
Abstract
In studies designed to evaluate the therapeutic window for treatment of traumatic brain injury, the caspase 3 inhibitor z-DEVD-fmk improved neurologic function and reduced lesion volumes when administered at 1 but not at 4, 8, or 24 hours after injury. Moreover, neither caspase 3 nor PARP, a caspase 3 substrate, were cleaved in injured, untreated cortex from 1 to 72 hours after injury. Few cortical neurons expressed active caspase 3 or were TUNEL positive from 6 to 24 hours after injury, and TUNEL staining was primarily Type I (necrotic). Nissl staining revealed extensive neuronal necrosis in the injured cortex from 6 to 24 hours after impact. Considered together, these data suggested that z-DEVD-fmk may reduce neuronal necrosis, so we used an in vitro model of necrotic cell death induced by maitotoxin to test this further and explore the potential mechanism(s) involved. Z-DEVD-fmk (1 nM-100 μM) significantly attenuated maitotoxin induced neuronal cell death and markedly reduced expression of the 145 kD calpain-mediated α-spectrin breakdown product after maitotoxin injury. Neither the 120 kD caspase-mediated α-spectrin cleavage product nor cathepsin B were expressed after maitotoxin injury. In a cell free assay, z-DEVD-fmk reduced hydrolysis of casein by purified calpain I. Finally, z-DEVD-fmk reduced expression of the 145 kD calpain-mediated α-spectrin cleavage fragment after traumatic brain injury in vivo. These data suggest that neuroprotection by z-DEVD-fmk may, in part, reflect inhibition of calpain-related necrotic cell death.
Traumatic brain injury (TBI) may cause either necrotic or apoptotic cell death. Apoptosis and necrosis have been classically distinguished by morphologic criteria (Clarke, 1990; Kerr et al., 1972). They have also been defined biochemically, with apoptosis often considered an active process that requires energy and protein synthesis and necrosis viewed as a passive process reflecting loss of ionic homeostasis secondary to energy depletion (Ankarcrona et al., 1995; Pang et al., 2003; Volbracht et al., 1999). However, hybrid forms of cell death showing mixed apoptotic/necrotic features have recently been identified, and some have coined the term aponecrosis to reflect a continuum between these phenotypes (Formigli et al., 2000; Kitanaka and Kuchino, 1999).
Studies suggest that both the type and temporal course of cell death after central nervous system (CNS) injury depends upon a variety of factors, including type and severity of the insult, as well as age, species, and sex of the subject (Kupina et al., 2003; Martin et al., 1998; Pohl et al., 1999; Prins and Hovda, 2003). Apoptotic neuronal death has been documented after head injury in humans, and in several of the most widely used models of TBI, including injury induced by lateral fluid-percussion (LFP) or controlled cortical impact (CCI) (Colicos et al., 1996; Conti et al., 1998; Fox et al., 1998; Newcomb et al., 1999; Rink et al., 1995). After LFP, DNA fragmentation (as detected by TUNEL staining) was observed in neurons with either “classic” apoptotic (Type II) or necrotic (Type I) features (Rink et al., 1995). Such neuronal apoptosis occurred both in the injured cortex, as well as in adjacent regions (thalamus and hippocampus), at later time points (Conti et al., 1998; Rink et al., 1995). After CCI, TUNEL-labeled neurons were Type II or Type I, and localized to the ipsilateral cortex (Fox et al., 1998; Newcomb et al., 1999), or to cortex and hippocampus, depending upon injury severity (Colicos et al., 1996; Fox et al., 1998; Newcomb et al., 1999).
Calpain and caspase 3 cysteine proteases have received considerable attention with regards to their potential role(s) in the pathophysiologic response to CNS injury. Both calpain and caspase 3 are activated after human head injury and after experimental TBI, where inhibitors of these proteases have also been shown to improve neurologic outcome and reduce tissue degradation (Beer et al., 2000; Buki et al., 1999, 2000; Clark et al., 1999, 2000; Harter et al., 2001; Kampfl et al., 1996, 1997; Keane et al., 2001; Knoblach et al., 2002; Kupina et al., 2001; McCracken et al., 1999; Posmantur et al., 1997; Saatman et al., 1996a,b; Yakovlev et al., 1997, 2001a; Zhao et al., 1998).
Active caspase 3 has been colocalized with positive TUNEL staining and DNA fragmentation in neuronal soma after CCI or LFP and is also increased within the axonal compartment in regions of diffuse injury associated with LFP (Beer et al., 2000; Buki et al., 2000; Clark et al., 2000; Knoblach et al., 2002; Yakovlev et al., 1997, 2001a). These studies, together with others showing activation of caspase-3 after cerebral hypoxia or ischemia, support a role for caspase-dependent apoptosis in acute neurodegeneration (Blomgren et al., 2001; Chen et al., 1998; Fink et al., 1998).
Z-DEVD-fmk is a tetrapeptide caspase inhibitor that is considered relatively selective for caspase-3 (Garcia-Calvo et al., 1998; Thornberry et al., 1997) and has been widely used in in vitro and in vivo models of acute injury to delineate roles for caspase 3 in neuronal cell death. Intracerebroventricular injections of z-DEVD-fmk improved function after LFP (Yakovlev et al., 1997). Intraparenchymal infusion of z-DEVD-fmk over several days after combined CCI and hypoxia reduced lesion size, although functional outcome was not significantly improved in this model (Clark et al., 2000).
Because we have previously described necrosis, apoptosis, and caspase 3 activation after CCI in the mouse (Clark et al., 1997; Fox et al., 1998; Newcomb et al., 1999), the present study evaluated the therapeutic window for z-DEVD-fmk treatment in this model. Z-DEVD-fmk was administered intracerebroventricularly, rather than intraparenchymally, to avoid the possibility of obtaining a local treatment effect and because in previous experiments intracerebroventricular injections improved behavioral outcomes (Knoblach et al., 2002; Yakovlev et al., 1997). Surprisingly, we observed substantial neuroprotective effects only after very early administraion (1 hour), at a time in which little apoptosis or caspase activation was present, and neuronal cell death was primarily necrotic. Additional in vivo and in vitro studies demonstrated that z-DEVD-fmk was a potent inhibitor of calpain and that improvement observed after treatment with z-DEVD-fmk may reflect, at least in part, this action.
MATERIALS AND METHODS
CCI injury and administration of z-DEVD-fmk
Male C57Bl/6 mice (20–25 g) (Taconic Farms, NY, U.S.A.) were maintained at constant temperature (22 ± 2°C) and a 12 hour light/dark cycle with food and water available ad libitum for 1 week before procedures. Details on the injury device, CCI injury model, and behavioral tests were previously reported (Fox et al., 1998). Briefly, animals were anesthetized with isoflurane in oxygen and placed on a heating pad to maintain core body temperature at 38 ± 0.2°C. A 4-mm craniotomy was performed on the central aspect of the left parietal bone, keeping the dura mater intact. The dura mater was moistened with sterile saline (37.5°C) and injury was induced (6.0 m/s velocity, 1 mm tissue deformation) with a pneumatic injury device. After injury, the incision was closed, anesthesia was discontinued, and mice were placed in a heated cage to maintain normothermia for 45 minutes. Animals were then monitored for 4 hours after surgery and daily thereafter. For treatment with z-DEVD-fmk or vehicle after CCI, mice were reanesthetized with isoflurane at various times after injury, placed in a stereotaxic apparatus, and the CCI wound was reopened for intracerebroventricular injection. Either Z-DEVD-fmk (160 ng in 2 μL DMSO) (ICN Biomedicals, Aurora, OH, U.S.A.), or DMSO vehicle (Sigma, St. Louis, MO, U.S.A.) was injected over a 5-minute period.
Live animal procedures were approved by the Georgetown University Animal Care and Use Committee and performed according to principles detailed in the Guide for the Care and Use of Laboratory Animals (DHEW publication NIH 85-23-2985).
Evaluation of motor and cognitive function
Recovery of motor function was evaluated using a beam walking task (Fox et al., 1998). To perform the task, mice traversed a narrow wooden beam (6 mm wide, 120 mm long) suspended 30 cm above a thick foam rubber pad. The number of footfaults (slips) made by the right hindlimb was recorded over 50 steps counted in either direction on the beam. A basal level of competence (<10 faults over 50 steps) was established before surgery. Cognitive function (spatial learning) was assessed using a Morris water maze paradigm. To perform the maze, mice were required to locate a resting platform hidden at a fixed location within a pool of opaque water, using extramaze visual cues. Mice experienced four trials per day, each 30 minutes apart, for 4 consecutive days. For each trial, mice were placed into the water at one of four randomly chosen locations, separated by 90°. The length of time to find the platform (latency) was recorded for each mouse. After 90 seconds, mice that failed to find the platform were assisted to it. Animals were allowed to remain on the platform for 15 seconds on the first trial and 10 seconds on subsequent trials. After completion of all trials, a black, clearly visible platform was placed into the pool at a new location. Mice were then reintroduced to the water tank, and the latency to locate the visible platform was recorded over three trials. This test was included as a control to verify the visuomotor status of injured mice. All behavioral data were expressed as means ± SEM, for each group.
Studies
For the study with behavioral/magnetic resonance imaging (MRI) outcomes, mice were subjected to CCI and then treated with z-DEVD-fmk either 1 hour (n = 19), 4 hours (n = 20), 8 hours (n = 16), or 24 hours (n = 16) later. Controls were treated with equivolume DMSO (n = 20) 1 hour after CCI. For the study to evaluate α-spectrin breakdown after z-DEVD-fmk treatment, animals were injured and then treated with either z-DEVD-fmk (n = 4) or vehicle (n = 3) 1 hour later as described above. Naive (n = 4) and sham-injured animals (n = 2) were also included. To assess expression of active caspase 3 subunits or PARP cleavage via immunoblot, untreated mice were killed at 1, 6, 15, 24, or 72 hours after injury (n = 5 for all groups except 72 hours, where n = 2), and their brains were removed and dissected on ice. Control cortical extracts were obtained from naive (n = 2) and sham-injured animals (n = 3). For the histologic studies, animals were subjected to CCI without treatment and then killed at 6, 12, 24, and 48 hours (n = 3/group) after injury. Sham-injured animals served as controls (n = 2). Study numbers were based on previous experience with each method and expected variability.
T2-weighted MRI to determine lesion volume
We have previously shown that the T2-weighted MRI signal days after TBI significantly correlates with histologic assessments of lesion volume taken from the same animals (Albensi et al., 2000; Faden et al., 2003). In addition, the protocol for T2-weighted imaging after TBI has been detailed (Albensi et al., 2000). Briefly, 21 days after TBI, animals were anesthetized with isoflurane (5% for induction, 1.5% for maintainence, in 70%/30% nitrous/oxygen) and placed in a 72 mm 1H birdcage resonator on a 37°C heating pad in a plexiglass MR probe. The probe was inserted into the bore of a horizontal 7T/21 cm Biospec-Avance magnet (Bruker, Germany) and positioned so that the animal's head was in the center of the magnet. A repiratory monitor was used for respiratory gating to reduce motion artifact. Field homogeneity across the brain was optimized and a sagittal scout image acquired (RARE image, FOV = 4 × 4 cm, 128 × 128 resolution, TR/TE = 1,500/10 ms with a rare factor of 8 making the effective TE = 40 ms). Eight contiguous multi-slice T2-weighted images were then acquired beginning from the end of the olfactory bulb and extending to the posterior base of the brain (FOV = 3 × 3 cm, slice thickness = 2 mm, 128 × 128 resolution, TR/TE = 1,500/20 ms, 4 echo images, and 2 averages). Lesion volume was estimated from the summation of areas of hyperintensity on each slice, multiplied by slice thickness, for the ipsilateral and contralateral hemispheres. Data are expressed as mean ± SEM (μL) lesion volume for each group.
Immunoblot Analyses
Cortical tissue samples were homogenized in RIPA buffer (60 mM Tris-HCl, pH 7.8, containing 150 mM NaCl, 5 mM EDTA, 10% glycerol, 2 mM Na3VO4, 25 mM NaF, 10 μg/mL leupeptin (Sigma), 10 μg/mL aprotinin (Sigma), 1 mM AEBSF (Sigma), 1 mM Pepstatin (Sigma), 0.1% SDS (Gibco BRL), 0.5% Deoxycholic Acid (Sigma) and 1% Triton X-100 (Calbiochem, La Jolla, CA, U.S.A.), or in extraction buffer K240-100 (BioVision, Mountainview, CA, U.S.A.). The supernatent was removed, aliquoted, and frozen at −80°C. Tissue culture samples were pooled (15 wells/group), and homogenized in Hepes buffer (10 mM Hepes/KOH, pH 7.4, 3 mM EDTA, 1% Chaps, 5 mM DTT, 10 μg/mL aprotinin, 1 mM AEBSF). The supernatant was removed, aliquoted and frozen at −80°C. Aliquots from either mouse cortex or tissue culture were thawed as needed, and the protein concentration in each determined with a Micro-BCA protein assay kit (Pierce), according to manufacturer's instructions. For caspase 3, poly-(ADP ribose) polymerase (PARP), and α-spectrin breakdown products, equal protein aliquots (50–100 mg) were resolved on 16% or 4% Tris-glycine gels in a 25 mM TRIS/192 mM Glycine/ 0.01% SDS, pH 8.3 running buffer. Proteins were transferred to Hybond-C Super nitrocellulose membranes (Amersham, Arlington Heights, IL, U.S.A.) in a 20% solution of methanol in running buffer, blocked for 16 to 24 hours in blocking buffer (5% BSA or 5% powdered milk /0.02% Tween in phosphate buffered saline [PBS]), and then probed with specific primary antibodies as follows: a) rabbit polyclonal antibody specific for the 17 and 19 kDa fragments of active caspase-3 (#9661) from Cell Signaling Technology (New England Biolabs, Beverly, MA, U.S.A.); b) rabbit polyclonal antibody specific for the 85 kDa caspase-mediated cleavage fragment of PARP (containing the carboxy-terminus) (sc-7150) from Santa Cruz Biotechnology; c) mouse monoclonal antibody to α-fodrin (αII-spectrin) from Affiniti Research Products Ltd. (Exeter, U.K.); or d) rabbit polyclonal antibody to cathepsin B (Upstate Cell Signaling Solutions, Lake Placid, NY, U.S.A.). Immune complexes were detected using horseradish peroxidase-linked secondary antibodies (Amersham), chemiluminescence reagent (Super Signal West-Dura, Pierce, Rockford, IL, U.S.A.), and Kodak Biomax MR-1 film (Sigma). Immunoblots were scanned with a GS-710 Imaging Densitometer (Bio-Rad, Hercules, CA, U.S.A.) and semiquantitative analysis was performed with Image Quant software. Data were expressed as the percentage of control (naive/sham-injury) relative density units.
Histologic procedures
Mice were anesthetized and transcardially perfused with saline and 4% paraformaldehyde. Their brains were removed, stored in fresh 4% paraformaldehyde overnight, protected in sucrose, frozen in O.C.T. media, sectioned (40 μm), and stored free-floating in 2% paraformaldehyde. Later, sections were placed in 4% paraformaldehyde for 5 minutes, rinsed in PBS and incubated with 10% goat serum/0.3% triton for 1 hour at room temperature. Primary antibody specific for the active subunit of caspase 3 (Cell Signaling, Beverly, MA) was diluted 1:100 in 2% goat serum/0.2% triton/PBS and applied overnight at 4°C. For cell-type specific double-labeling experiments, antineuronal nuclear protein (NeuN) (Chemicon, Temecula, CA, U.S.A.) was diluted 1:100 in solution with anti-caspase primary antibody. The next day, sections were rinsed twice in PBS and incubated with secondary antibodies (1:50) for 1 hour at room temperature. Goat anti-rabbit FITC (Sigma) and goat anti-mouse Texas Red (Accurate Chemicals, Westbury, NY, U.S.A.) were used as secondary antibodies. After incubation with secondary antibody, sections were rinsed twice in cold PBS, mounted on glass slides, coverslipped with antifade mounting medium, and visualized with an Olympus IX-70 laser confocal scanning microscope. Standardized parameters were used to allow valid comparisons between experimental groups. For negative controls, either primary or secondary antibody was omitted. In sections colabeled with TUNEL, Tdt reaction mix was applied after primary antibodies were rinsed off. Tdt reaction mix contained biotinylated dNTPs, Tdt buffer, and Tdt enzyme (Gibco, Gaithersburg, MD, U.S.A.) in ultrapure H2O. After 1 hour (37°C), slides were rinsed in 0.1 M EDTA, pH 8.0 for 2 to 5 minutes, PBS briefly, and then incubated with secondary antibody plus DN-Avidin-FITC (Vector Labs, Burlingame, CA, U.S.A.) for 1 hour at room temperature. Finally, sections were rinsed in PBS, coverslipped, and viewed as described previously in this report. For cresyl violet staining, sections were dehydrated in ethanol and chloroform, stained with cresyl violet (10 minutes), rinsed and cleared in ethanol and xylenes, coverslipped with Permount (Fisher Scientific, Hanover Park, IL, U.S.A.), and viewed with an Olympus BH-2 light microscope.
Neuronal glial cocultures and maitotoxin injury
The preparation of neuronal glial cocultures has previously been detailed (Mukhin et al., 1998). Briefly, cortices from 1- to 5-day-old Sprague-Dawley (Taconic, Germantown, NY, U.S.A.) rat were freed from the meninges and triturated in magnesium and calcium free Hank's Balanced Salt Solution supplemented with 1% 1 M HEPES (pH 7.0) and 1% 100 mM sodium pyruvate. Dissociated cells were seeded on 96-well cell + plates (Sarstedt, Newton, NC, U.S.A.) in minimal essential medium (MEM) with Earle's salts that contained 10% each of fetal bovine and equine serums, 25 mM HEPES (pH 7.2), 2 mM glutamine, 20 mM glucose, 10 ng/mL murine epidermal growth factor (EGF), and 1% antibiotic/antimycotic. At 9 days in vitro (DIV), cortices of day 17 to 19 gestation rat embryos were dissociated and seeded onto plates in media as described previously in this report, except that serums were reduced to 5%, and no EGF was added. Cultures were fed twice weekly thereafter until used for experiments on 18 to 21 DIV. Cytosine arabinoside (3 mM) was added at the first feeding, and bovine serum was deleted from all feeding media. After 11 DIV, all serum was excluded from the feeding medium. For maitotoxin injury, cultures were washed with MEM plus 10 mM HEPES (pH 7.2–7.4), 1 mM glutamine, and 20 mM glucose, and then preincubated with drugs for 15 minutes before addition of maitotoxin. Maitotoxin (Alexis Biochemicals, San Diego, CA, U.S.A.) was added to a final concentration of 0.1 nM. One hour later, cultures were washed, and fresh media, with or without drugs, was added as appropriate. At various times after injury, as described for each experiment, media was removed for lactate dehydrogenase (LDH) measurement using a CytoTox 96 cytotoxicity assay kit (Promega, Madison, WI, U.S.A.). To stain nuclei, Hoechst 33258 was added to a final concentration of 0.2 ng/μL, and nuclear morphology was visualized with a Nikon TE3000 inverted fluorescent microscope. Some cultures were washed and then incubated with 0.3 μM staurosporine for 16 hours to serve as positive controls for apoptotic morphology. Data were expressed as the percentage of LDH release in treated cultures compared to that induced by injury alone, with LDH from uninjured controls subtracted from all values. Bars represent the means ± SEM for n = 12 to 20 wells per condition.
Cell-free digestion assay
Lyophilized BODIPY TR-X conjugated casein was reconstituted in 100 mM NaHCO3, pH 8.3 to a 10 μg/mL stock solution (4°C). The stock was diluted 1:200 in Tris-HCL buffer, pH 7.8 that contained 1 U of purified calpain I with or without z-DEVD-fmk (0.01–100 μM). Triplicates of each condition were incubated in the dark for 1 hour at room temperature, and fluorescence was measured with a Cytofluor Series 4000 fluorometer (Perseptive Biosystems, Framingham, MA, U.S.A.) using ex/em of 590/645 nm. The assay was performed twice, and data from one assay were expressed as the percentage of calpain activity observed in the absence of z-DEVD-fmk.
Data analysis
Beam walking and Morris water maze data were analyzed by two-way ANOVA with time or group as factors, followed by post hoc Tukey's pairwise comparison of each time of treatment versus control. MRI data were analyzed by one-way ANOVA, followed by post hoc Tukey's pairwise comparison of each treatment time versus control. LDH and casein digestion data were analyzed by one-way ANOVA, followed by post hoc t-tests of each concentration versus control, with Bonferroni correction for multiple comparisons. Immunoblot data (%) were subjected to square root transformation and then analyzed by t-test with Bonferroni correction. Individual data points greater than 3 standard errors from the mean were considered outliers and not included for statistical analysis. For all statistics, P < 0.05 was considered statistically significant.
RESULTS
Early treatment with z-DEVD-fmk improved neurologic function and reduced lesion volume
Z-DEVD-fmk (160 ng) was injected intracerebroventricularly at either 1, 4, 8, or 24 hours after CCI. To assess motor recovery, mice were tested for the ability to traverse a narrow, suspended beam during recovery over a 21-day period (Fig. 1A). Mice treated 1 hour after CCI performed significantly better than did vehicle controls on days 7, 14, and 21 after injury. Mice treated 4 hours after CCI performed significantly better than controls only on day 21 after injury, but this was an isolated observation, as they did not show a trend toward better performance compared with other treatment groups on any other testing day. Groups treated at other times did not differ significantly from controls on any testing day. Cognitive recovery was assessed with a Morris water maze (MWM) test. Mice treated 1 hour after CCI performed significantly better than vehicle controls on the third and fourth days of the MWM test (days 16 and 17 after injury) (Fig. 1B). Groups treated at other times did not differ significantly from controls. At the end of the 21-day recovery period, mice were evaluated via T2-weighted MRI to determine the volume of the lesion in the hemisphere ipsilateral to impact. Treatment at 1 hour after CCI significantly reduced lesion volume, in comparision with controls (Figs. 2A and 2B). Groups treated at other times were not significantly different than controls.
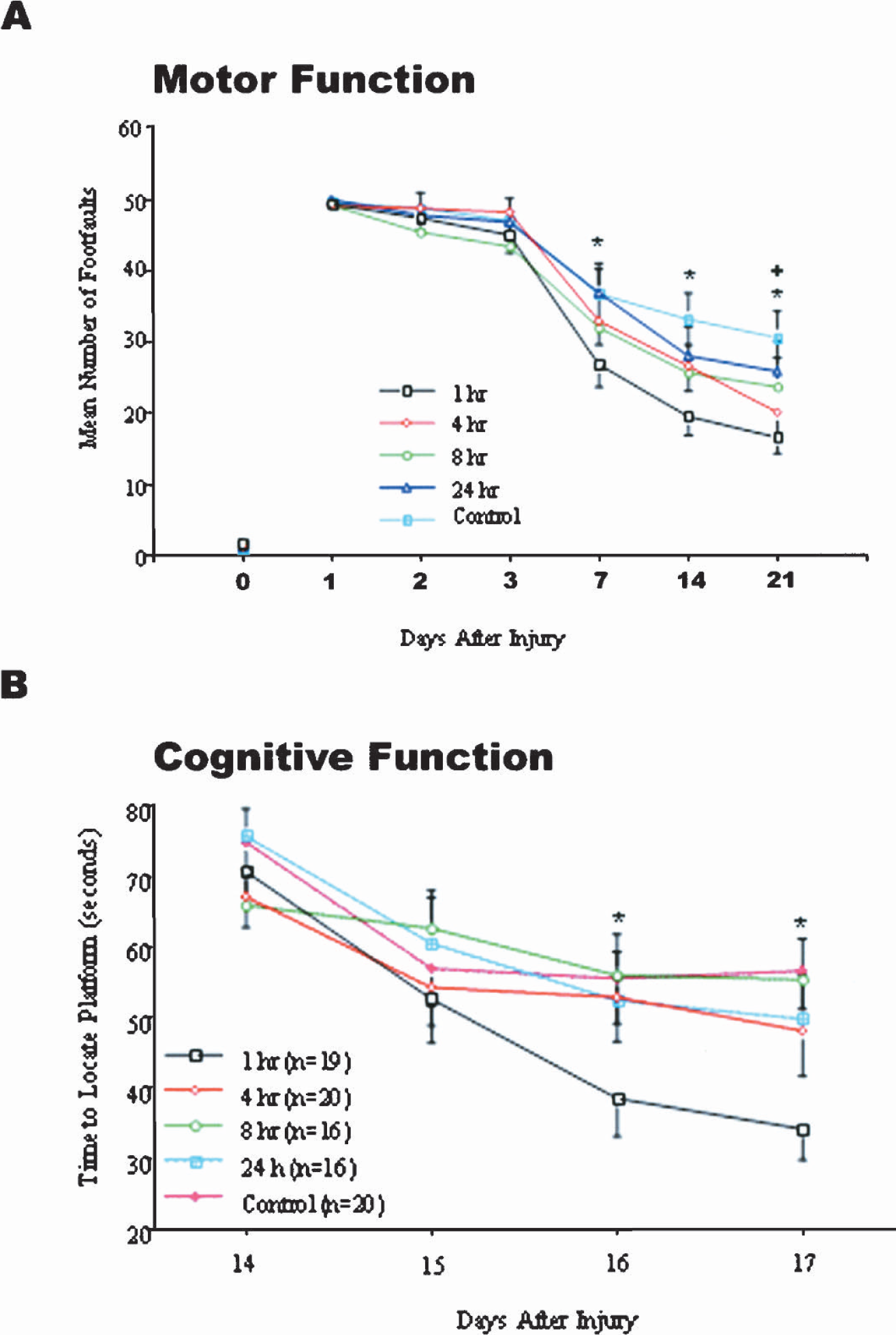
Early z-DEVD-fmk treatment improved motor and cognitive function after traumatic CNS injury induced by severe controlled cortical impact (CCI) in the mouse. Mice were subjected to CCI and then treated with z-DEVD-fmk (160 ng, intracerebroventricularly) at 1 hour (n = 19), 4 hours (n = 20), 8 hours (n = 16), or 24 hours (n = 16) after injury. Controls received equivolume DMSO vehicle at 1 hour (n = 20) after injury. (A) Motor function: footfaults (missteps) made by injured mice were counted as they traversed a narrow beam 1, 2, 3, 7, 14, or 21 days after injury as indicated on the x-axis. Dots represent the mean ± SEM number of footfaults for each group as shown in the legend. The 0 indicates footfaults made by all mice before injury. *P < 0.05 (at 7 and 14 days) or *P < 0.01 (at 21 days) for 1 hour treatment vs. control. +P < 0.05 for 4 hours treatment vs. control (at 21 days). (B) Cognitive function: animals experienced four training trials 30 minutes apart on a Morris water maze test on days 14 to 17 after injury as indicated on the x-axis. Dots represent the mean ± SEM time required to find a hidden platform (goal latency) averaged over four trials on each day. *P < 0.05 for 1 hour treatment vs. control. Data were analyzed by two-way ANOVA with time or group as Factors followed by post hoc Tukey's test.
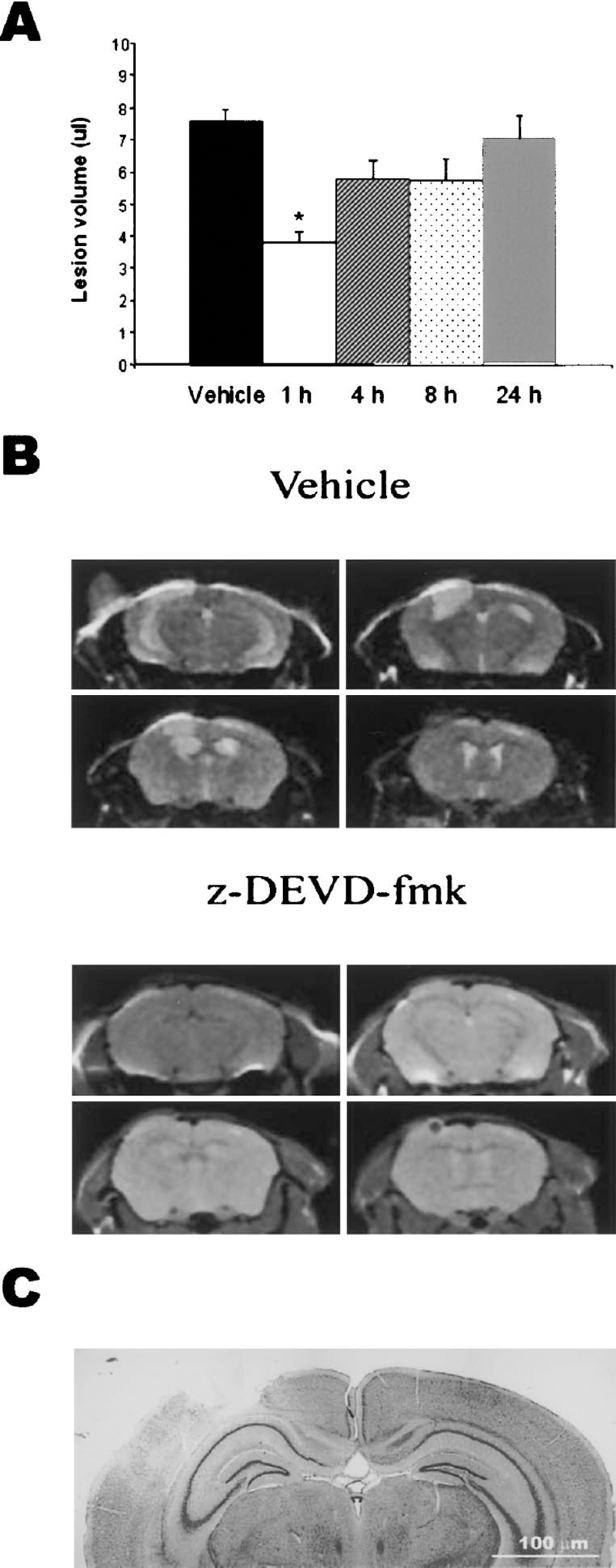
Early z-DEVD-fmk treatment reduced lesion volume after traumatic CNS injury induced by severe controlled cortical impact (CCI) in the mouse. (A) Lesion volume of the injured/ipsilateral cortex at 21 days after CCI as assessed by T2 weighted MRI. Bars represent the mean lesion volume for each z-DEVD-fmk treatment group, as shown on the x-axis. *P < 0.05 vs. control. Data were analyzed by 1-way ANOVA followed by post hoc Tukey's test. (B) Representative MRI scans from rats treated either with vehicle or z-DEVD-fmk, as indicated. (C) Photomicrograph of a cresyl violet stained section showing a representative lesion from an untreated mouse.
Limited caspase 3 activation in injured cortex and marked neurodegeneration
A separate group of animals was concurrently prepared to evaluate caspase-3 activity and apoptotic cell death after CCI. Neither cleaved caspase 3 subunits nor cleaved PARP fragments were detected in immunoblot assays of injured cortical homogenates from these animals at 1, 5, 12, 24, or 72 hours after CCI (Figs. 3A and 3B).
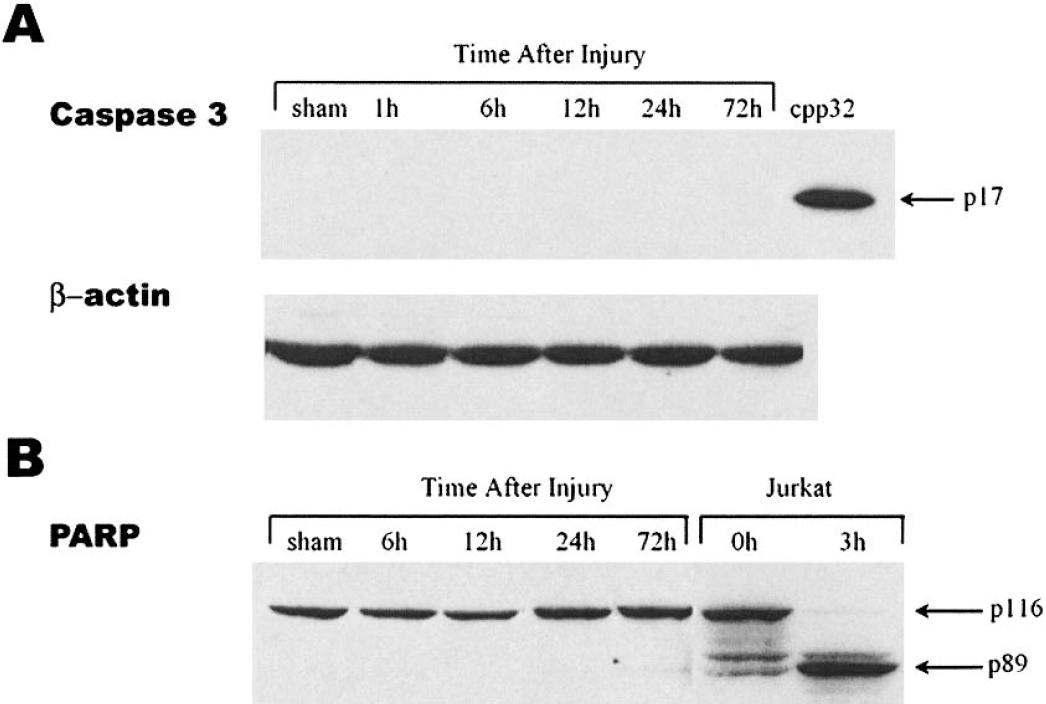
Cleavage of caspase 3 (A), or of poly-ADP-ribose-polymerase (PARP) was not detected by immunoblot analysis of the injured cortex at 1, 6, 12, 24, or 72 hours after CCI. Protein extracts from sham control or traumatized mouse cortex at indicated times after TBI were subjected to SDS-PAGE and immunoblot analysis. In (A) the positive control is from cleaved rat recombinant caspase 3. A representative β-actin control for procedures is shown directly below. In (B), the positive control for the 89 kDa fragment of cleaved PARP is from Fas-stimulated Jurkat cells. Representative blot is from n = 5 per group, except 72 hours, where n = 2.
Because immunoblotting procedures may lack the sensitivity to detect limited expression in selected cells, additional studies used cell-type specific double-labeling methods to qualitatively assess whether caspase 3 activation and DNA fragmentation via TUNEL labeling. A few cells in the injured cortex and hippocampus (dentate gyrus) expressed active caspase 3 at 6, 12, and 24 hours after injury (Figs. 4 and 5) (data for 24 hours not shown). Some of these cells were colabeled with anti-NeuN, indicating that they were neurons (Figs. 4 and 5). TUNEL labeling had a similar pattern, although TUNEL-positive cells were more frequently colabeled with NeuN than were anti-active caspase 3 positive cells (Fig. 6A). TUNEL labeling was predominately Type I (necrotic) (Rink et al., 1995), as very few TUNEL-positive cells showed any evidence of nuclear condensation or fragmentation. There was less NeuN staining in the injured cortex, compared with the contralateral side. Patchy clumps of reduced staining were evident as early as 6 hours after injury and had expanded to include most of the ipsilateral cortex by 12 hours after injury.
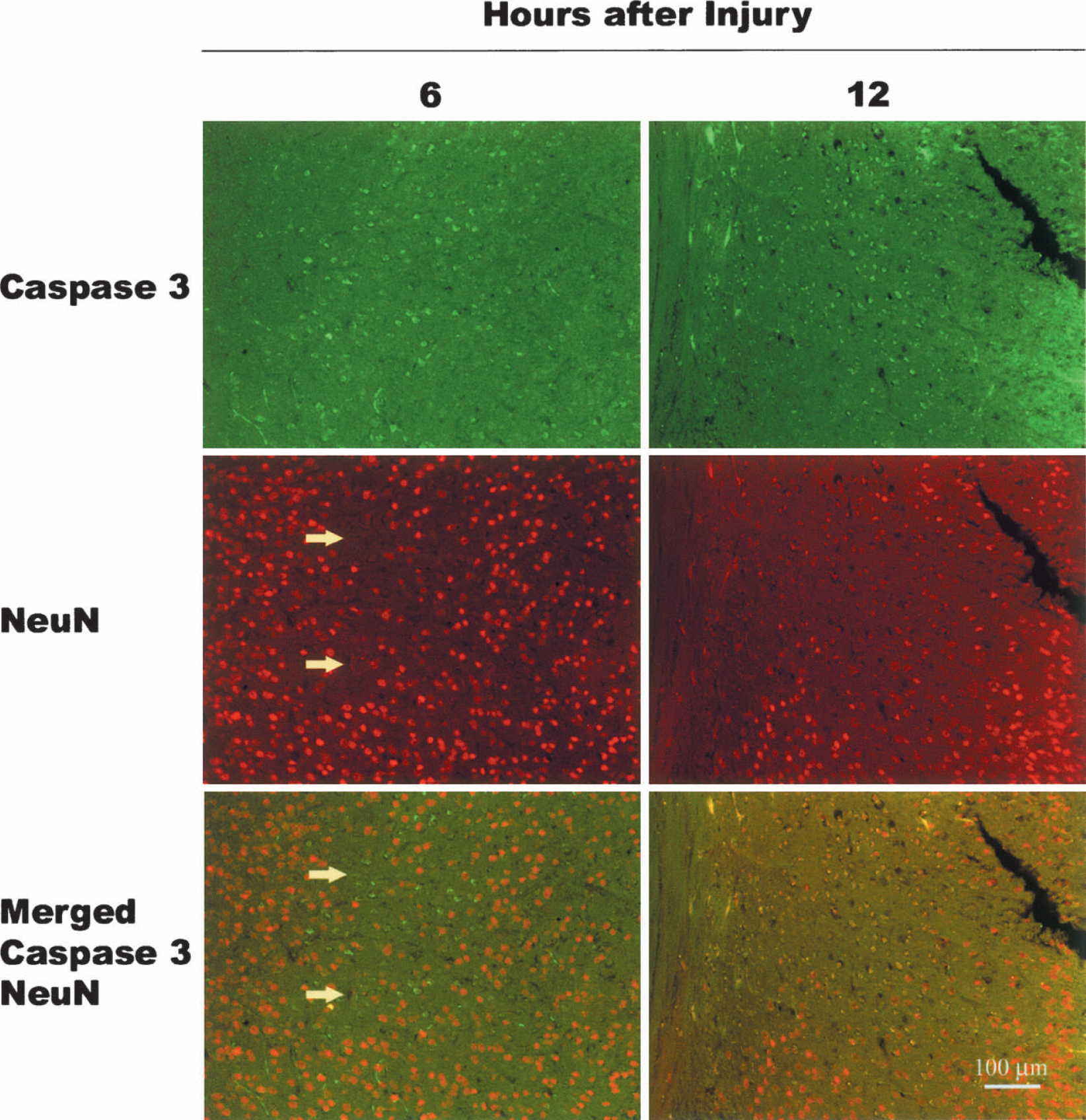
Active caspase 3 was not widely expressed in cortical neurons at 6 or 12 hours after CCI. Double-label immunocytochemical staining for anti-active caspase 3 (FITC/green), anti-neuronal nuclear protein (NeuN) (Texas Red/red), or merged images of both proteins are shown. Antiactive caspase 3 positive cells were thinly scattered throughout the injured cortex. Some of these were neuronal, as indicated by coexpression of NeuN. Anti–NeuN-positive cells were distinctly absent from some areas of injured cortex at 6 hours after injury (arrows on 6 hour images). By 12 hours after injury, these regions had expanded throughout the contused cortex. Images are representative of data obtained from n = 3 animals per timepoint.
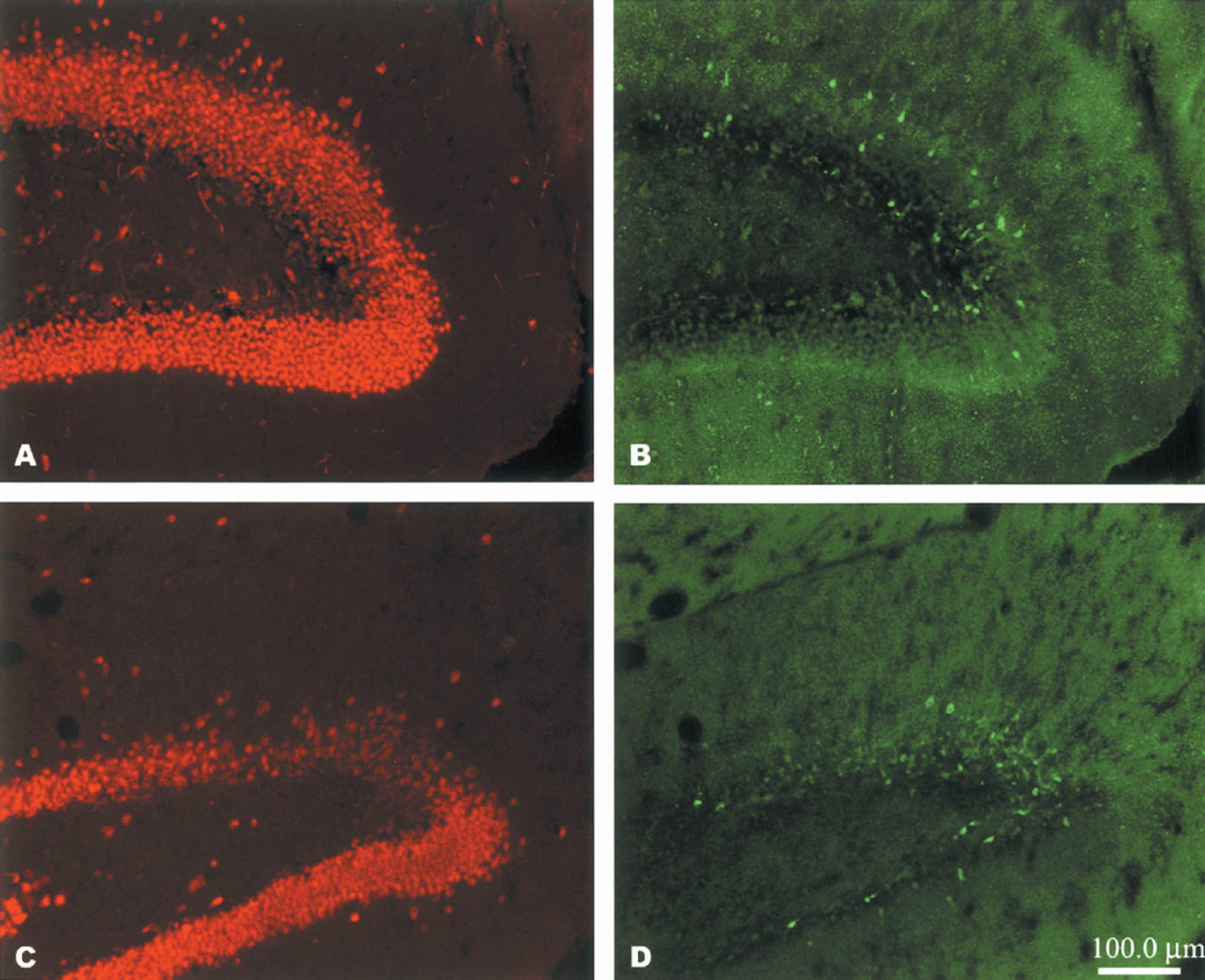
Active caspase 3 was expressed in the dentate gyrus of the hippocampus at 24 (A, B) and 48 (C, D) hours after CCI. Double-label immunocytochemical staining for anti-active caspase 3 (FITC/green) (B, D) or anti-neuronal nuclear protein (NeuN) (Texas Red/red) (A, C) are shown. Note positive staining for active caspase 3 in areas where NeuN staining is reduced. Images are representative of data obtained from n = 3 animals per timepoint.
To characterize degeneration that was indirectly evident as reduced staining with anti-NeuN, alternate sections were stained with cresyl violet and visualized by light microscopy. Using this method, marked neurodegeneration was observed thoughout the cortex 6 hours after CCI (Fig. 6B), as well as at 12 or 24 hours after injury. Most remaining cells were swollen with condensed nuclei (necrotic), but some cells with reduced cytoplasmic volumes and punctate or fragmented nuclei (apoptotic) were also present.
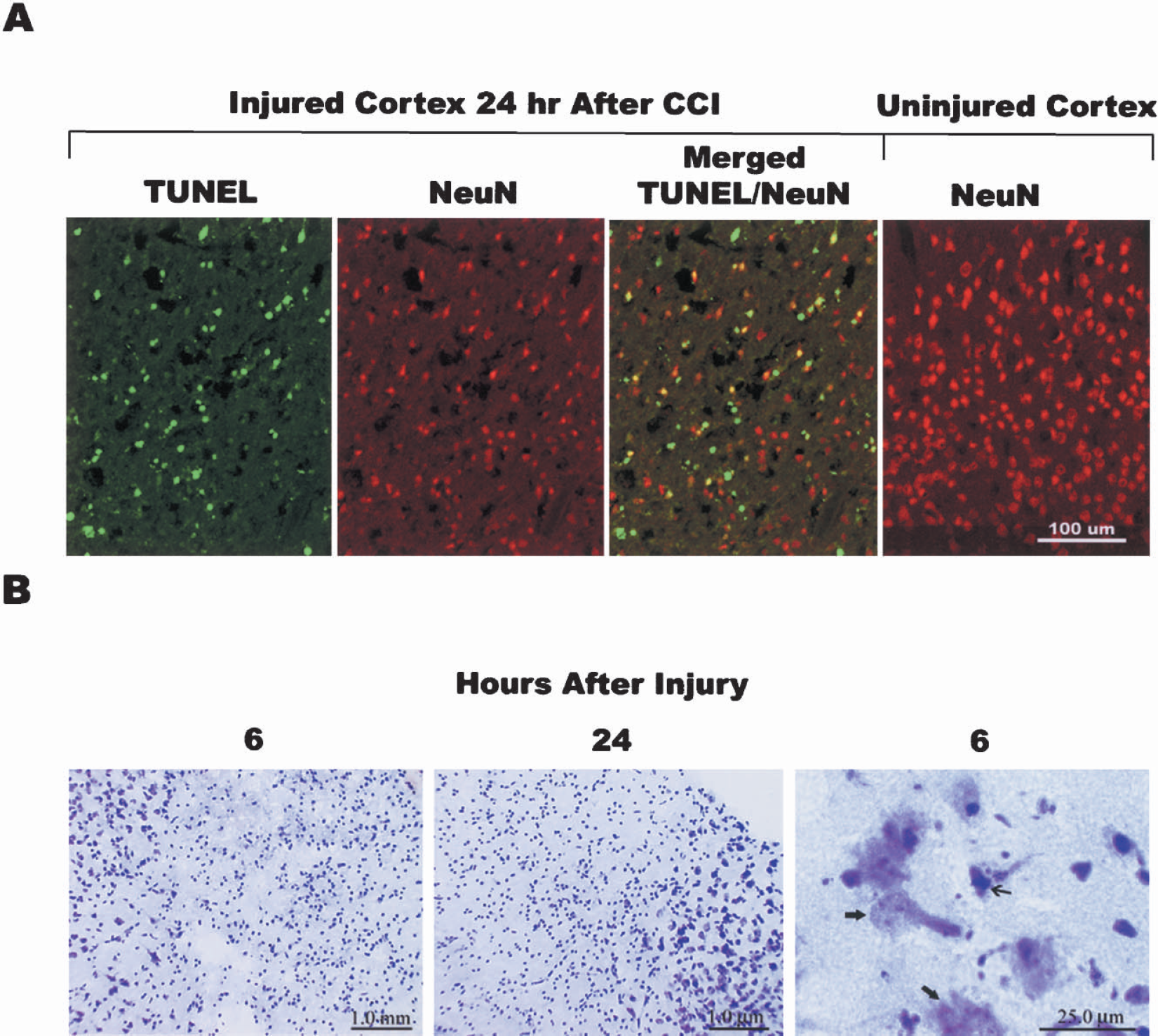
Few TUNEL labeled neurons were observed in the injured cortex, despite cell loss. (A) Colocalization of TUNEL labeling (FITC/green) and anti-NeuN staining (Texas Red/red) in injured mouse cortex 24 hours after CCI. Images shown represent the outer injured cortex or contralateral, uninjured side, as indicated. In injured cortex, approximately 30% of anti-NeuN positive cells were also TUNEL positive (merged frame). In most of these cells TUNEL staining was diffuse and cytoplasmic (Type I), rather than condensed or punctate/fragmented (Type II). There were many more anti-NeuN stained cells in the contralateral (uninjured) cortex than in injured cortex. (B) Nissl stain of injured mouse cortex at 6 and 24 hours after injury. At low magnification, patchy areas of extensive cell loss were visualized at 6 and 24 hours after injury. High magnification of these areas revealed degenerating neurons with necrotic (wide arrows) and apoptotic (narrow arrow) features.
Z-DEVD-fmk reduces cell death and inhibits calpain in a model of in vitro necrosis and a cell free assay
Maitotoxin evokes necrotic cell death in neurons and glia through calcium influx and activation of calpain (Gusovsky and Daly, 1990; Legrand and Bagnis, 1984). Nevertheless, z-DEVD-fmk (1 nM to 100 μM) was neuroprotective in this model, as it both reduced LDH release and attenuated morphologic evidence of neuronal cell death (Figs. 7A–7D). Staining with Hoechst 33258 confirmed that cells injured with maitotoxin had rounded nuclei and showed no indication of DNA fragmentation or apoptotic bodies (Figs. 7E and 7F). Calpain inhibitors E64D, ALLN, and calpastatin were used as positive controls and were neuroprotective in this model, as well (data not shown). Calpain and caspase cleave α-spectrin at unique sites, generating enzyme-specific α-spectrin breakdown products (SBDs) that indicate calpain or caspase 3 activation (Vanderklish and Bahr, 2000). Therefore, additional cultures were injured with maitotoxin in the presence or absence of z-DEVD-fmk, and LDH release, active caspase 3 fragments, and SBDs were assessed over time. Initial elevations in LDH release were observed 1 and 3 hours after maitotoxin injury and were not altered by z-DEVD-fmk (Fig. 8A). However, LDH release at 6, 9, and 12 hours after maitotoxin was substantially higher than that observed initially, and this delayed release was significantly reduced in the presence of z-DEVD-fmk. The calpain-mediated 145 kD SBD increased from 1 to 12 hours after maitotoxin injury but was abolished or reduced at all times in the presence of z-DEVD-fmk (Fig. 8B). In contrast, the caspase-mediated 120 kDa SBD was not increased after maitotoxin nor altered by z-DEVD-fmk treatment. In agreement with this observation, caspase 3 cleavage fragments could not be detected at any time after maitotoxin injury (Fig. 8C). However, analysis of PARP cleavage or of caspase 3 activation at the cellular level was not performed.
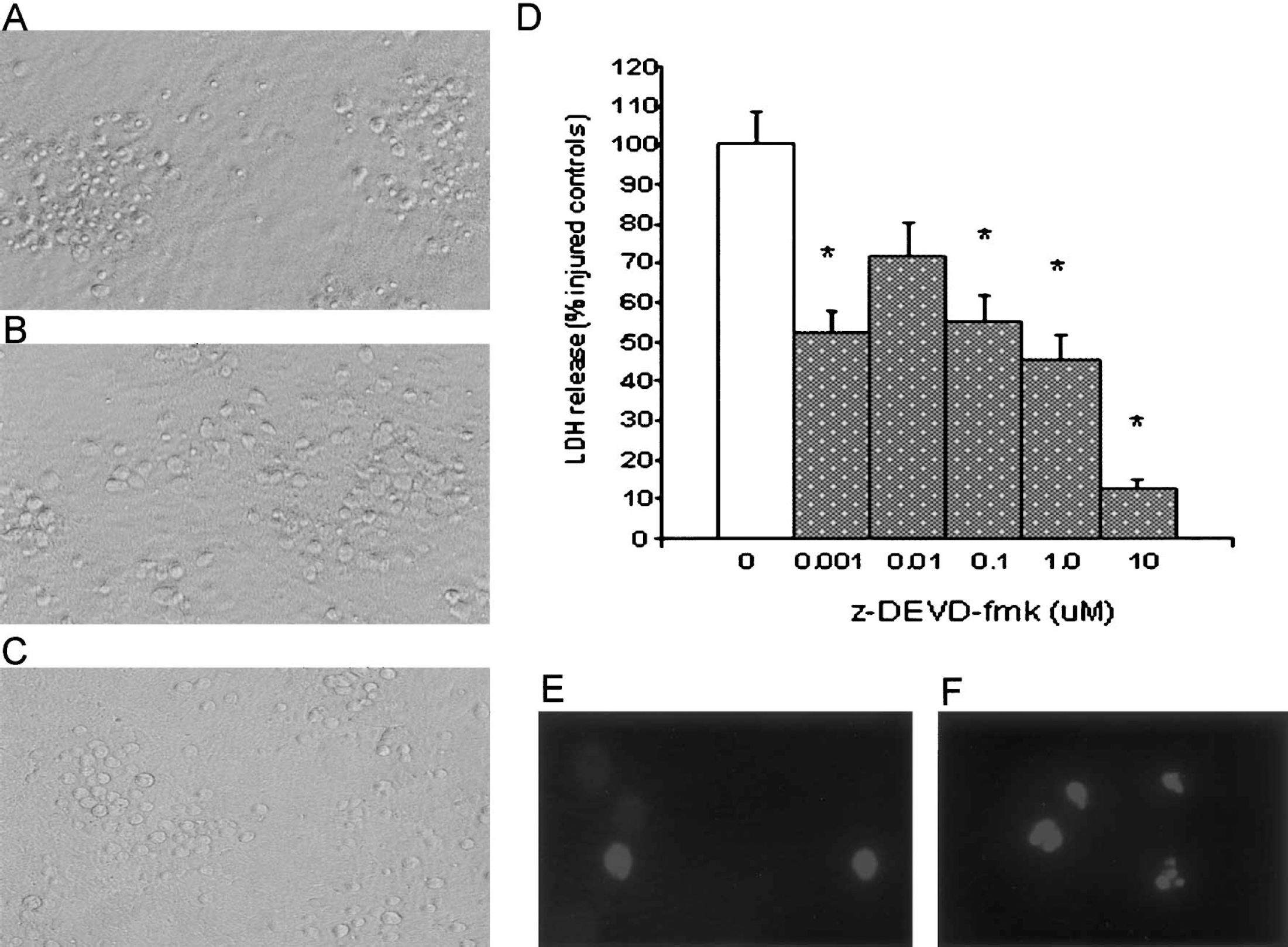
Z-DEVD-fmk prevented cell death in neuronal glial cocultures exposed to maitotoxin, an inducer of necrotic cell death. (A) Representative phase-contrast images of cocultures 16 hours after injury induced by incubation with 0.1 nM maitotoxin for 1 hour or (B) maitotoxin in the presence of z-DEVD-fmk (10 μM). (C) Control cultures received identical procedures but no drugs. (D) LDH release 14 to 16 hours after maitotoxin injury in the presence of increasing concentrations (as shown on x-axis) of z-DEVD-fmk. DMSO served as the vehicle control (0 μM). Bars represent the mean ± SEM (n = 12–15 wells per condition) from one experiment of two performed. *P < 0.01 vs. control by ANOVA and post hoc t-tests with Bonferroni correction.
Z-DEVD-fmk has recently been shown to inhibit cathepsin B (Schotte et al., 1999). Therefore, expression of the active form of this enzyme was evaluated, but it was not increased by maitotoxin-induced cell death, nor altered by z-DEVD-fmk (Fig. 8D).
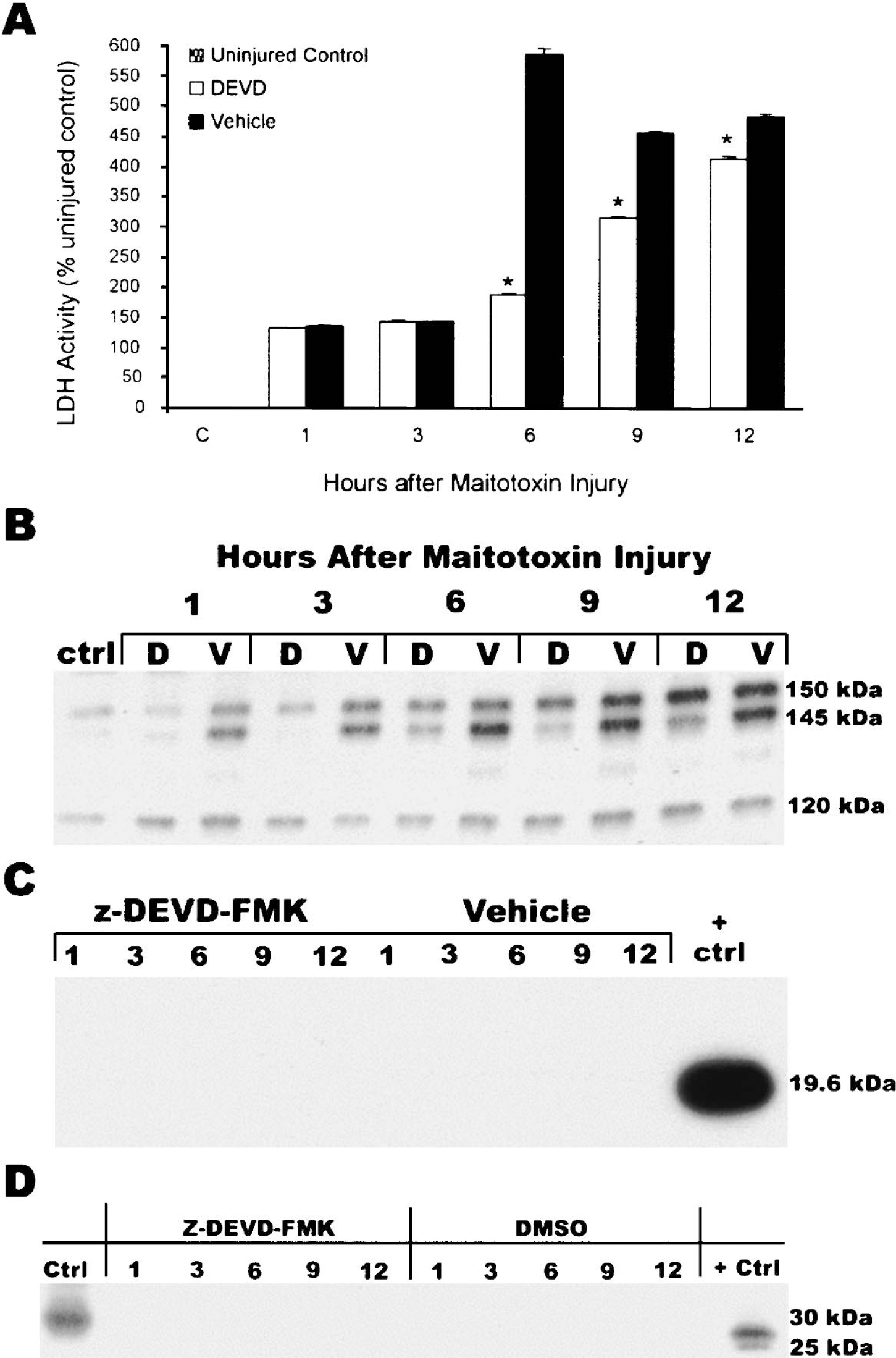
Z-DEVD-fmk reduced cell death and calpain-mediated proteolysis of α-spectrin in neuronal glial cocultures exposed to maitotoxin. (A) LDH release over time after maitotoxin injury. Media was sampled from different cultures over time as indicated on the x-axis. *P < 0.05 vs. vehicle treatment at same time by Student's t-test comparison. (B) Expression of α-spectrin breakdown products (SBDs) over time from cultures represented in A. Time course is indicated above the blot. D, z-DEVD-fmk; V, equivalent volume of DMSO vehicle, ctrl, uninjured control. The 150 kD SBD is produced by both calpain and caspase 3, the 145 kD SBD is specifically generated by calpain, and the 120 kD SBD is specifically generated by caspase 3. (C) No expression of active caspase 3 over time in cultures represented in A and B. Active recombinant caspase 3 served as a positive control. (D) No expression of cathepsin B over time in cultures represented in A, B, and C. Ctrl, uninjured cultures; +Ctrl, mouse spleen. Data shown in A–D represent one of four total experiments performed: two with 100 μM z-DEVD-fmk and two with 50 μM z-DEVD-fmk. Results from all experiments were identical.
A cell-free casein digestion assay was also used to assess inhibition of calpain by z-DEVD-fmk directly. In this experiment, calpain I activity was reduced more than 95% by 100 or 10 uM z-DEVD-fmk, and more than 50% by 1 uM z-DEVD-fmk (Fig. 9). Z-DEVD-fmk did not inhibit calpain at concentrations less than or equal to 0.01 uM.
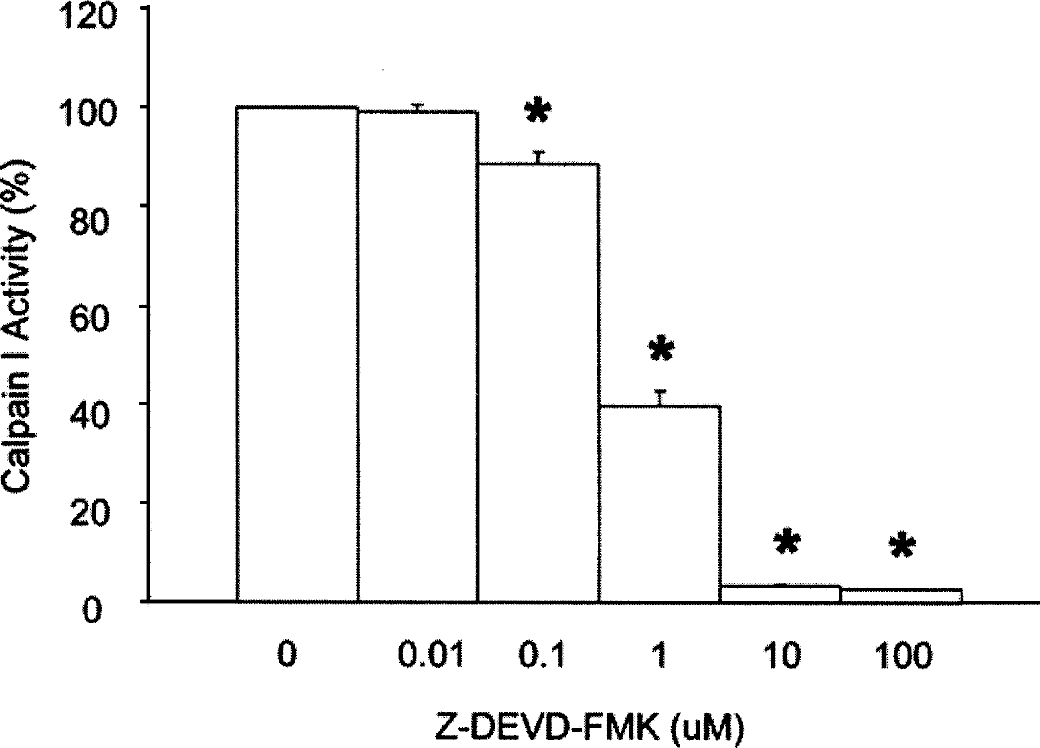
BODIPY-labeled casein assay directly shows inhibition of calpain I by z-DEVD-fmk. BODIPY-labeled fluorescent casein was incubated with 10 units of calpain I and increasing concentrations of z-DEVD-fmk as indicated on the x-axis. Bars represent the mean ± SEM from triplicates prepared at each concentration of z-DEVD-fmk. *P < 0.05 vs. 0 μM z-DEVD-fmk, by one-way ANOVA followed by t-tests with Bonferroni correction. Data are representative of one experiment from two repetitions.
Z-DEVD-fmk treatment inhibits calpain activity after TBI in vivo
To test whether z-DEVD-fmk may inhibit calpain activity after TBI in vivo, mice were injected with z-DEVD-fmk or vehicle 1 hour after CCI, as in the original experiment which determined the optimal therapeutic window for treatment with z-DEVD-fmk, and killed 1 hour later. Expression of SBD in the injured cortex was then analyzed by immunoblot. The 145 kDa SBD was substantially increased after injury in vehicle controls, but such expression was completely abolished in animals treated with z-DEVD-fmk (Figs. 10A and 10B). In contrast, injury had no effect upon expression of the caspase-mediated 120 kDa SBD band, indicating that no caspase activation was present at this time.
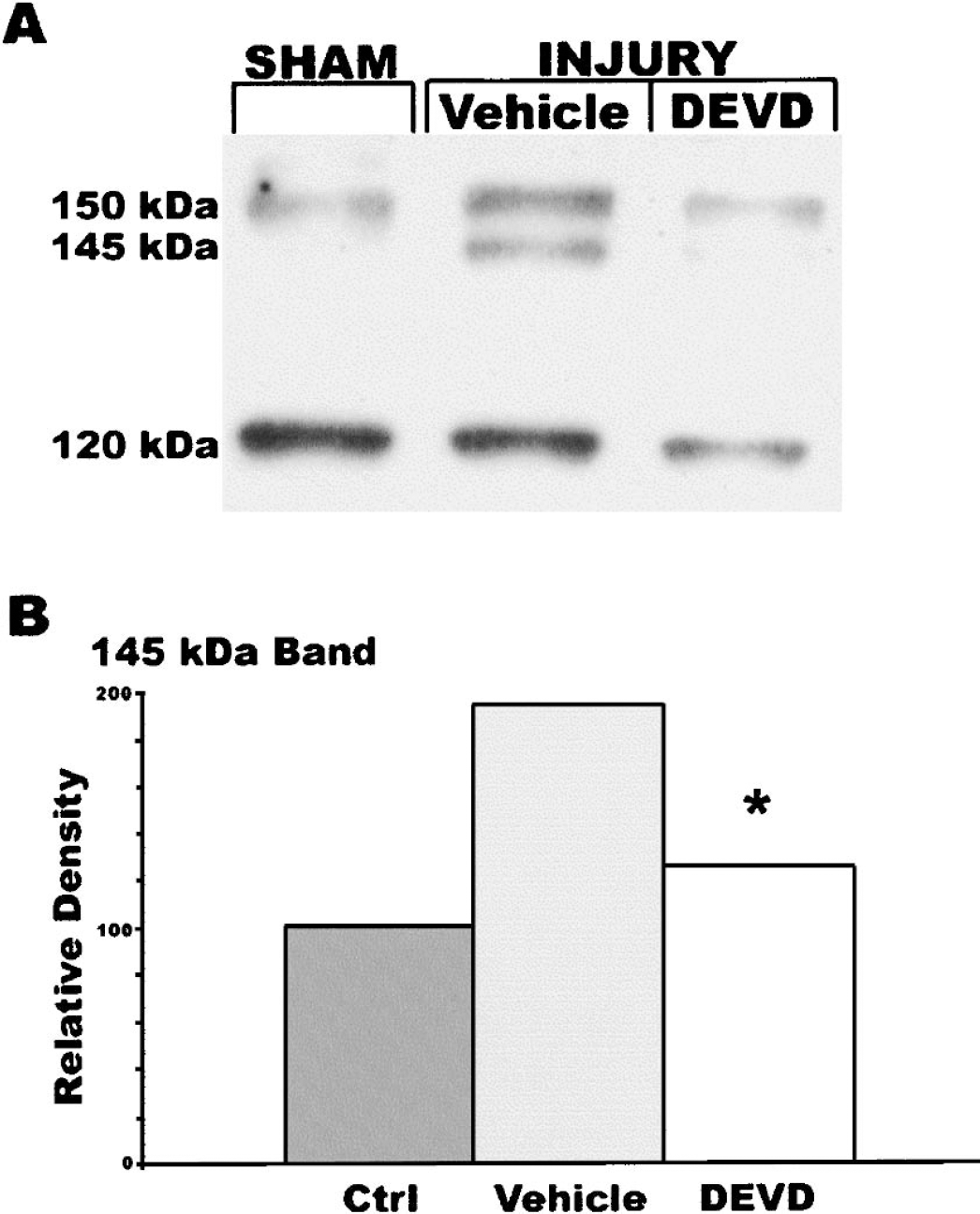
Z-DEVD-fmk treatment reduces accumulation of the 145 kD SBD 2 hours after CCI. Mice were subjected to CCI and treated with 160 ng z-DEVD-fmk 1 hour later as detailed in Fig. 1. Two hours after injury (1 hour after z-DEVD-fmk treatment), the injured cortex was removed and homogenized, and α-spectrin proteolysis was assessed by immunoblotting methods. (A) Representative blot. (B) Semiquantitative analysis of 145 kD band from n = 6 sham or naive mice, n = 4 injured, z-DEVD-fmk treated mice, or n = 3 injured, vehicle control mice. *P < 0.05 vs. vehicle. Bars represent the percent of control relative density units. Percent data were subjected to square root transformation and then analyzed by t-tests with Bonferroni correction.
DISCUSSION
The present studies were designed to evaluate the therapeutic window for z-DEVD-fmk treatment in a mouse model of CCI. We found that z-DEVD-fmk improved neurologic function and reduced tissue damage at an injury severity that showed predominantly necrotic neuronal cell death with minimal evidence of caspase 3 activation. The therapeutic window for treatment with z-DEVD-fmk in our model was less than 4 hours and thus most consistent with an antinecrotic mechanism. This conclusion was supported by both biochemical and histologic outcome measures: active caspase 3 subunits or caspase 3-mediated cleavage fragments of PARP were undetectable in immunoblots of cortical extracts taken from 1 to 72 hours after injury; TUNEL staining was primarily Type I (necrotic); active caspase 3 expression was limited in the injured cortex and adjacent hippocampus; and Nissl staining of the injured cortex revealed primarily necrotic morphology. Moreover, effective treatment with z-DEVD-fmk was associated with reduced calpain-mediated α-spectrin degradation. Z-DEVD-fmk was also neuroprotective, at concentrations lower than those routinely used to inhibit caspase 3, in an in vitro model of necrotic neuronal cell death induced by maitotoxin. After maitotoxin injury, as in the in vivo study, neuroprotection was associated with inhibition of calpain rather than caspase 3, which was not activated in this model. Finally, in a cell-free assay, z-DEVD-fmk directly inhibited calpain-mediated hydrolysis of casein at a concentration range that extended below 1 uM, and was similar to the concentration range that protected against maitotoxin injury.
Previous work by our group and others using the rat LFP model suggests that caspase activation and apoptotic cell death are important delayed events after TBI (Conti et al., 1998; Knoblach et al., 2002; Rink et al., 1995; Yakovlev et al., 1997, 2001a, b ). In addition, initial characterization of the impact device used in the present study indicated that delayed apoptosis (24 hours), primarily confined to the ipsilateral cortex, was a feature of mouse CCI (Fox et al., 1998). To date, however, caspase inhibition strategies have only been evaluated in preinjury or repetitive administration paradigms. Therefore, the goals of the present study were both to extend previous observations to a mouse model of CCI and to evaluate the therapeutic window for caspase inhibition. Although the results we obtained were unexpected, both in terms of injury severity and in response to z-DEVD-fmk treatment, they are consistent with the view that cell death mechanisms and phenotypes may vary with species, model, and injury severity. In general, the CCI model tends to have a larger necrotic component than does LFP, and the proportion of necrosis after injury in either model has been shown to increase with severity (Kochanek et al., 1995; Newcomb et al., 1999; Rink et al., 1995).
Calpains are activated by micromolar (calpain I) or millimolar (calpain II) elevations of calcium, whereas caspases are activated in response to specific signals from external (cell death receptors) or internal (mitochondria, endoplasmic reticulum) sources (for review see Chan and Mattson, 1999; Ray and Banik, 2003). Caspase 3 activation has been used as a confirmatory marker for ongoing apoptosis. In contrast, calpains are frequently associated with necrotic cell death, although they may also be involved in neuronal apoptosis (Pike et al., 1998). Calpain inhibitors have been shown to improve neurologic outcome and reduce neurodegeneration in several models of TBI, although such benefits did not always correlate precisely with calpain inhibition (Kupina et al., 2001; Saatman et al., 1996b, 2000). Caspase inhibition strategies may also be beneficial. Repeated injection of z-DEVD-fmk from just before injury until 24 hours later improved function after LFP (Yakovlev et al., 1997). Intracerebroventricular injection of the pan-caspase inhibitor z-VAD-fmk just after injury also improved functional recovery in this model, as well (Knoblach et al., 2002). Although these studies did not address the effect of treatment upon lesion volume, other work supports that use of caspase inhibitors may be neuroprotective. For example, intracerebroventricular pretreatment with z-VAD-fmk, or the caspase 1 selective inhibitor Ac-YVAD-cmk, reduced lesion volume after CCI (Fink et al., 1999). Intraparenchymal infusion of z-DEVD-fmk over several days after combined CCI + hypoxia reduced lesion size, although functional outcome was not improved (Clark et al., 2000). In contrast to these results, intracerebroventricular pretreatment with z-Boc-fmk, another pan-caspase inhibitor, was reported to show no effect upon lesion volume after CCI (Sullivan et al., 2002).
To circumvent complex issues of injury model, severity and mechanism(s) related to in vivo studies, z-DEVD-fmk was tested in an in vitro model of neuronal cell death caused by maitotoxin. Even mild injury induced by maitotoxin produces relatively pure necrotic cell death (Kutty et al., 1989; McGinnis et al., 2003; Wang et al., 1996). This is characterized by a rapid increase in cytosolic free calcium, opening of a cytolytic pore on the plasma membrane, ATP depletion, calpain activation, and release of cytochome c, but not activation of caspase 3 or generation of caspase 3-specific cleavage fragments of PARP or α-spectrin (Kutty et al., 1989; McGinnis et al., 2003; Wang et al., 1996). Thus, the demonstration that z-DEVD-fmk is neuroprotective in this model supports a role for it as an inhibitor of caspase-independent or necrotic cell death. In addition, the data showing that expression of calpain-mediated SBD is abolished or reduced by z-DEVD-fmk after maitotoxin injury strongly support inhibition of calpain as the mechanism of protection, particularly as there was no increase in caspase-mediated SBD (consistent with the model). Although the present studies focused on calpain as a potential mechanism, the data do not preclude additional effects of z-DEVD-fmk on other mechanisms or factors. For example, lysosomal proteases have historically been considered to be passively involved in necrotic cell death, and z-DEVD-fmk inhibits cathepsin B (Schotte et al., 1999). However, active cathepsin B was not detected after maitotoxin injury; therefore, it is unlikely that the neuroprotective effect of z-DEVD-fmk involves this protease.
A direct effect of z-DEVD-fmk on calpain was supported by data from the BODIPY-labeled casein digestion assay, where the activity of purified calpain I was inhibited 57% by 1 uM z-DEVD-fmk. In seperate experiments, we have confirmed that z-DEVD-fmk inhibits calpain I at a Ki of 1 uM (Knoblach et al., unpublished data; August, 2004). Notably, z-DEVD-fmk showed neuroprotection against maitotoxin at even lower concentrations, suggesting the possible existence of additional cell death factors that are activated by maitotoxin and inhibited by z-DEVD-fmk.
Despite their efficacy in improving outcomes in models of ischemia and CNS trauma, it has been known for some time that most presently available calpain inhibitors have poor cell permeability. Many also lack specificity and inhibit other cysteine proteases, such as cathepsins (B and L), as well as the proteosome (Inoue et al., 2003). Tetrapeptide-based caspase inhibitors like z-DEVD-fmk have generally been considered to be selective for caspase cysteine proteases, albeit only relatively selective for individual caspases (Garcia-Calvo et al., 1998; Thornberry et al., 1997). For this reason, they have been used extensively as “proof of principle” tools to implicate caspase activation or ongoing apoptotic cell death in physiologic or pathophysiologic processes. Recently, however, data have emerged that indicate that these drugs effectively inhibit other cysteine proteases of the papain superfamily. Specifically, inhibition of calpain(s) and cathepsins by z-VAD-fmk is supported by several studies from different groups, as is inhibition of cathepsin B by z-DEVD-fmk (Blomgren et al., 2001; Schotte et al., 1999; Vancompernolle et al., 1998; Waterhouse et al., 1998; Wolf et al., 1999). To our knowledge, the present data are the first to show inhibition of calpains by z-DEVD-fmk. It is important to point out that the concentration of z-DEVD-fmk that showed neuroprotection in the maitotoxin model and that inhibited calpain directly was much lower than that which is widely used to study caspase-mediated cell death in vitro.
Ultimately, it may be most effective clinically to develop broad-spectrum protease inhibition therapeutic strategies, rather than to use selective approaches, because both calpain and caspase may contribute critically to the injury response. In addition, these proteases participate in cross-talk pathways that are still not understood (Nakagawa and Yuan, 2000; Neumar et al., 2003). Further, inhibition of one cell death mechanism (or protease), in general, may cause cells to switch to another (Lemaire et al., 1998; Schwab et al., 2002). Finally, both calpain and caspase cleave and inactivate PARP, and this may influence noncaspase mediated apoptotic cell death induced by apoptosis inducing factor, which appears to be both activated by PARP and involved in cell death after TBI (McGinnis et al., 1999; Yu et al., 2002; Zhang et al., 2002) (Clark et al., 1997; Kampfl et al., 1997).
CONCLUSION
The present data show that treatment with z-DEVD-fmk improves behavioral recovery, reduces tissue damage and prevents accumulation of calpain-mediated α-spectrin breakdown products when administered not later than 1 hour after injury in a TBI model that primarily shows necrosis. Z-DEVD-fmk also reduces necrotic neuronal cell death in vitro, and such neuroprotection is associated with inhibition of calpain, but not caspase 3 or cathepsin B. In addition, z-DEVD-fmk reduces calpain-mediated hydrolysis of casein, which indicates that z-DEVD-fmk can directly inhibit calpain. This nonspecific property of z-DEVD-fmk may account, at least in part, for its neuroprotective actions.
Footnotes
Acknowledgment:
The authors thank Dr. Xiao Di and Ms. Sadia Aden for expert technical assistance.