
Abstract
Neurotrophins play a protective role during cerebral ischemia, and mice lacking both alleles for neurotrophin 4 (Nt4−/-) or deficient in a single allele for brain-derived neurotrophic factor (Bdnf−/-) have increased susceptibility to cerebral ischemia. This study directly compared the biologic activities of brain-derived neurotrophic factor (BDNF) and NT4 by replacing the Bdnf coding sequence with the Nt4 sequence (Bdnf+/nt4–ki). Mice expressing one Nt4 allele in place of Bdnf develop 61% bigger lesions after 1-hour middle cerebral artery occlusion compared with wild-type littermates. Physiologic parameters did not contribute to ischemia susceptibility. In conclusion, NT4 is less potent than BDNF in promoting brain survival after stroke.
Neurotrophins are a family of growth factors that are required for the survival and maintenance of neurons. Members include nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT3), and neurotrophin-4/5 (NT4). The diverse functions of neurotrophins are mediated by binding to high-affinity tyrosine kinase receptors and low-affinity p75NTR receptor with distinct specificities: nerve growth factor signals through the TrkA receptor, both BDNF and NT4 bind to the TrkB receptor, and NT3 interacts primarily with TrkC (Bothwell, 1995; Lewin and Barde, 1996).
Given their role as neuronal survival factors, much attention has focused on neurotrophin involvement in models of central nervous system injury, such as cerebral ischemia. Indeed, application of BDNF, NT-3, and NT4 have all decreased infarct volume in models of cerebral ischemia (Lindvall et al., 1992; Beck et al., 1994; Chan et al., 1996; Zhang et al., 1999). Although in these studies large amounts of neurotrophins were applied to circumscribed areas, we have addressed the role of endogenous NT4 and BDNF after cerebral ischemia: mice lacking both alleles for NT4 (Nt4−/- mice) or deficient in a single allele for BDNF (Bdnf+/- mice) develop significantly bigger cerebral infarcts than their wild-type littermates (Endres et al., 2000).
To directly compare the biologic activities of NT4 and BDNF during cerebral ischemia in vivo, we replaced the Bdnf coding sequence with the Nt4 sequence in mice (Bdnfnt4-ki). We compared lesion volume and neurologic outcome after middle cerebral artery occlusion in mice expressing NT4 instead of BDNF with wild-type littermate control mice.
MATERIALS AND METHODS
Knock-in mice
Bdnfnt4-ki mice were generated through gene targeting in embryonic stem (ES) cells and blastocyst injections of targeted ES cells as reported (Fan et al., 2000). A targeting vector was constructed that introduced in frame all NT4 coding sequences after the first ATG of the BDNF coding sequences at the fifth exon of the Bdnf gene. A positive/negative selection cassette (CMV-hygro-TK) flanked by two loxP sites was inserted directly behind the stop codons of the inserted NT4, and the targeting vector was transfected into J1 ES cells for homologous recombination. By transient transfection with pMC-Cre, a plasmid that expresses Cre-recombinase, the loxP-flanked selection cassette was deleted yielding a new Bdnf allele, Bdnfnt4-ki. Two independent ES clones carrying the Bdnfnt4-ki allele were originally used to produce transgenic mice as described (Fan et al., 2000). To best control the genetic background, we backcrossed Bdnfnt4-ki heterozygous with 129/SVJae wild-type mice, and only the littermate animals were used in experiments. Bdnf+/nt4-ki mice are healthy and fertile, although their body weight tends to be less than the littermate wild-type mice. Gross and microscopic anatomy of the brains revealed no overt abnormalities.
Model of cerebral ischemia
All animal experiments were performed in accordance with the National Institutes of Health guide (NIH publications N0. 80–23) and institutional guidelines. Mice (18 to 20 g) were first anesthetized by halothane anesthesia (1.5% for induction and 0.8% for maintenance) in 70% nitrous oxide (N2O) and 30% oxygen (O2) with a face mask. Left focal cerebral ischemia for 1 hour followed by reperfusion was produced with a siliconecoated monofilament as described (Endres et al., 1997, 2000). In all animals, regional cerebral blood flow was measured by means of a flexible probe and laser-Doppler monitoring (Perimed, Järfälla, Stockholm, Sweden).
Physiologic parameters
Recording of physiologic parameters (mean arterial, blood pressure, blood gases, temperature) was essentially performed as previously described (Endres et al., 1997; 2000).
Determination of infarct size
Infarct areas were quantified on 20-μm hematoxylin/eosin—stained cryostat sections with an image analysis system (M4; Imaging Research, St. Catherines, Ontario, Canada) as described (Endres et al., 1997).
Determination of neurologic deficits
Mice were tested for neurologic deficits and scored from 0 to 3 as described by Bederson et al. (1986) with minor modifications as described (Endres et al., 1997).
Statistical analysis
All experiments were performed in a blinded fashion. Values are presented as mean ± standard deviation. Statistical comparisons were performed by unpaired Student's t-test (infarcts), analysis of variance followed by Tukey test (physiology) or Mann-Whitney rank sum test (neurologic deficits); P values smaller than 0.05 were considered statistically significant.
RESULTS
Bdnf+/nt4-ki mice and wild-type littermate control mice were subjected to 1-hour filamentous occlusion of the left middle cerebral artery followed by reperfusion for 23 hours. Infarcts were measured on hematoxylin and eosin—stained cryostat sections. Bdnf+/nt4-ki mice had significantly bigger infarcts (direct lesion size) than wild-type littermate control mice (83.3 ± 29.0 mm3 versus 54.4 ± 15.6 mm3, P < 0.005). When infarction volume was corrected for brain swelling (i.e., calculated by the indirect method), infarcts in the Bdnf+/nt4-ki mice were significantly increased by 61% (Fig. 1A). Significantly bigger lesions were evident in two of the five standardized coronal brain sections (Fig. 1B).
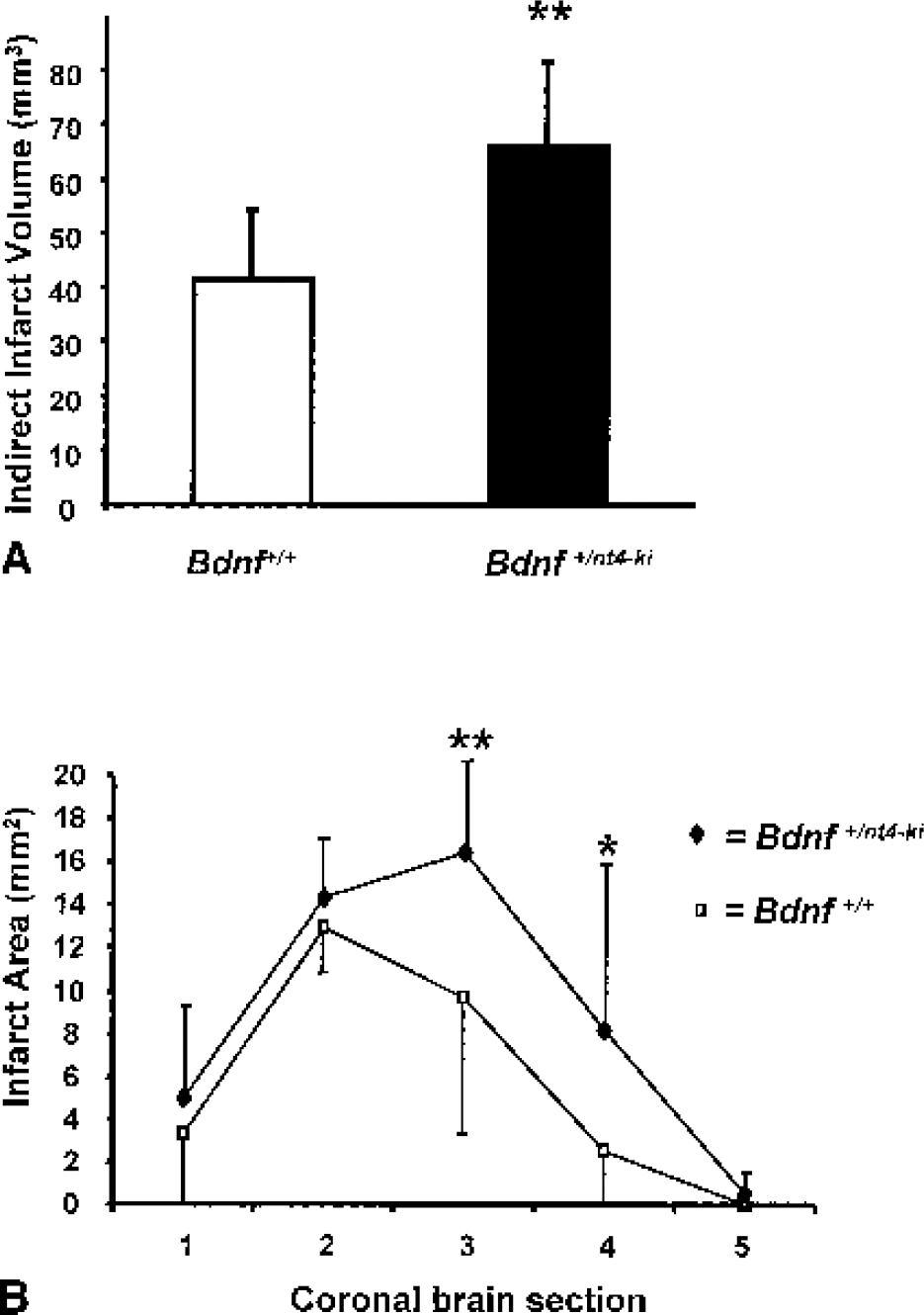
Effects of 1-hour middle cerebral artery occlusion and 23 hours of reperfusion in littermate Bdnf+/+ and Bdnf+/nt4-ki mice.
All animals exhibited a neurologic deficit score of 2 or higher (i.e., moderate or severe deficit) 30 minutes after onset of cerebral ischemia. At 24 hours, the deficits tended to be higher in the Bdnf+/nt4-ki mice, but the differences between groups did not reach statistical significance (Table 1).
Neurologic sensory—motor deficit score
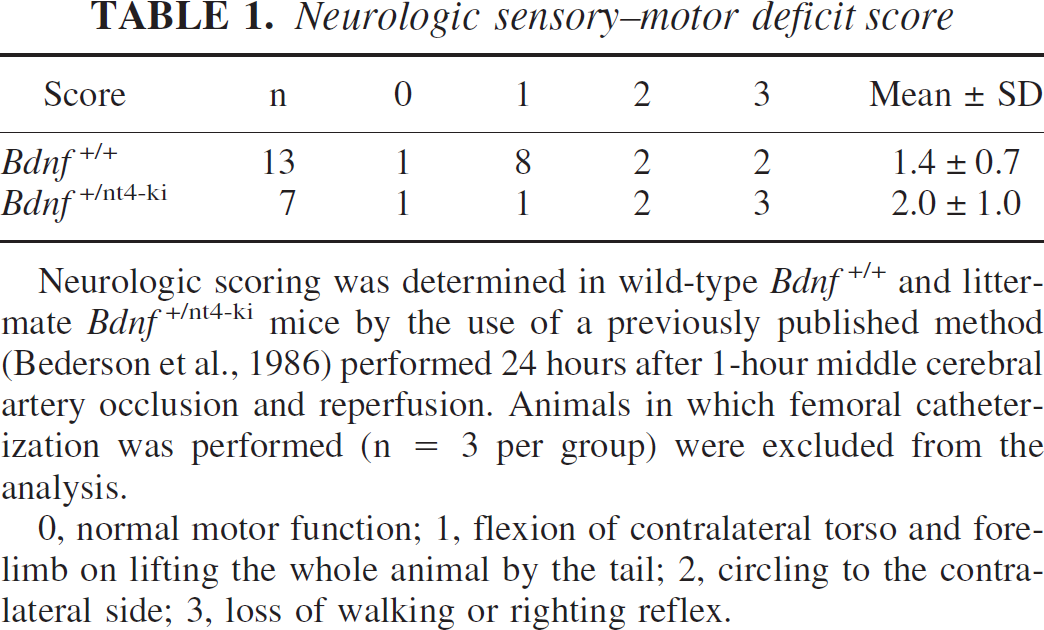
Neurologic scoring was determined in wild-type Bdnf+/+ and littermate Bdnf+/nt4-ki mice by the use of a previously published method (Bederson et al., 1986) performed 24 hours after 1-hour middle cerebral artery occlusion and reperfusion. Animals in which femoral catheterization was performed (n = 3 per group) were excluded from the analysis.
0, normal motor function; 1, flexion of contralateral torso and forelimb on lifting the whole animal by the tail; 2, circling to the contralateral side; 3, loss of walking or righting reflex.
We carefully monitored physiologic parameters in randomly selected animals before, during, and after brain ischemia. As indicated in Table 2, we did not observe any differences arterial blood pressure or blood gases (i.e., pH, PO2, PCO2). Moreover, there was an equivalent drop in regional cerebral blood flow to less than 20% of baseline at filament insertion and an equivalent rise in regional cerebral blood flow at filament withdrawal.
Physiologic parameters in littermate Bdnf+/+ and Bdnf+nt4-ki mice
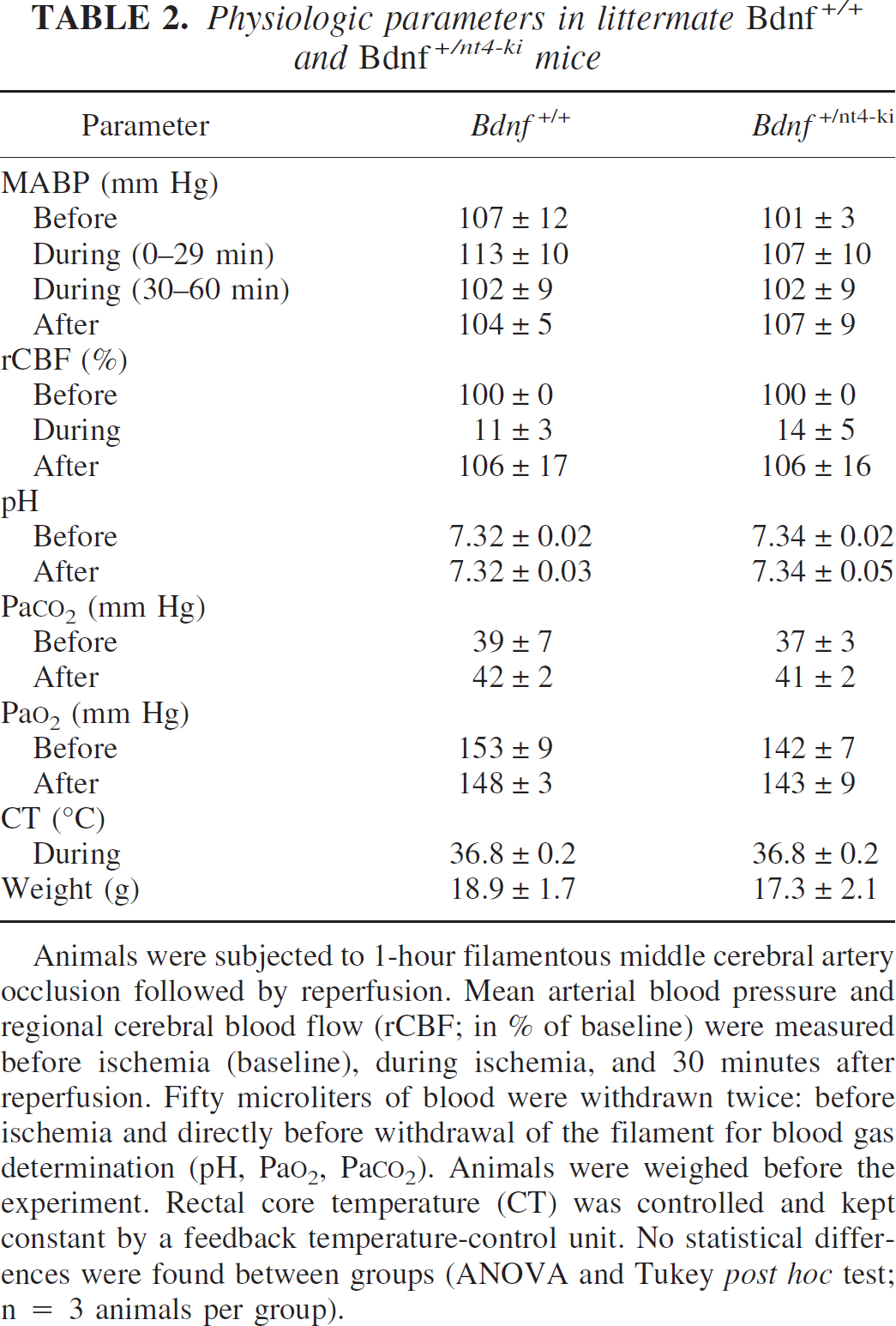
Animals were subjected to 1-hour filamentous middle cerebral artery occlusion followed by reperfusion. Mean arterial blood pressure and regional cerebral blood flow (rCBF; in % of baseline) were measured before ischemia (baseline), during ischemia, and 30 minutes after reperfusion. Fifty microliters of blood were withdrawn twice: before ischemia and directly before withdrawal of the filament for blood gas determination (pH, PaO2, PaCO2). Animals were weighed before the experiment. Rectal core temperature (CT) was controlled and kept constant by a feedback temperature-control unit. No statistical differences were found between groups (ANOVA and Tukey post hoc test; n = 3 animals per group).
DISCUSSION
This study provides evidence that NT4 is less potent than BDNF in protecting the brain from focal ischemic brain insults. We evaluated stroke outcome in transgenic animals in which one allele of Bdnf was replaced by a copy of the Nt4 gene (Bdnf+/nt4-ki). Bdnf+/nt4-ki mice had increased infarct volumes, indicating that the knock-in of NT4 cannot compensate for the loss of one Bdnf allele. Bigger infarcts were accompanied by a trend to worse neurologic scores, suggesting that increases in infarct volumes corresponded to functional outcome. No alterations in systemic physiologic parameters were demonstrated; therefore, it seems unlikely that the observed differences in ischemia susceptibility can be explained by obvious effects on cardio- or cerebrovascular parameters but rather by a direct neuroprotective effect.
TrkB receptor activation is neuroprotective in situations of focal and global cerebral ischemia: (1) Neurons that upregulate TrkB (but not TrkA and TrkC) are located in the penumbra zone (focal ischemia) or CA3 subsector of the hippocampus (global ischemia) and survive the insult. Neurons, however, that do not show such an upregulation of TrkB, such as in the ischemic core after focal ischemia or in the CA1 subsector of the hippocampus after global ischemia, are likely to die (Merlio et al., 1993; Takeda et al., 1993; Kokaia et al., 1996). (2) Exogenous administration of BDNF and NT4 protects against focal and global cerebral ischemia (Beck et al., 1994; Chan et al., 1996; Schäbitz et al., 1997). (3) Moreover, endogenous expression of BDNF and NT4 at normal levels in brain confers resistance to brain ischemia (Endres et al., 2000).
NT4 and BDNF share a 54% amino acid identity and both bind to the TrkB (and p75NTR) receptors with equal affinity (Klein et al., 1991, 1992). NT4 and BDNF mutant mice, however, exhibit contrasting phenotypes: NT4 knockout mice are viable and fertile with only mild sensory deficits (Liu et al., 1995), whereas BDNF knockout mice die postnatally with a severe neurologic phenotype (Ernfors et al., 1994; Jones et al., 1994). Different in vivo functions of NT4 and BDNF could be caused by different expression patterns or, alternatively, by distinct biologic activities in vivo. Of note, NT4 and BDNF may be at limiting amounts in vivo that may contribute to their different activities at particular low concentrations. Our results demonstrated that when NT4 expression was under Bdnf gene regulatory control, NT4 could not compensate for the loss of one BDNF allele. Hence, our data favor the hypothesis that, with regard to stroke protection, BDNF and NT4 have different biologic activities, rather than that the differences merely stem from divergent expression patterns.
Although BDNF seems to be more effective than NT4 in protecting against cerebral ischemia, we observed different phenotypes in other neural systems in Bdnf+/nt4-ki mice (Fan et al., 2000). On the one hand, NT4 rescues BDNF-deficient mice and supports many BDNF neurons in vivo, which is in favor of the idea that the contrasting phenotypes of BDNF- and NT4-deficient mice are mostly caused by their divergent expression patterns. Conversely, NT4 has a more potent activity than BDNF in promoting survival of sensory neurons and synapse formation in the hippocampus. A divergence in TrkB-receptor signaling might be responsible for a difference between NT4 and BDNF (Fan et al., 2000). Hence, the TrkB receptor seems to distinguish between binding of NT4 or BDNF in vivo, which may also explain why NT4 knock-in mice have increased susceptibility to cerebral ischemia. In addition to TrkB, p75NTR expression is also changed on ischemia and different effects on p75NTR signaling may also add to the effects of NT4 and BDNF in vivo (Kokaia et al., 1998).
Our study demonstrates for the first time that BDNF is more effective than NT4 in protecting the brain from focal ischemic insults. Because both neurotrophins bind TrkB, the generation of NT4 knock-in mice proved useful to detect different biologic activities of NT4 and BDNF during focal cerebral ischemia in vivo. Brain-derived neurotrophic factor may be a better candidate than NT4 for possible treatment of stroke in humans.
Footnotes
Acknowledgments
The authors thank Michael A. Moskowitz and Ulrich Dirnagl for advice and for providing laboratory facilities.