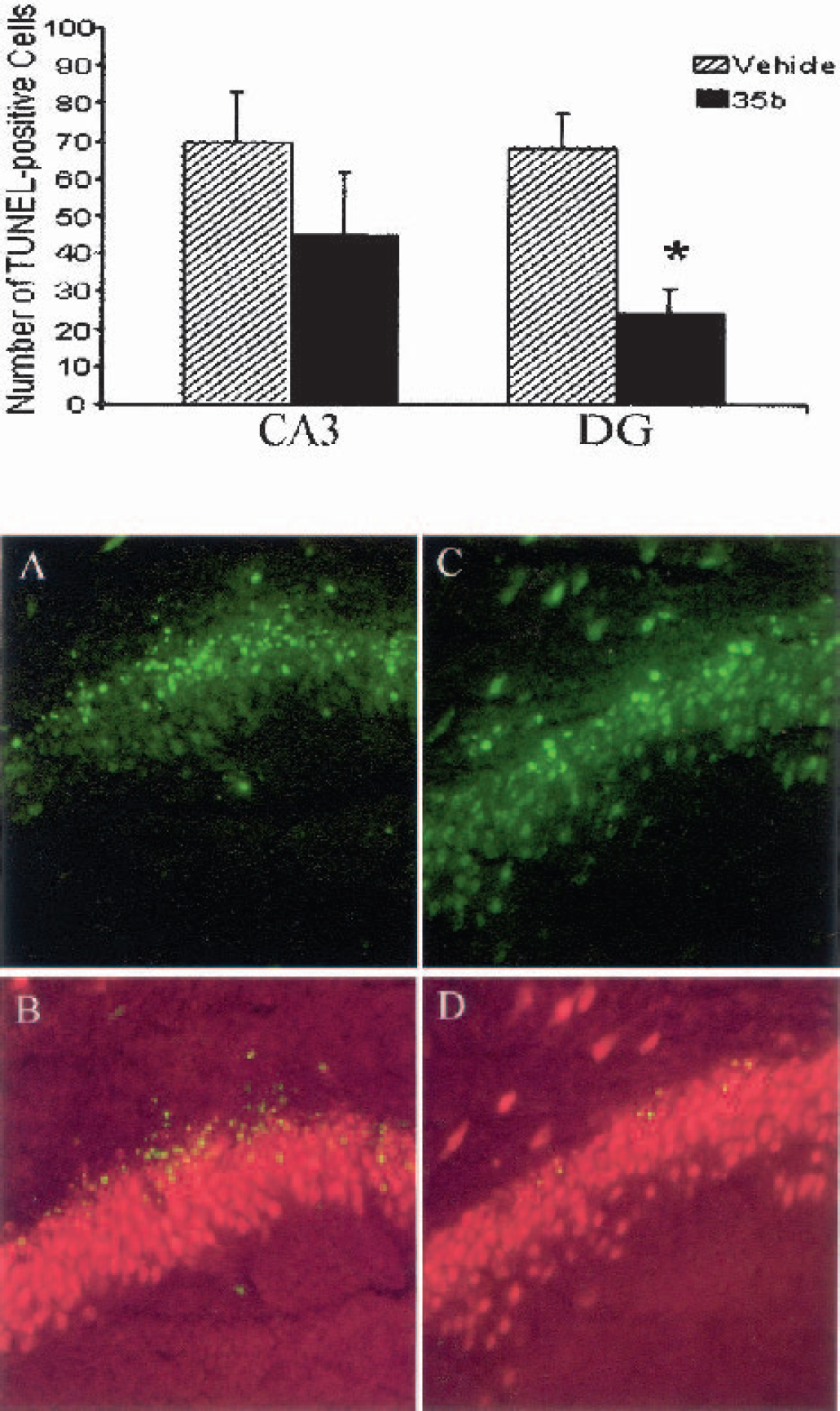
Effect of 35b on apoptotic cell death after traumatic brain injury. 35b (1.0 mg/kg or vehicle) was injected 30 minutes after moderately severe lateral fluid percussion injury. The number of TUNEL-positive cells in the ipsilateral hippocampus was then quantified 24 hours later. Bars on graph indicate the mean ± SEM of TUNEL-positive cells in the CA3 and dentate granule (DG) cell layers for each treatment group as indicated.
Autonomic and analeptic studies
After 35b treatment, there were no significant effects on blood pressure, body temperature, TSH activity, or latency to recover the righting reflex (Figs. 8 to 11). Blood pressures were indistinguishable between 35b- and vehicle-treated, uninjured animals examined over a 2-hour period, whereas YM-16743 caused a rapid (<10-minute) and sustained (>2-hour) pressor effect. Animals subjected to TBI showed a transient pressor response (30–50 mm Hg for 10–20 seconds), followed by a transient drop in pressure below the original baseline (20–40 mm Hg for 5–15 minutes). In all TBI studies, drugs were injected 30 minutes after injury, when the initial TBI-induced changes had stabilized. Nevertheless, the blood pressure was monitored for an additional 30 minutes after injection in approximately one third of the animals (selected at random). Only one of these animals, a vehicle-treated injured control, showed transient pressor response (5 minutes) after injection. It was not possible to assess the effect of 35b on core body or brain temperature after TBI, because injured animals were routinely maintained on feedback-controlled heating pads to exclude the effect of temperature on TBI-associated outcomes.
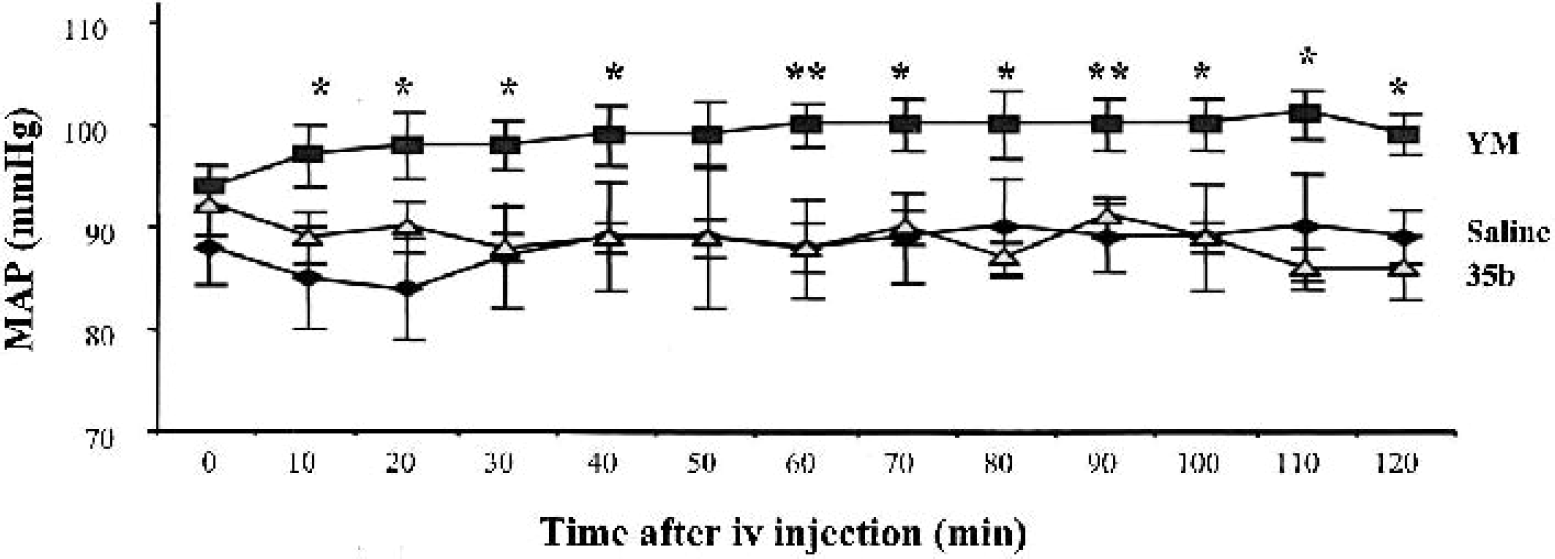
Graph of mean arterial blood pressure (MAP) up to 120 minutes after vehicle or drug administration (1 mg/kg) in the fully conscious and unrestrained rat. Rats treated with 35b (n = 6) did not differ significantly from saline-treated controls (n = 6). Rats treated with YM-14673 (n = 6), however, maintained a significantly higher MAP over the 120-minute testing period (P < 0.05).
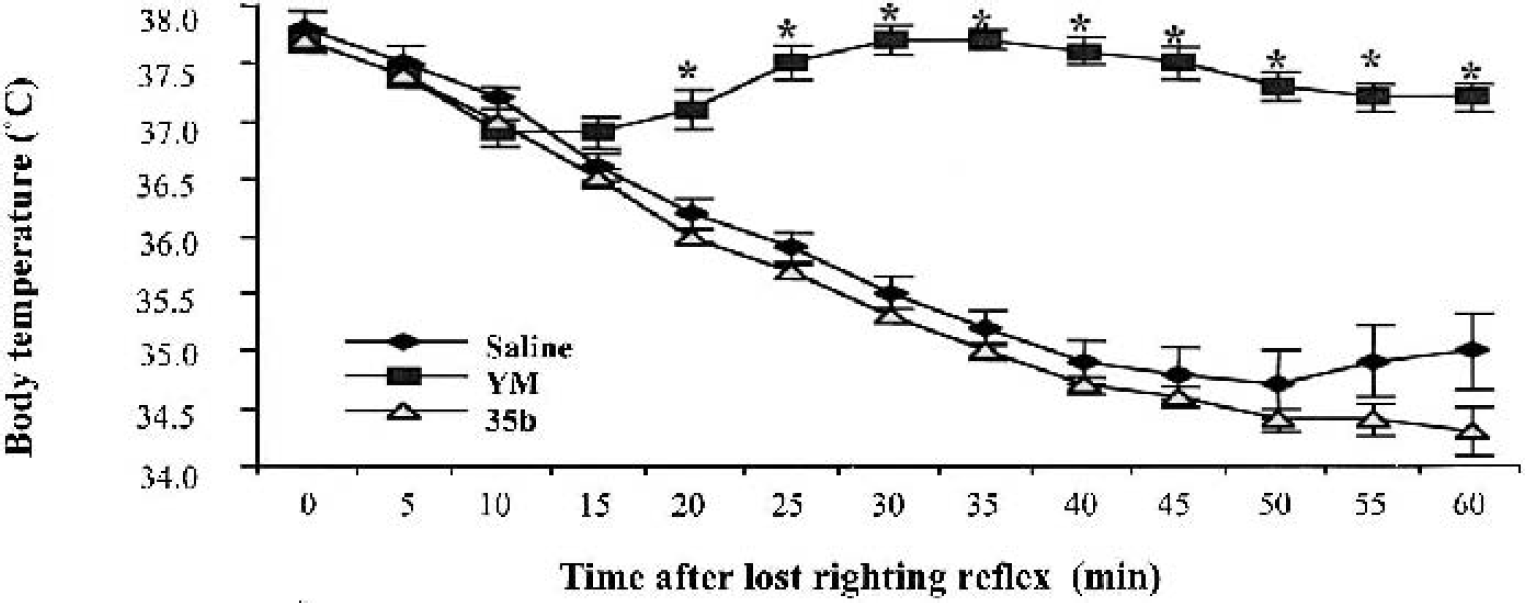
Effect of vehicle and thyrotropin-releasing hormone analogs (1 mg/kg; n = 6 for each group) on core body temperature in the lightly anesthetized rat. All treatment groups, with the exception of animals receiving YM-14673, exhibited a pronounced decrease in body temperature up to 60 minutes after loss of righting reflex. The YM-14673–treated group maintained normothermia for the duration of testing, with temperatures differing significantly from vehicle- or 35b-treated animals 20 to 60 minutes after loss of righting reflex (P < 0.01).
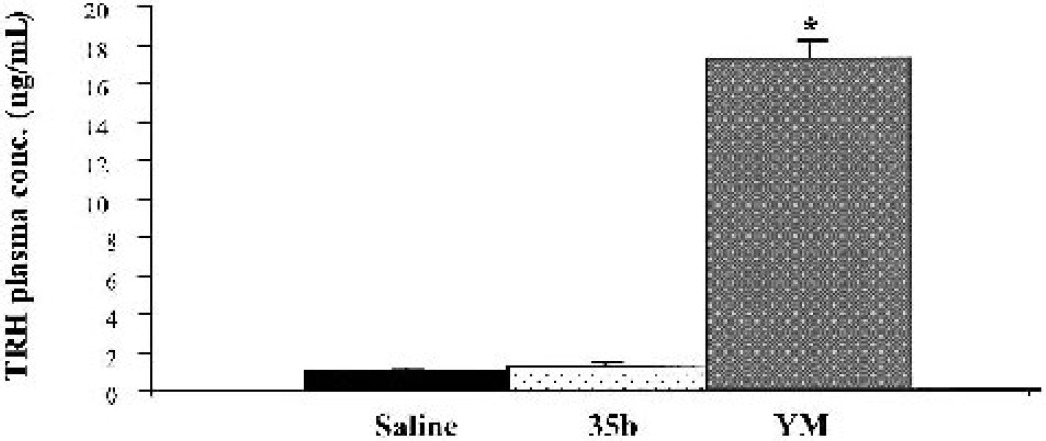
Graph of mean (± SEM) plasma thyrotropin-releasing hormone (TRH) levels 30 minutes after saline or drug (1 mg/kg) administration. Rats treated with YM-14673 exhibited significantly elevated levels of TRH when compared with saline controls. TRH levels in 35b-treated rats did not differ significantly from those of saline controls (*P < 0.0001 versus saline control).
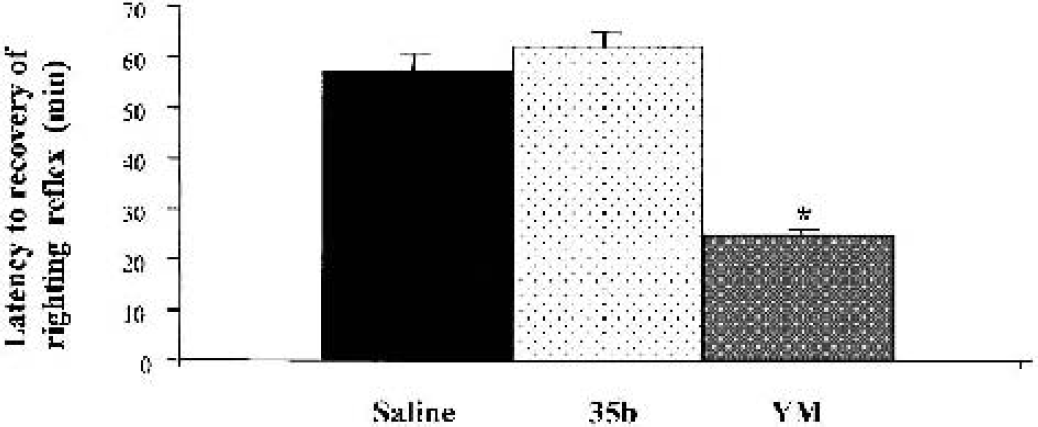
Effect of vehicle and thyrotropin-releasing hormone (TRH) analogs (1 mg/kg) on latency to recovery of righting reflex after light anesthesia. Rats treated with YM-14673 recovered this function significantly earlier than any of the other treatment groups (*P < 0.0001). Rats treated with 35b did not differ significantly from saline treated controls.
Magnetic resonance spectroscopy studies
After injury, there were no significant changes in pH or ATP concentration as determined from the magnetic resonance spectra (Fig. 12), which is consistent with previous results using this severity of injury (Vink et al., 1996). There was, however, a marked decline in brain free-magnesium concentration after injury in both groups. Before injury, brain free-magnesium in all animals (n = 10) was 0.47 ± 0.05 mmol/L. After injury, magnesium levels declined in both groups to reach a minimum value of 0.30 ± 0.04 (P < 0.05) by 2 hours after trauma. The free-magnesium concentration did not change significantly from this value for the remainder of the 4-hour magnetic resonance monitoring period. Similarly, cytosolic phosphorylation potential declined in both treatment groups from a preinjury value of 41 ± 5 mmol/L−1 (n = 10) to 21 ± 4 mmol/L−1 (P < 0.05) by 2 hours after trauma. There was no significant change from this value for the remainder of the magnetic resonance monitoring period. At no point was there a significant difference between the two treatment groups.
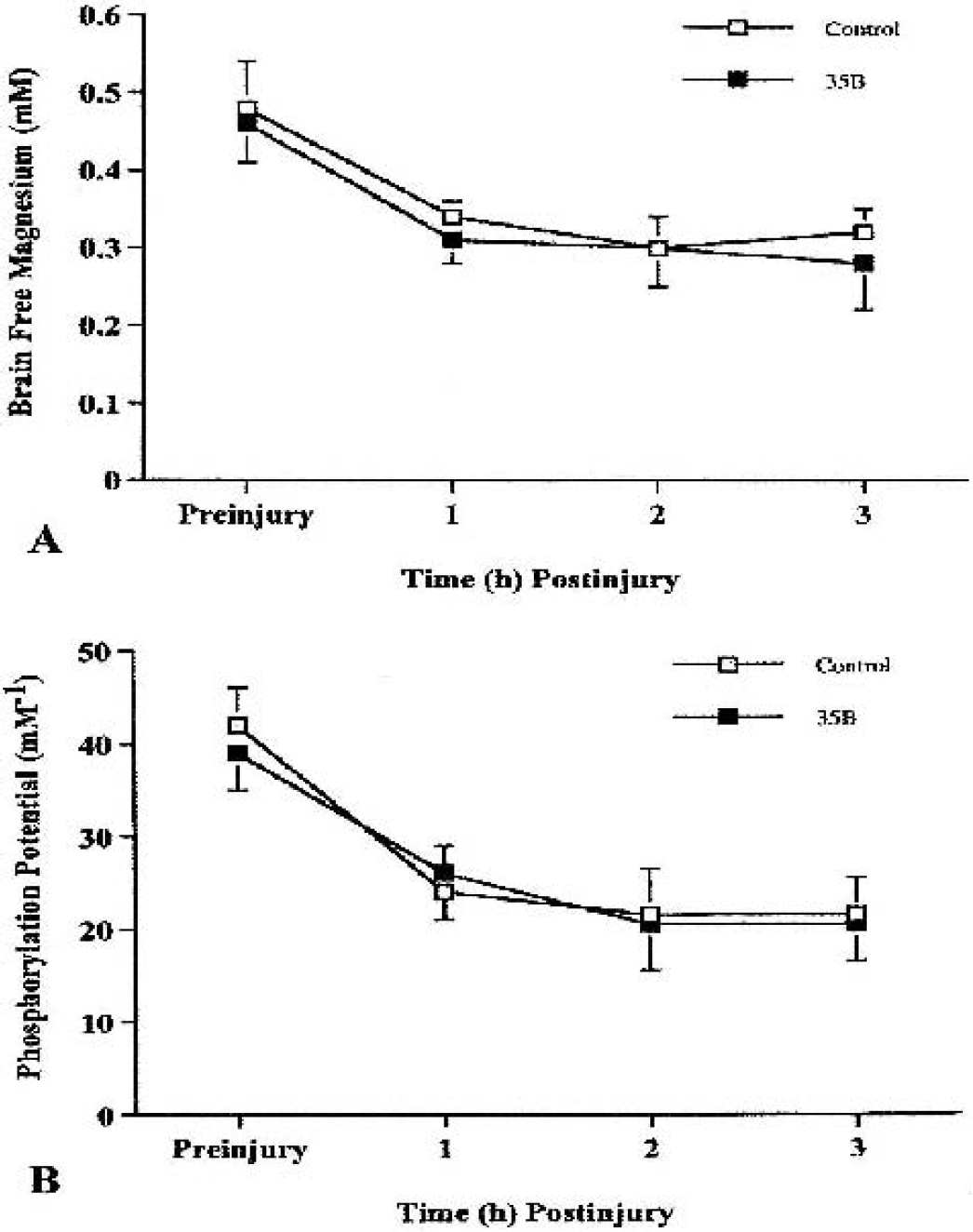
Effect of 35b on cerebral metabolism and free magnesium after moderately severe (2.6 to 2.7 atmospheres) traumatic brain injury (TBI). Change in cytosolic phosphorylation potential from before injury up to 3 hours immediately after TBI is shown in
In vitro studies
As shown in Fig. 13, 1 μmol/L 35b significantly attenuated cell death, as reflected by LDH release in three different models of neuronal cell death. Greatest neuroprotective actions (approximately 5%) were found with maitotoxin-induced neuronal death, a model of cellular necrosis (Zhao et al., 1999). Lactate dehydrogenase release was reduced by approximately 30% after mechanical trauma in a model that causes predominantly necrotic cell death (Mukhin et al., 1998). Approximately 30% protection was also found for treatment of staurosporine in cleaved cell death, a classical model of neuronal apoptosis (Koh et al., 1995).
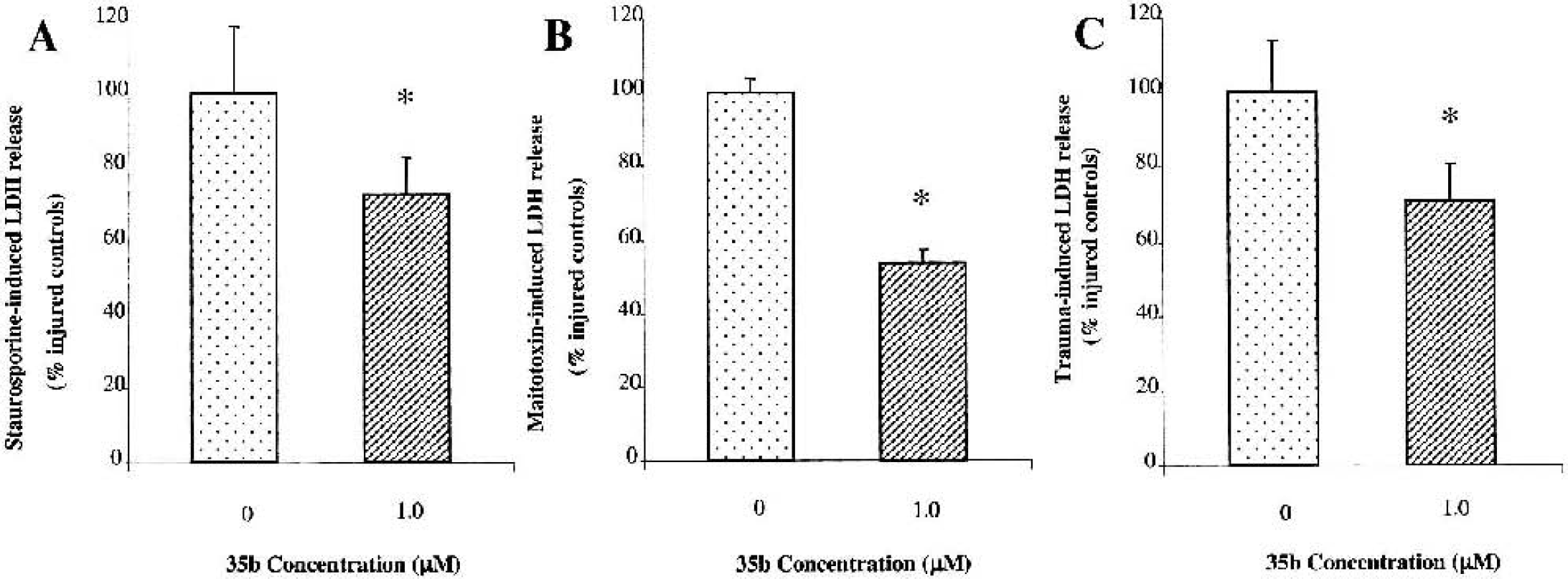
Effect of 35b on lactate dehydrogenase (LDH) release in three different in vitro models of cell death: apoptotic death induced by staurosporine
Receptor-binding profiles for 35b
35b was screened at a total of 50 receptors, channels, and transporters using the resources of the NIMH Psychoactive Drug Screening Program (Table 1) at a 10-μmol/L concentration. At only three cloned receptor preparations (M1, M3, and M4-muscarinic receptors) was significant inhibition measured, though the Ki values for 35b were still quite low (Table 1) and likely to be insignificant. These results indicate that 35b does not interact with high affinity at any of the tested receptors, channels, or transporters.
Receptor-binding profile of 35b
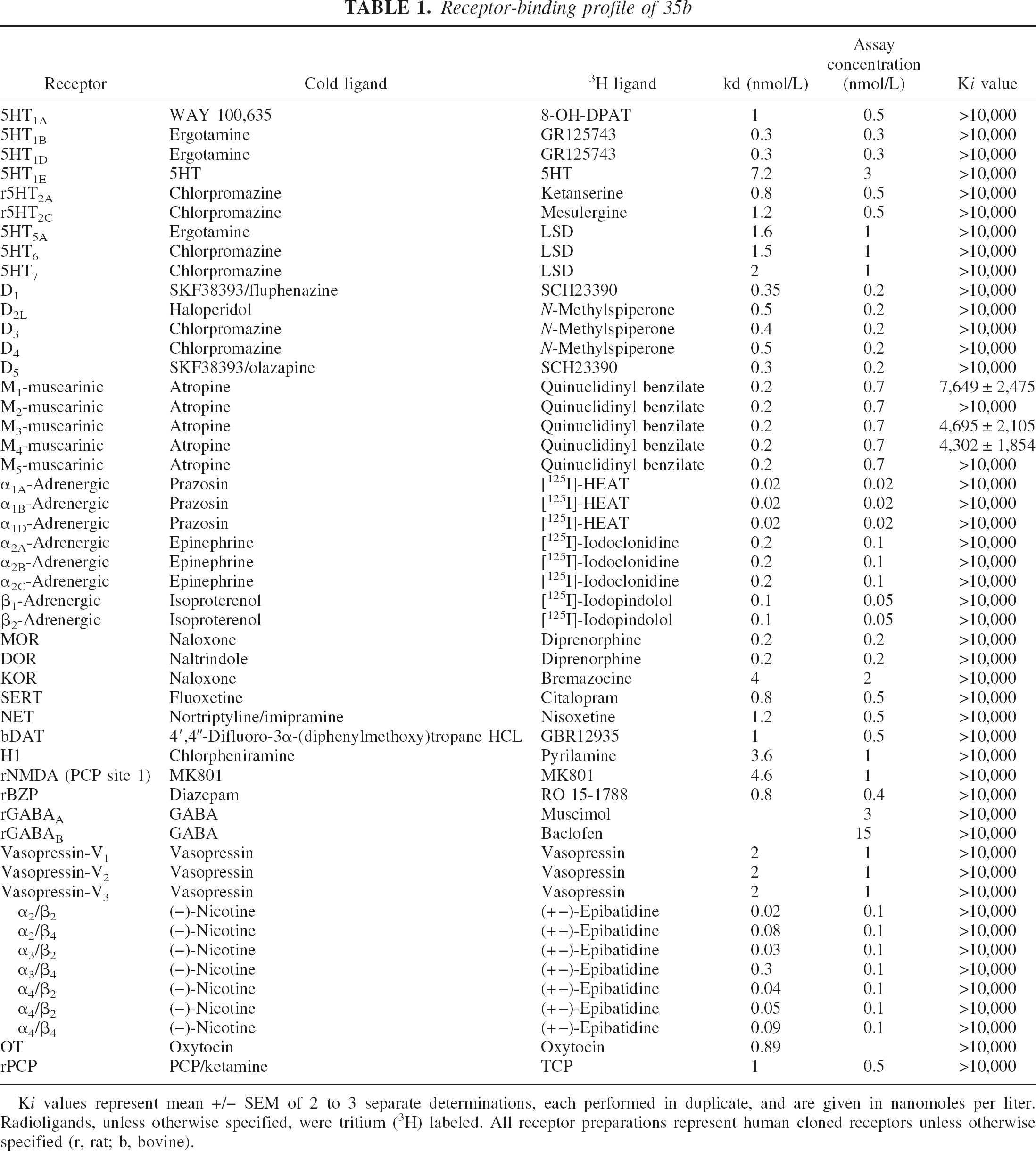
Ki values represent mean +/− SEM of 2 to 3 separate determinations, each performed in duplicate, and are given in nanomoles per liter. Radioligands, unless otherwise specified, were tritium (3H) labeled. All receptor preparations represent human cloned receptors unless otherwise specified (r, rat; b, bovine).
DISCUSSION
The present studies show that a novel diketopiperazine, derived from a modified TRH analog, exhibits substantial neuroprotective effects in vitro and in vivo. Fluid percussion–induced TBI models certain components of human head injury (McIntosh et al., 1987) and has been used extensively both to study pathobiologic mechanisms and to evaluate neuroprotective treatment strategies (Faden, 1996a). Treatment with 35b, administered systemically after the insult, significantly enhanced both motor and cognitive recovery in rats subjected to TBI. Using the Morris water maze as an assessment tool, 35b-treated rats showed reduced latencies to find the submerged platform, decreased path length, and greater swim time near the platform. These findings did not reflect differences in swim speed, which did not differ between groups. Treatment with 35b reduced lesion volumes, as shown by T2-weighted MRI, and decreased the number of TUNEL-positive neurons in ipsilateral hippocampus, which indicate the 35b is neuroprotective in vivo. Such neuroprotective effects were reproduced in vitro using a well-characterized trauma (punch) model in rat neuronal–glial cocultures (Mukhin et al., 1996, 1998).
Compound 35b is one of a class of diketopiperazines derived from modified TRH analogs. Thyrotropin-releasing hormone itself is metabolized to form CHP, a diketopiperazine that has been shown to have considerable physiologic activity (Metcalf, 1982). It has been reported to have thermoregulatory actions, anorectic effects, and analgesic activity; some of these properties may reflect opioid and dopaminergic mechanisms (Prasad, 1995). Although there are no reports that CHP has neuroprotective activity, it is not clear to what extent this issue has been directly addressed. Interestingly, CHP may increase cerebral blood flow (Koskinen, 1986; Prasad, 1995), and it may have antioxidant activity (Prasad, 1995; Shvachkin et al., 1989). This dipeptide, however, does not appear to have any nootropic activity and may even disrupt some forms of learning (Andrews and Sahgal, 1983, 1984; Prasad, 1995).
In our studies, 35b did not alter hypothermia associated with pentobarbital administration, nor did it show analeptic or endocrine actions exhibited by TRH or traditional TRH analogs. These observations are consistent with the finding that 35b does not bind to high-affinity TRH receptors (M. Gershengorn, unpublished observation, 2001) and indicates that this diketopiperazine may have substantial advantages over TRH or traditional TRH analogs as potential neuroprotective agents. For example, the absence of pressor effects may be an advantage in a patient with significant posttraumatic hemorrhage, and the absence of euthermic actions would permit associated hypothermia treatment. Moreover, the lack of analeptic actions would permit the use of sedative or anesthetic agents.
It is noteworthy that 35b administration significantly attenuated both necrosis-related and apoptosis-related neuronal cell death in vitro. It strongly antagonized the neurotoxic effects of maitotoxin, a marine toxin that causes a marked influx of calcium and has recently been used as a model of neuronal necrosis in vitro (Zhao et al., 1999). Significant neuroprotective effects for 35b were also found in our mechanical trauma culture model (Mukhin et al., 1998), which causes necrotic cell death that is largely mediated by glutamate receptors (Mukhin et al., 1997, 1998). In contrast, staurosporine has been commonly used to induce apoptotic cell death in cultured neurons (Koh et al., 1995). In our staurosporine-treated cultures, neurons showed morphologic features of apoptosis. Treatment with 35b significantly reduced such cell death. Both necrosis and apoptosis occur after TBI and appear to contribute to the subsequent neurologic dysfunction. Moreover, studies in vitro and in vivo suggest that attenuation of posttraumatic necrosis may exacerbate apoptosis (Pohl et al., 1999) and that blockade of both cell death pathways may have additive or synergistic neuroprotective effects (Allen et al., 1999). Therefore, the ability of 35b to attenuate each type of cell death may explain its striking neuroprotective activity.
Unlike TRH or traditional TRH analogs, 35b did not affect cellular bioenergetic state or magnesium homeostasis, and does not affect cerebral blood flow (L. O. Koskinen, unpublished observation, 2001). The lack of effect on blood flow may relate to its lack of pressor action in a subject that has lost its autoregulatory ability. That 35b did not modulate magnesium homeostasis or cellular bioenergetic after trauma was unexpected because these have been postulated to be major mechanisms by which TRH/TRH analogs exert their neuroprotective activity (Faden et al., 1990; McIntosh et al., 1988; Vink et al., 1988).
35b did not show appreciable receptor binding to a large number of other classical neurotransmitter or neuromodulator receptors, including glutamate, opioid, cholinergic, adrenergic, or serotonergic receptors (Table 1). Many of these receptors have been implicated in secondary injury after trauma. The ability, however, of TRH to antagonize the effects of many factors linked to secondary injury (e.g., endogenous opioids, platelet-activating factor, leukotrienes, and perhaps glutamate) may reflect so-called physiologic antagonism and has not been linked to specific receptor systems. Another possibility that should be addressed is whether these diketopiperazines modulate the regulation of genes or gene products associated with necrotic or apoptotic cell death.
In summary, we report the development of a novel cyclic dipeptide that has substantial neuroprotective actions in vitro and that enhances both motor and cognitive recovery in vivo. Neuroprotective mechanisms associated with this compound are likely to be multifactorial and may involve attenuation of both necrotic and apoptotic pathways of cell death.
Footnotes
Acknowledgments:
The authors thank Mike O'Reilly, Elena Kazanova, Alejandro Hidalgo, and Svetlana Ivanova for technical assistance.