
Abstract
Using homozygous human apolipoprotein E2 (apoE2) (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice, we aimed to examine whether an apoE isoform-specific exacerbation of delayed infarct expansion occurs after permanent middle cerebral artery occlusion (pMCAO). Compared with 2/2- or 3/3-KI mice, 4/4-KI mice exhibited significantly larger infarct volumes and worse neurologic deficits after pMCAO, with no significant differences between the latter two groups. Infarct volume in 4/4-KI mice was significantly increased from 1 to 5 days after pMCAO, whereas that in 2/2- or 3/3-KI mice was not significantly altered. DNA fragmentation in the peri-infarct area as detected by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphatenick end-labeling was increased to a similar degree in all of the KI mice by 5 days after pMCAO, with no significant differences among the mouse groups. At every time-point examined, human apoE was most markedly expressed in the peri-infarct area, with similar immunoreactivity among the three lines of KI mice. The glial fibrillary acidic protein immunoreactive burden in the peri-infarct area was progressively increased through 7 days in 4/4-KI mice, but not in 2/2- or 3/3-KI mice. Taken together, these data show that the apoE4 isoform acts to aggravate delayed infarct expansion and peri-infarct reactive astrocytosis during the subacute phase of pMCAO in genetically engineered apoE-KI mice.
For human apolipoprotein E (APOE denotes the gene; apoE denotes the protein), three isoforms (apoE2, apoE3, or apoE4) are encoded by three alleles (ɛ2, ɛ3, or ɛ4) at a single APOE locus on the long arm of chromosome 19. Numerous clinical studies have documented that the presence of the APOE ɛ4 allele worsens the outcome of acute neurologic insults such as cerebral trauma (Friedman et al., 1999; Nicoll et al., 1995; Sorbi et al., 1995; Teasdale et al., 1997), intracerebral hemorrhage (Alberts et al., 1995), or subarachnoid hemorrhage (Leung et al., 2002). Glial cells (particularly astrocytes) are the main producers of apoE in the CNS (Fujita et al., 1999), and recent evidence suggests that apoE can modulate the endogenous CNS inflammatory response (Laskowitz et al., 2001; Lynch et al., 2001, 2002; Styren et al., 1998). Bystander neuronal injury after acute CNS insult and chronically progressive brain injury caused by neurode-generative disease may be aggravated by inflammatory responses triggered by glial activation and subsequent formation of a damaging “cytokine cycle” (Barone and Feuerstein, 1999; Griffin et al., 1998; Weninger and Yankner, 2001).
Regarding ischemic cerebrovascular disease, an association between the APOE ɛ4 allele and exacerbation of cerebral infarction has been supported by clinical studies (Kim et al., 2003; Margaglione et al., 1998; McCarron et al., 1999), although contradictory results have also been reported (Basun et al., 1996; Frikke-Schmidt et al., 2001; Zhu et al., 2000). Based upon a reduced frequency of the APOE ɛ4 allele in patients who had survived a previous stroke (Basun et al., 1996), Laskowitz and colleagues (1998) surmised that the APOE genotype may not alter risk for stroke, but it may alter recovery once it occurs. In conformity with this view, previous studies using different types of genetically engineered mice have revealed that the apoE4 isoform retards acute recovery after a variety of brain injuries (Buttini et al., 1999, 2000; Horsburgh et al., 2000; Sheng et al., 1998; White et al., 2001). Furthermore, our previous study using homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice demonstrated that the apoE4 isoform acts to exacerbate acute ischemic brain damage (24 hours after the onset of ischemia) in an isoform-specific fashion (apoE4 > apoE3 = apoE2), whereas the apoE2 isoform did not show any protective effect (Mori et al., 2003). Although the previously mentioned studies establish a deleterious effect of the apoE4 isoform on acute brain damage, attention needs to be directed towards more long-term effects of brain injury, because the infarct continues to expand for a much longer period than 1 day after the insult.
Infarct volume rapidly increases during the acute phase of ischemia (<12–24 hours) (Garcia et al., 1993). Du and colleagues (1996) reported that when the degree of ischemia was mild, infarction could develop in a surprisingly delayed fashion after transient focal ischemia. In this regard, we have shown that the infarct volume caused by permanent focal ischemia in the rat increases in two phases; namely, the acute expansion during the initial 24 hours, and the delayed expansion during the succeeding 144 hours (Asano et al., 1999; Matsui et al., 2002). In stroke patients, an analogous form of delayed infarct expansion has been noted (Baird et al., 1997; Beaulieu et al., 1999), indicating that this phenomenon is a viable therapeutic target for the disease. There is ample reason to suspect that the mechanisms causing infarction are substantially different between acute and delayed infarct expansion. A major difference is that the former primarily involves acute necrosis caused by energy failure within the ischemic core (Dirnagl et al., 1999), whereas the latter is earmarked by an increase in the number of apoptotic cells within the peri-infarct area along the outer border of the infarct in the midst of prominent reactive astrocytosis (Stoll et al., 1998). As has been shown in other neurodegenerative disorders such as Alzheimer's disease, proinflammatory glial activation may promote or oppose brain cell demise depending on the stimulus (Wyss-Coray and Mucke, 2002). Regarding infarct expansion after focal cerebral ischemia, we hypothesized that reactive astrocytosis plays a detrimental role given the previously mentioned tight association between apoptotic cells and reactive astrocytes in the peri-infarct area (Stoll et al., 1998).
It should be noted that, whereas some studies have indicated a protective role of reactive astrocytes (Chen and Swanson, 2003; Giulian, 1993), others are suggestive of a detrimental influence of astrocytosis during the subacute phase (1–7 days) of stroke. The latter view is supported by the following findings: 1) astroglial S-100β induces neuronal cell death by promoting nitric oxide release from astrocytes (Hu et al., 1996; Murphy 2000); and 2) the occurrence of apoptosis, reactive astrocytosis [hallmarked by hyperplasic and hypertrophied glial fibrillary acidic protein (GFAP)-expressing astrocytes] and delayed infarct expansion was suppressed by administration of ONO-2506, which is known to inhibit S-100β synthesis in activated astrocytes (Tateishi et al., 2002).
Thus, the aim of the present study was twofold. First, we wished to determine whether delayed infarct expansion after permanent middle cerebral artery occlusion (pMCAO) occurs in an isoform-specific manner, using 2/2-, 3/3-, or 4/4-KI mice. Second, we sought to evaluate whether there is a correlation between the severity of delayed infarct expansion and peri-infarct astrocytic activation as assessed by GFAP immunoreactive burden. Our findings provide evidence that a significant delayed infarct expansion occurs exclusively in 4/4-KI mice from 1 to 5 days after pMCAO, and it is accompanied by pronounced peri-infarct reactive astrocytosis from 1 to 7 days after pMCAO in an isoform-specific fashion.
MATERIALS AND METHODS
Animals
Homozygous mice expressing the apoE4 isoform in place of mouse apoE were generated by the gene-targeting technique taking advantage of homologous recombination in embryonic stem cells as previously described (Hamanaka et al., 2000). Both 2/2- or 3/3-KI mice were produced using the same strategy except that the transgenes carried apoE2 or apoE3 cDNA in place of apoE4 cDNA. The nucleotide sequences of the transgene apoE cDNAs were confirmed by sequencing cDNAs prepared from liver polyA+ RNAs of the three homozygous strains. Ten-week-old weight-matched male mice were verified to be homozygous KIs using allele-specific oligonucleotide primers and polymerase chain reaction analysis, as described previously (Mori et al., 2003). In addition, immunoblotting has confirmed the expression of human apoE (as opposed to mouse apoE) from separate sets of 2/2-, 3/3-, or 4/4-KI mice according to previously described methods (Mori et al., 2003).
Surgical procedures
The procedures followed were in accordance with the guidelines of the Animal Use Ethics Committee of the Saitama Medical Center/School and NIH guidelines [DHHS publication No. (NIH) 85–23, revised 1985]. All efforts were made to minimize animal suffering and to reduce the number of animals used. Animals were housed in a virus-free barrier facility under a 12 hours/12 hours light-dark cycle, with ad libitum access to food and water. All mice were subjected to fasting overnight (12 hours) with free access to water before surgical procedures. Anesthesia was induced and maintained with halothane (1.5–2% and 0.5%, respectively) in a mixture of 70% nitrous oxide and 30% oxygen under spontaneous ventilation. Rectal temperature was monitored throughout the experiment, and nor-mothermia was maintained with a homeothermic blanket control unit that was preset to 37°C. The distal segment of the middle cerebral artery (MCA) crossing over the rhinal fissure was exposed for induction of permanent middle cerebral artery occlusion (pMCAO) as described previously (Brint et al., 1988). Briefly, a 1-cm skin incision was made approximately midway between the left outer canthus and anterior pinna. Incision and retraction of the temporalis muscle were performed to expose the squamous portion of the temporal bone. Under a surgical microscope, a craniotomy (2-mm in diameter) was created at the juncture of the zygomatic process and the temporal bone. The dura mater was opened with a fine curved needle to expose the MCA. The left common carotid artery (CCA) was ligated with 8–0 silk, and then a 2-mm segment of the MCA was electrocauterized. The coagulated MCA segment was then transected with microscissors. Thereafter, the wound was closed after applying lidocaine to the operative field. After halothane was discontinued, mice were returned to their cages and allowed free access to food and water. Evaluation of neurologic deficits was performed at 24 hours intervals after pMCAO (until the animals were killed) as follows: score 0, no neurologic deficit; score 1, forelimb flexion; score 2, decreased resistance to lateral push and forelimb flexion without circling; score 3, same behavior as grade 2, with circling; and score 4, inability to walk spontaneously (Mori et al., 2003). Evaluation was performed by a single investigator, blinded to animal genotype.
Determination of infarct volume
A total of 72 mice (2/2-, 3/3-, or 4/4-KI mice; n = 6 at each time-point for the three lines of KI mice) were randomly allocated for death 1, 3, 5, and 7 days after pMCAO. After mice were reanesthetized with halothane as described above, they were killed by transcardial perfusion of 200 mL of 10 U/mL heparin in saline followed by 200 mL of 4% paraformaldehyde in 0.1 mol/L (pH 7.4) phosphate-buffered saline. The brain was removed and fixed in the same fixative at 4°C for 48 hours. Then, the bilateral cerebral hemispheres were embedded in paraffin with 48 hours of processing. Serial sections (5-μm in thickness) of the cerebral hemispheres at six predetermined coronal planes separated by 1-mm intervals were sequentially labeled as sections 1 to 6 and stained with hematoxylin and eosin or cresyl violet. The infarct area in each section was measured using a computer-based image analyzer (Scion Image beta 4.02 for Windows, Scion Corporation, Frederic, MD, U.S.A.). To exclude the effects of brain edema, the infarct area was corrected by the ratio of the entire area of the ipsilateral hemisphere to that of the contralateral hemisphere. Because the interval between sections was 1 mm, infarct volumes (mm3) were calculated as running sums of corrected infarct area in all six of the slices. Measurements were performed in a blinded manner by a single investigator.
Evaluation of physiologic parameters and regional cerebral blood flow
The repeated blood withdrawal or continuous monitoring of regional CBF for 60 minutes is likely to affect the outcome of pMCAO. All parameters (PaO2, PaCO2, pH, MABP, blood glucose, body temperature, and regional CBF) were examined in separate sets of 2/2-, 3/3-, or 4/4-KI mice (n = 6 each) under halothane anesthesia with spontaneous ventilation as described previously. Animals were subjected to fasting overnight (12 hours) with free access to water before blood sampling. A femoral artery was cannulated to monitor MABP and to collect a blood sample. The body temperature was monitored using a rectal probe. The previously mentioned physiologic parameters were recorded at 15 minutes before (except for PaO2, PaCO2, and pH) and 60 minutes after pMCAO. These physiologic parameters were maintained within their normal ranges throughout the operative procedures; they showed no significant differences among 2/2-, 3/3-, or 4/4-KI mice. Before ligation of the left CCA, regional CBF was continuously monitored with two laser-Doppler flow probes (OMEGAFLO FLO-C1, Omegawave Co., Tokyo, Japan) at two points of the dorsolateral cerebral cortex of the ischemic hemisphere, as described previously with modifications (Mori et al., 2003). The skull was exposed and thinned at the core (1-mm caudal to bregma and 4-mm lateral to the midline) and the periphery (1.7-mm rostral to lamda and 1.5-mm lateral to the midline) of the ischemic area. Next, the head of the mouse was fixed in a stereotaxic frame (Narishige, Tokyo, Japan), and the tip of the fiberoptic laser-Doppler flow probe (0.5-mm in diameter) was fixed perpendicular to the skull surface with surgical adhesive. After baseline values were obtained, regional CBF was measured continuously and recorded at the point of ligation of the left CCA every 5 minutes during 60 minutes after pMCAO.
Immunohistochemistry
In brains excised from 2/2-, 3/3-, or 4/4-KI mice at 1, 3, 5, and 7 days after pMCAO, additional sections adjacent to the coronal brain slice at the level of the anterior commissure (section No. 3) were obtained for the following immunohistochemical study. For detecting human apoE in situ, sections were deparaffinized and pretreated by hydrolytic autoclaving in 10 mmol/L citrate buffer (pH 6.0) for 15 minutes at 121°C to retrieve antigens. Detection of human apoE and GFAP was done according to the manufacturer's protocol using a Vectastain ABC elite kit (Vector Laboratories, Burlingame, CA, U.S.A.) coupled with the diaminobenzidine reaction. Rabbit polyclonal anti-human apoE antibody (diluted 1:200; incubated at 4°C overnight; IBL, Gunma, Japan), or rabbit polyclonal anti-cow GFAP antibody (diluted 1:1,000; incubated at 4°C overnight; DAKO, Carpinteria, CA, U.S.A.) was used as primary antibodies. Phosphate-buffered saline (0.1 mol/L; pH 7.4) was used instead of primary antibody or ABC reagent as a negative control. TUNEL [terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)-nick end-labeling] staining was performed according to the manufacturer's protocol using an ApopTag Kit (Intergen Company, Purchase, NY, U.S.A.) coupled with the diaminobenzidine reaction. Controls for the specificity of the signal included omission of TdT reagent.
Image analysis
Images were acquired using an Olympus BX60 microscope with an attached digital camera system (DP-50, Olympus, Tokyo, Japan), and the digital image was routed into a Windows PC for quantitative analysis using SimplePCI software (Compix, Inc. Imaging Systems, Cranberry Township, PA, U.S.A.). Images of five different fields (604,800 pixels per field) in regions of interest were captured via a ×20 objective lens, and a threshold optical density was obtained that discriminated staining from background. Each field was manually edited to eliminate artifacts. To determine the number of TUNEL-positive cells in the peri-infarct area, the positive signals of labeled nuclei were manually plotted and counted in each image. To evaluate human apoE immunoreactivity, after the mode of all images was converted to gray scale, the average intensity of the neural and glial immunoreactivity in each image was quantified in the three predetermined regions (nonischemic or peri-infarct areas in ischemic hemispheres, and symmetrical areas in contralateral hemispheres). The GFAP immunoreactive burden in the peri-infarct area is presented as the percentage of immunolabeled area captured (positive pixels) divided by the full area captured (total pixels). A single investigator performed each analysis and compared the images taken from the three lines of KI mice at each time-point after pMCAO in a blinded manner.
Statistical analysis
Data are presented as the mean ± 1 SD. The Kolmogorov-Smirnov test was used to determine the normality of the data. If data were not found to be normally distributed or were ordinal level, the nonparametric Kruskal-Wallis H test was performed followed by post-hoc testing using the Mann-Whitney U test. If data were found to be normally distributed and were interval or ratio level, statistical analysis was performed using parametric one-way analysis of variance (ANOVA), followed by post-hoc testing using Bonferroni's or Dunnett's T3 methods as appropriate (where appropriateness was determined using Levene's test for equality of the variance). In instances of single comparisons of the means, t-test for independent samples was used to assess significance. For comparison of regional CBF levels among 2/2-, 3/3-, or 4/4-KI mice, repeated measure ANOVA was performed with time as the repeated measure. For correlation of GFAP immunoreactive burden with infarct area, bivariate correlations were performed using the Pearson's product-moment correlation coefficient. P values of less than 0.05 were considered to be significant. All analyses were performed using the Statistical Package for the Social Sciences, release 11.0 (SPSS, Chicago, IL, U.S.A.).
RESULTS
Brain damage and neurologic deficits after pMCAO in apoE-KI mice
At post-mortem examination, the absence of subarachnoid hemorrhage or intracerebral hematoma was visually confirmed in all of the KI mice subjected to pMCAO. In all of the KI mice, the infarct was restricted to the cortices. Mortality was not encountered before the end of the experiment. Infarct volumes (Fig. 1A) generally showed a tendency to increase in all three lines of KI mice until 5 days after pMCAO [results expressed as mean infarct volume (mm3) ± 1 SD in 2/2-, 3/3-, or 4/4-KI mice after pMCAO: at 1 day, 14 ± 2, 13 ± 2, or 17 ± 3; at 3 days, 14 ± 3, 14 ± 2, or 20 ± 3; at 5 days, 15 ± 3, 15 ± 2, or 21 ± 3; at 7 days, 14 ± 2, 13 ± 2, or 18 ± 2]. Notably, delayed infarct expansion was most marked in 4/4-KI mice, reaching a peak value at 5 days after pMCAO that was significantly different from the value at 1 day (P < 0.05). Furthermore, at each time-point, the infarct volume in 4/4-KI mice was significantly larger than that in 2/2- (P ≤ 0.01 at 3, 5, and 7 days) or 3/3-KI mice (P < 0.05 at 1 day; P < 0.01 at 3, 5, and 7 days). No significant differences in the infarct volumes were present between 2/2- and 3/3-KI mice at any time-point after pMCAO.
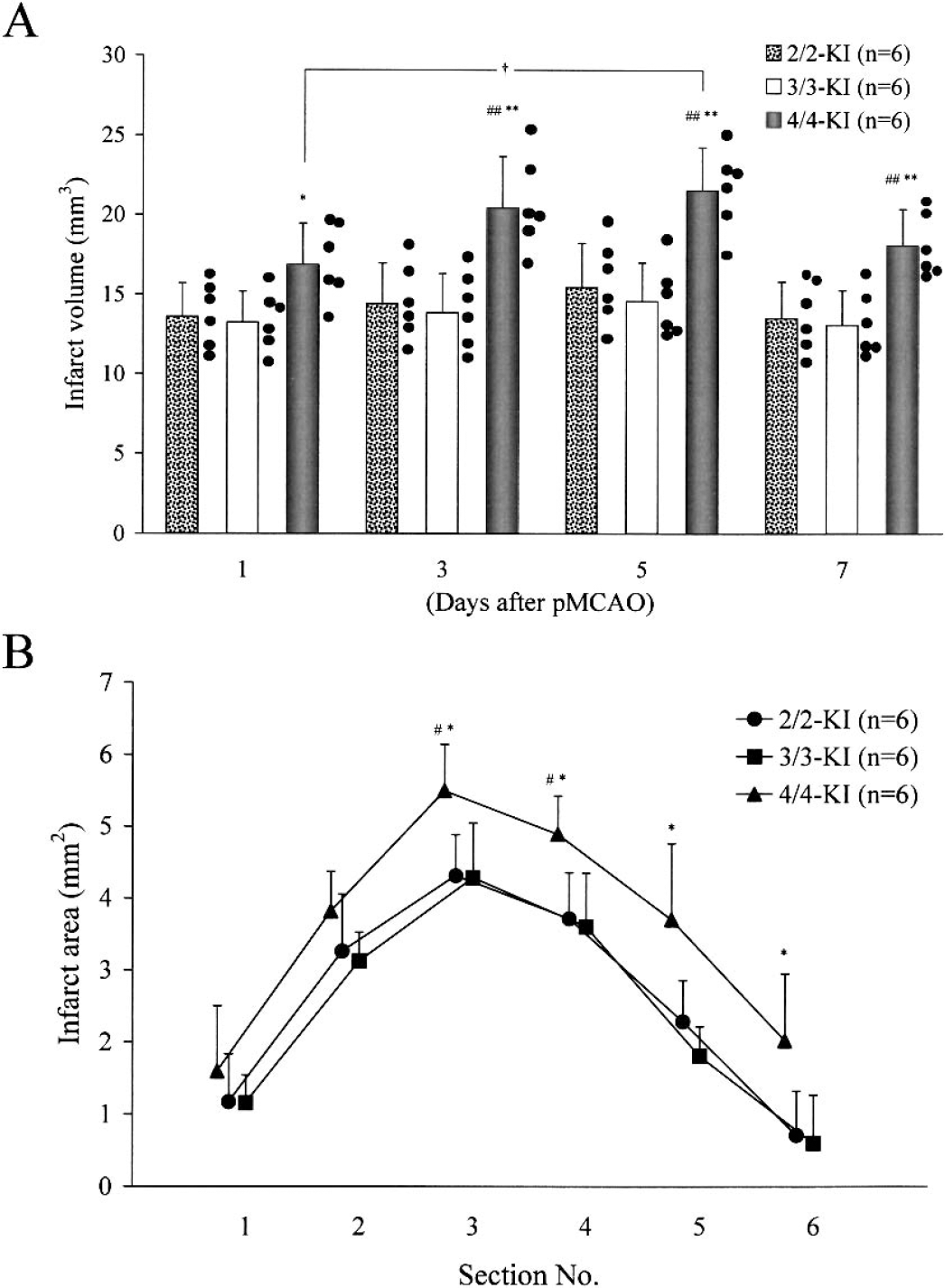
ApoE isoform-specific infarct expansion after pMCAO. Infarct volumes at 1, 3, 5, and 7 days after pMCAO in the three lines of KI mice (A). Infarct areas in each coronal plane section at 5 days after pMCAO in the three lines of KI mice (B). Each value represents the mean + 1 SD. Individual scores are plotted as closed circles (A). Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. (A) A significant difference of †P < 0.05 (vs. 1 day) was found at 5 days after pMCAO in 4/4-KI mice. Significant differences of *P < 0.05 vs. 3/3-KI mice at 1 day, ##P ≤ 0.01 vs. 2/2-KI mice at 3, 5, and 7 days, and **P ≤ 0.01 vs. 3/3-KI mice at 3, 5, and 7 days were found. (B) Significant differences of #P < 0.05 vs. 2/2-KI mice in sections 3 and 4 and *P < 0.05 vs. 3/3-KI mice in sections 3 to 6 was found.
In ipsilateral hemispheres, extensive pallor, highlighted in hematoxylin and eosin- and cresyl violet-stained specimens, was present only in the cortex at 1, 3, 5, and 7 days after pMCAO. At 5 days after pMCAO (when infarct volume was the largest in each of 2/2-, 3/3-, or 4/4-KI mice), section No. 3 exhibited the largest infarct area (Fig. 1B) of all the sections. In some of the sections, infarct areas were significantly larger in 4/4-KI mice than in 2/2- (P < 0.05 in sections 3 and 4) or 3/3- (P < 0.05 in sections 3 to 6) KI mice. No significant differences were found in the infarct areas between 2/2- and 3/3-KI mice in any of the sections examined. The significant enlargement of both infarct area and volume in 4/4-KI mice clearly indicates apoE isoform-specific vulnerability in these mice during both the acute and delayed stages of ischemic brain injury. The edema index, calculated as the ratio of the ipsilateral to the contralateral hemispheric area, showed no significant differences in any of the section planes among the experimental mouse groups at any time-point after pMCAO. On neurologic examination, atypical neurologic deficits (torsion of the neck or rolling fits) or severe neurologic scores (score 4) were not encountered throughout the experiment. At every time point, neurologic scores were significantly higher in 4/4-KI mice than in 2/2- (P ≤ 0.001 at 1, 2, and 3 days; P < 0.01 at 4 and 5 days; P < 0.05 at 6 and 7 days) or 3/3-KI mice (P ≤ 0.001 at 1, 2, 3, 4, and 5 days; P < 0.05 at 6 and 7 days), whereas no significant differences were detected between 2/2- and 3/3-KI mice (Table 1).
Neurologic scores after pMCAO in the three lines of KI mice
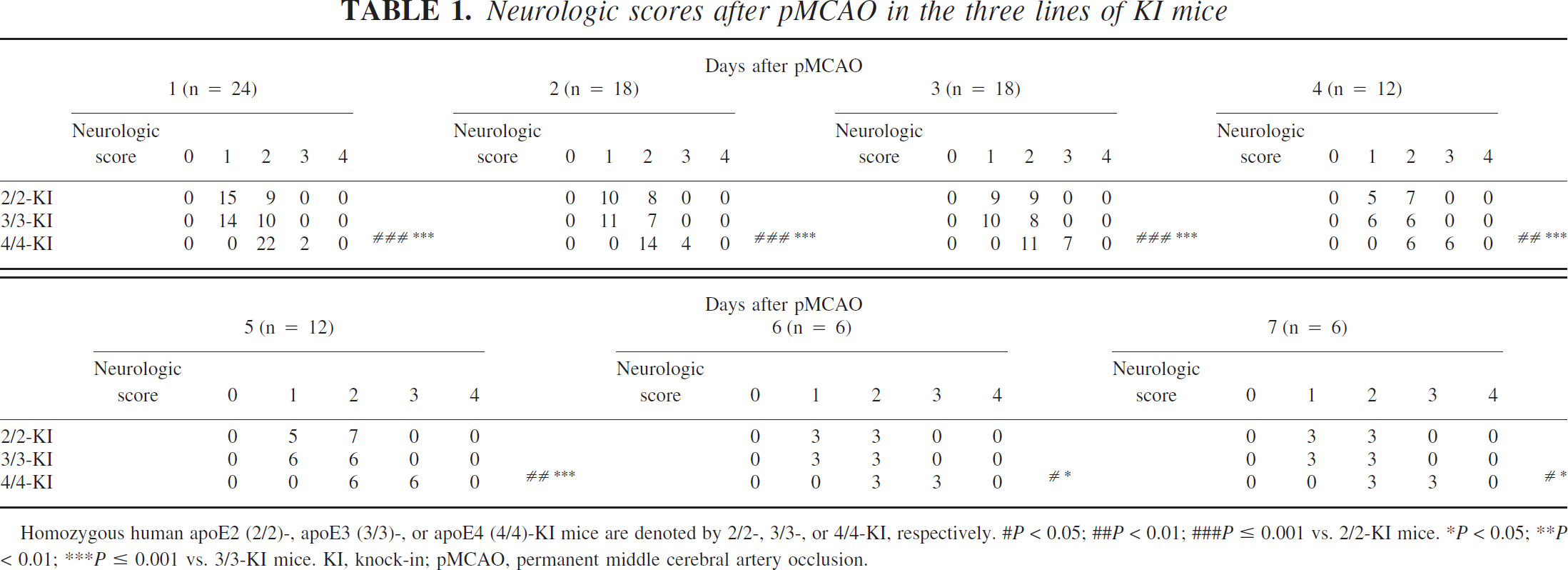
Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively.
P < 0.05;
P < 0.01;
P ≤ 0.001 vs. 2/2-KI mice.
P < 0.05;
P < 0.01;
P ≤ 0.001 vs. 3/3-KI mice. KI, knock-in; pMCAO, permanent middle cerebral artery occlusion.
Evaluation of regional CBF after pMCAO in apoE-KI mice
In all the experimental groups, the alterations in the regional CBF in the core or the periphery of the ischemic area after pMCAO were estimated by the laser-Doppler flow meter, and the results are shown in Fig. 2. Repeated measures ANOVA was used to test for significant differences in regional CBF between apoE-KI mouse groups. This analysis strategy revealed a significant main effect of time after pMCAO within apoE-KI mouse groups when considering either the core (P < 0.001) or the periphery (P < 0.01) of the ischemic area. Thus, pMCAO produced a significant decrease in regional CBF that was time dependent. However, there was no significant interaction between time after pMCAO and apoE-KI mouse group when considering either the core or the periphery of the ischemic area, showing that there were no significant differences in regional CBF among the three lines of KI mice during the time-period monitored (Fig. 2).
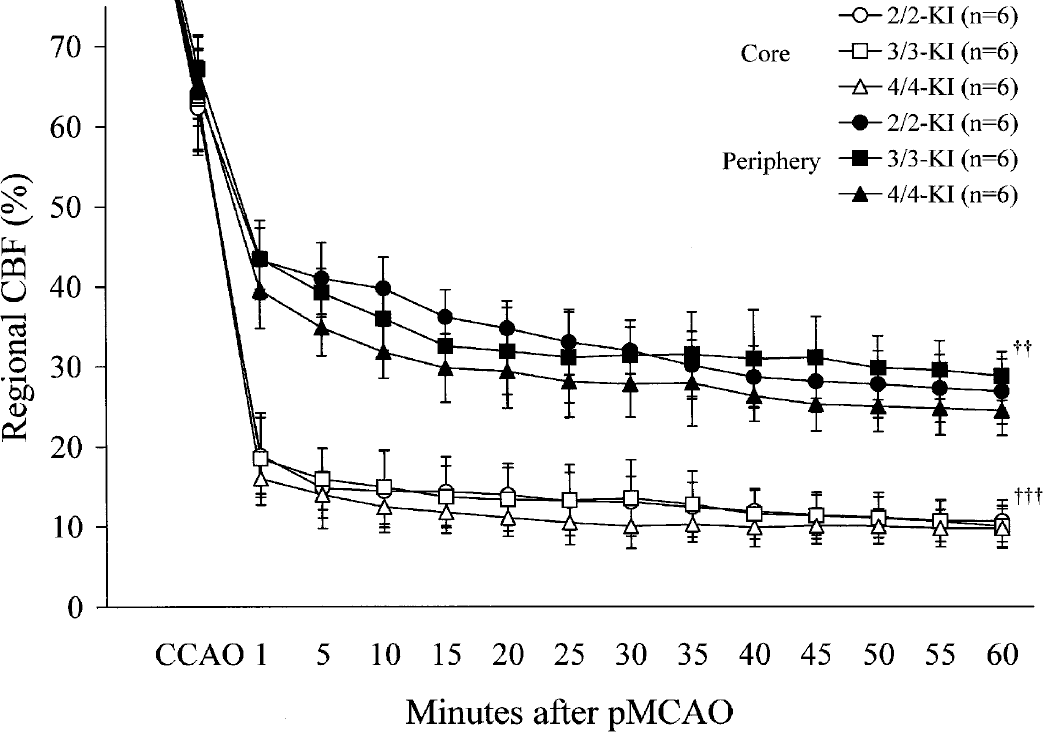
Regional CBF measured in the core and the periphery of the ischemic area after pMCAO in the three lines of KI mice. Each value represents the mean ± 1 SD. Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Statistical analysis revealed a within-apoE-KI mouse group main effect of time after pMCAO when considering either the core (†††P < 0.001) or the periphery (††P < 0.01) of the ischemic area. However, there was no significant interaction between time after pMCAO and apoE-KI mouse group (which would indicate an apoE isoform-specific effect) when considering either the core or the periphery of the ischemic area.
Evaluation of TUNEL-positive cells after pMCAO in apoE-KI mice
At each of the time-points examined, TUNEL-positive cells were preferentially located within a narrow rim along the outer border of infarct, and they were clearly greater in number within the border of the infarct compared with the surrounding region outside of the infarct border. However, because the purpose of this study was to examine the outward expansion of infarct, only TUNEL-positive cells outside the infarct border (peri-infarct area) were counted (Fig. 3). Whereas TUNEL-positive cells were only sparsely present in the region outside the infarct border at 1 day after pMCAO, their numbers significantly increased in each of the 2/2-, 3/3-, or 4/4-KI mouse groups (for each apoE-KI mouse group, P < 0.001 at 3, 5, and 7 days vs. 1 day) to peak at 5 days after pMCAO. However, we did not detect significant differences among 2/2-, 3/3, or 4/4-KI mice at any time-point examined.
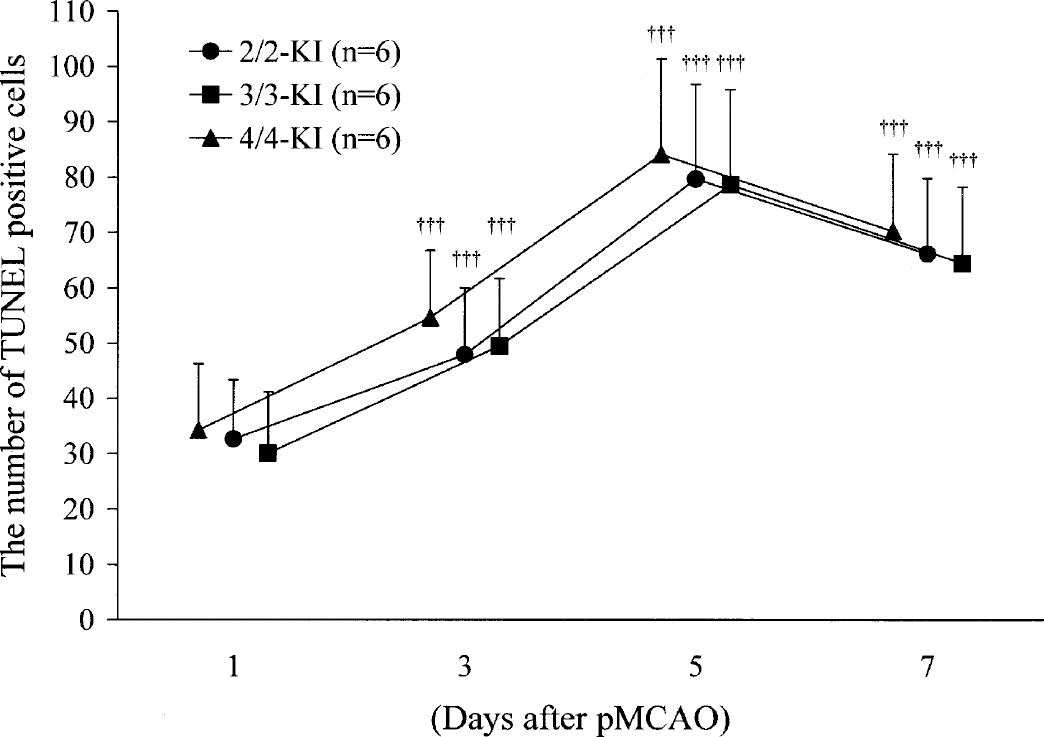
The number of TUNEL-positive cells at 1, 3, 5, and 7 days after pMCAO in the three lines of KI mice. Each value represents the mean + 1 SD. Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Statistical analysis revealed a difference of P < 0.001 (†††vs. 1 day) at 3, 5, and 7 days in each line of KI mice. There were no significant between-mouse groups differences at any time-point.
Human apoE immunoreactivity after pMCAO in apoE-KI mice
In the infarct areas of the ischemic hemispheres of 2/2-, 3/3-, or 4/4-KI mice, the expression of human apoE in neurons and astrocytes was markedly diminished throughout the experiment. These areas were present only in association with a minimal number of residual neurons and astrocytes until 3 days after pMCAO (Fig. 4A) and in a diffuse or a plaque-like pattern at 5 and 7 days after pMCAO (Fig. 4D). In the noninfarct areas of both ischemic and contralateral hemispheres of 2/2-, 3/3-, or 4/4-KI mice, numerous neurons in the entire cortices and in the striatum showed weak expression of human apoE in their somata and neurites during the time interval from 1 (Fig. 4B) to 7 days (Fig. 4E) after pM-CAO. Also, a small number of resting astrocytes in the neocortex (layer I) and in the white matter showed weak expression of human apoE in their somata and processes at every time-point examined. By contrast, more intense expression of human apoE in some neurons and astrocytes was invariably detected in the peri-infarct areas of 2/2-, 3/3-, or 4/4-KI mice from 1 (Fig. 4C) to 7 days (Fig. 4F) after pMCAO. This result suggests that human apoE is functionally expressed under endogenous regulatory control in the brains of the three lines of KI mice.
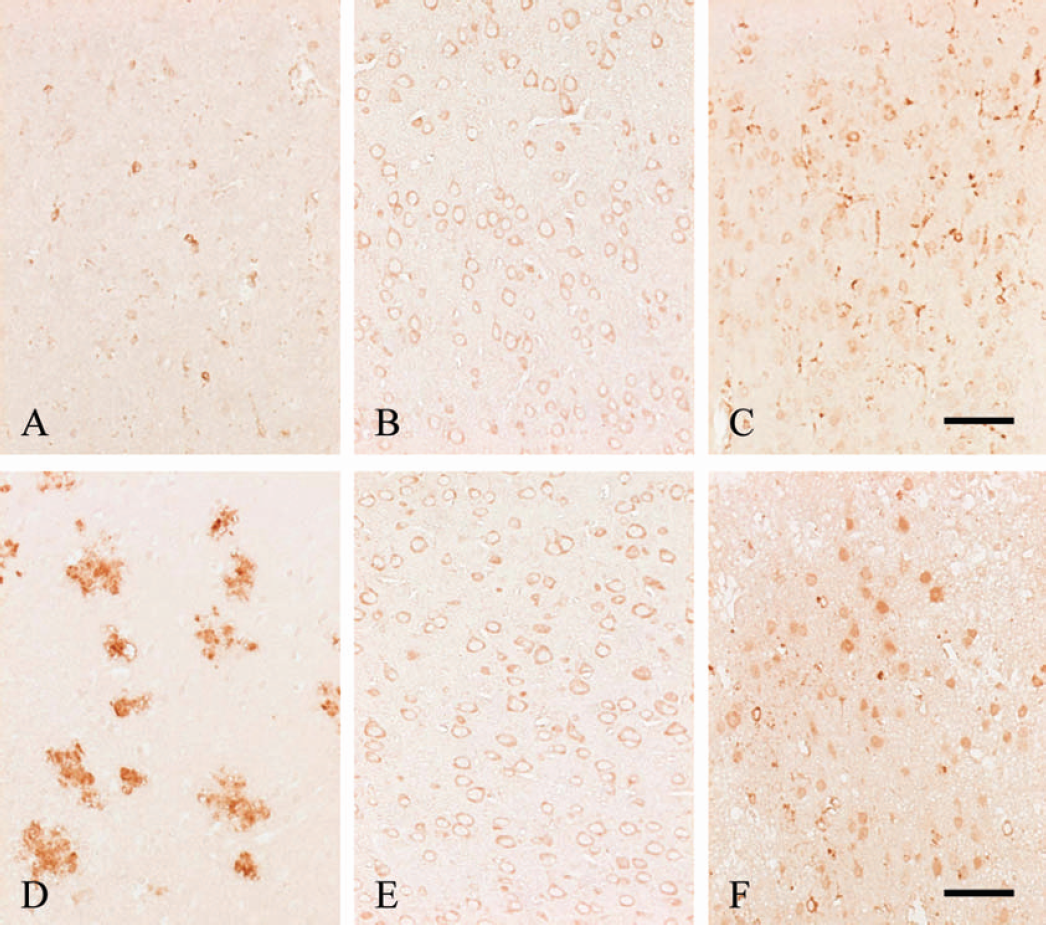
Expression of human apoE in brains from 4/4-KI mice at 1 or 7 days after pMCAO. In the infarct area of the ischemic hemisphere, human apoE immunoreactivity in neurons and astrocytes is markedly reduced except in association with a minimal number of residual neurons and astrocytes at 1 day after pMCAO (A) and is only present in a diffuse pattern at 7 days after pMCAO (D). In the noninfarct area of the ischemic hemisphere at 1 (B) and 7 (E) days after pMCAO, a considerable number of neurons show weak expression of human apoE in somata and neurites in the neocortex. Also, a lesser number of resting type astrocytes in the neocortex (layer I) show weak expression of human apoE in their somata and processes. Notably, more intense expression of human apoE in the peri-infarct area is detected in some neurons and astrocytes at 1 (C) and 7 (F) days after pMCAO. Scale bar = 50 μm.
Quantitative image analysis showed no significant differences in intensity of human apoE expression among 2/2-, 3/3-, or 4/4-KI mice in any region or at any time-point examined. In all of the experimental groups, however, neural and astroglial expression of human apoE was significantly more intense in the peri-infarct areas than in either the nonischemic areas of the ischemic hemispheres or the contralateral hemispheres at every time-point examined (P < 0.001 for each of 2/2-, 3/3-, or 4/4-KI mice). No significant differences were noted between nonischemic areas of the ischemic and contralateral hemispheres (Table 2).
Mean intensity of human apoE immunoreactivity after pMCAO in the three lines of KI mice

Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Each value represents the mean ± SD.
P < 0.001 for each of 2/2-, 3/3-, or 4/4-KI mice vs. the nonischemic areas of the ischemic hemispheres.
P < 0.001 for each of 2/2-, 3/3-, or 4/4-KI mice vs. the nonischemic areas of the contralateral hemispheres. KI, knock-in; pMCAO, permanent middle cerebral artery occlusion.
Increased reactive astrocytosis in the peri-infarct area of 4/4-KI mice
Both in the nonischemic areas of the ipsilateral hemispheres and the contralateral hemispheres of 2/2-, 3/3-, or 4/4-KI mice subjected to pMCAO, GFAP-expressing astrocytes were detected in the vicinity of the pia mater and the small penetrating vessels in the cortex, as well as in the white matter. Whereas reactive astrocytosis (as hallmarked by increased GFAP expression) was always most pronounced in the immediate vicinity of the infarct border (peri-infarct area), it was scarcely observed within the ischemic core. Astrocytic GFAP expression was particularly confined to swollen astrocytic processes and was slightly enhanced at 1 day after pMCAO in 2/2-, 3/3-, or 4/4-KI mice, likely reflecting the early stage of activation (Figs. 5A, 5E, and 5I). At 3 days after pMCAO, astroglia were observed in the peri-infarct area (Figs. 5B, 5F, and 5J) and were strongly GFAP-positive in their somata and processes, thus representing typical reactive astrocytes. At 5 (Figs. 5C, 5G, and 5K) and 7 days (Figs. 5D, 5H, and 5L) after pMCAO, reactive as-trocytes in 2/2-, 3/3-, and 4/4-KI mice showed enhanced immunoreactivity for GFAP, particularly in their somata and processes, and their processes were interwoven to form a glial barrier around the infarct. Such morphologic alterations, namely anisomorphic gliosis, are probably the result of functional changes in reactive astrocytes, and these alterations were more prominent in 4/4-KI mice than in 2/2- or 3/3-KI mice from 3 to 7 days after pMCAO.
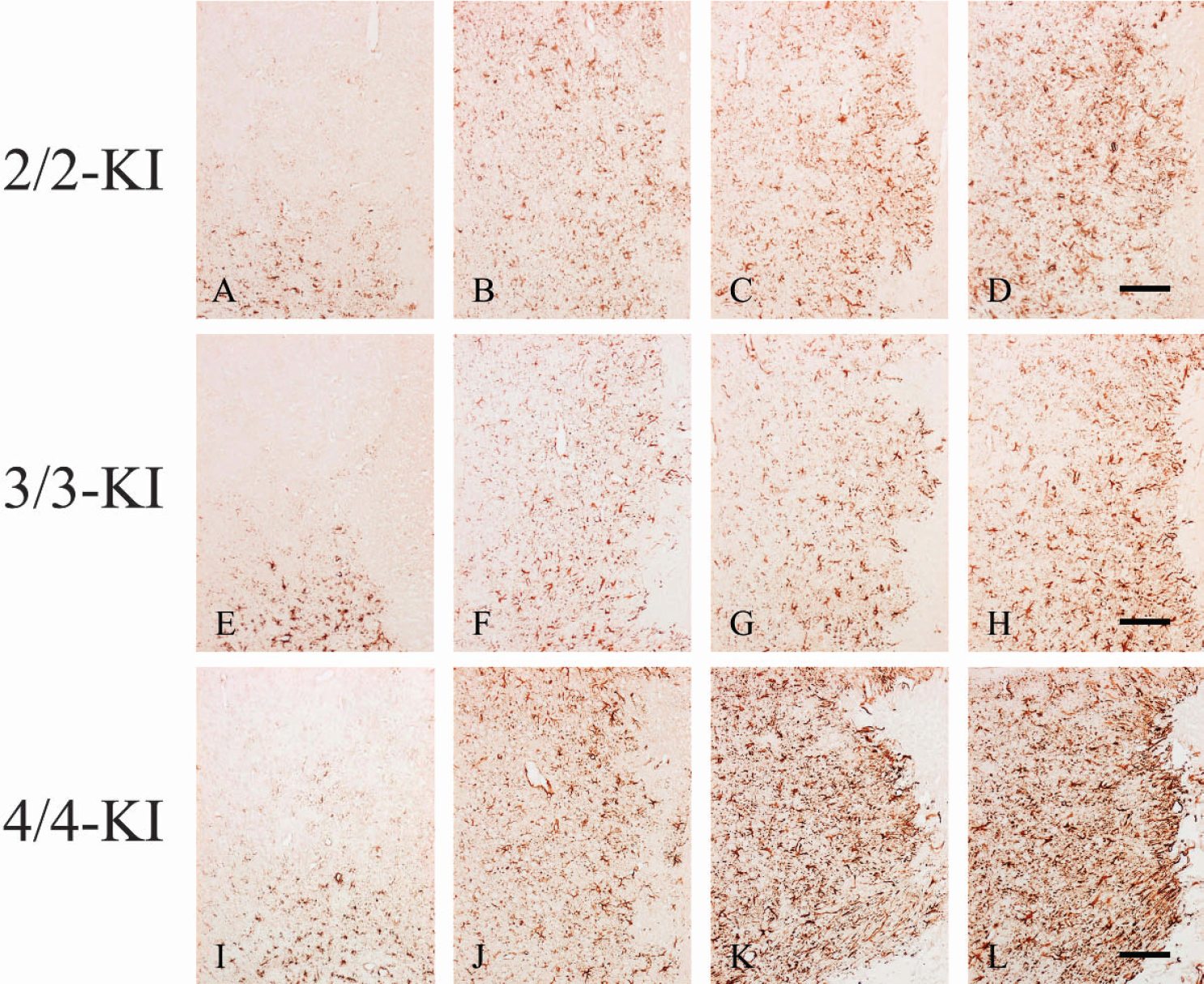
Expression of GFAP in the peri-infarct areas at 1 (A, E, I), 3 (B, F, J), 5 (C, G, K), and 7 (D, H, L) days after pMCAO in the three lines of KI mice. Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Astrocytic activation appears to be enhanced at 3, 5, and 7 days compared with day 1 after pMCAO in the three lines of KI mice. Most importantly, astrocytic activation appears to be more prominent in 4/4-KI mice compared to 2/2- or 3/3-KI mice at 5 and 7 days after pMCAO, creating a barrier close to the infarct area where anisomorphic gliosis is observed. Scale bar = 100 μm.
The result of quantitative assessment of astrocytic activation by the use of GFAP immunoreactive burden is shown in Fig. 6. Reactive astrocytosis in the peri-infarct areas was significantly enhanced at 3, 5, and 7 days compared with 1 day after pMCAO in all of the experimental groups (P < 0.001 for each of 2/2-, 3/3-, or 4/4-KI mice). Most importantly, GFAP immunoreactive burden in 4/4-KI mice was significantly larger than in 2/2- or 3/3-KI mice (P < 0.001 vs. 2/2-KI mice at 3, 5, and 7 days; P < 0.001 vs. 3/3-KI mice at 1, 3, 5, and 7 days). Bivariate correlation analyses between infarct area (as determined in section No. 3) and GFAP immunoreactive burden (as determined in the coronal section adjacent to section No. 3) across all days did not reach statistical significance in 2/2- or 3/3-KI mice, whereas it was significant in 4/4-KI (r = 0.44, P < 0.05).
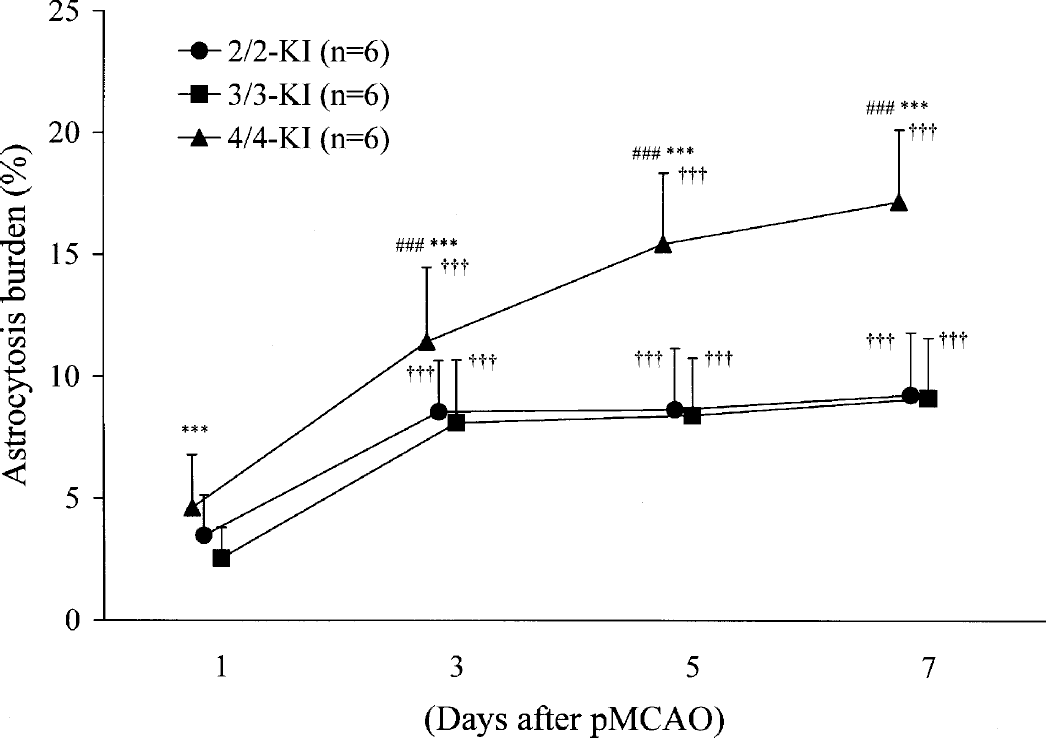
Astrocytosis burden in the peri-infarct areas at 1, 3, 5, and 7 days after pMCAO in the three lines of KI mice. Each value represents the mean + 1 SD. Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-KI mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Statistical analysis revealed a difference of P < 0.001 (†††vs. 1 day) at 3, 5, and 7 days in each line of KI mice. When comparing within days between KI mouse groups, statistical analysis showed a difference of P < 0.001 (###vs. 2/2-KI mice at 3 to 7 days; ***vs. 3/3-KI mice at 1 to 7 days).
DISCUSSION
Previous experiments using genetically engineered mice have shown that the apoE4 isoform aggravates brain damage within 24 hours after transient (Sheng et al., 1998) or permanent focal ischemia (Mori et al., 2003). However, the infarct volume can significantly increase after the initial 24 hours, as was shown with the transient (Du et al., 1996) or the permanent focal ischemia (Asano et al., 1999) model in the rat. Delayed infarct expansion is considered to be a viable target for stroke therapy. The present study aimed to clarify the long-term effect of the apoE4 isoform on infarct volumes, neurologic deficits, and magnitude of reactive astrocytosis in the peri-infarct area for up to 7 days after pMCAO, using genetically engineered apoE-KI mice as in our previous study (Mori et al., 2003). The results obtained show that each of delayed infarct expansion, peri-infarct reactive astrocytosis, and neurologic deficits during the time interval between 1 and 7 days were significantly aggravated in 4/4-KI mice versus 2/2- or 3/3-KI mice.
It is important to ascertain whether the above differences among 2/2-, 3/3-, or 4/4-KI mice represent apoE isoform-specificity as opposed to epiphenomenon. In this regard, immunoblot analysis of brain homogenates has shown that human apoE expressed in 2/2-KI mice was greater in amount than human apoE expressed in 3/3- or 4/4-KI mice (Mori et al., 2003). However, such a difference in the amount of protein expressed as above is considered to exert little influence on the evolution of ischemic brain damage for the following reasons: 1) the three lines of KI mice exhibited no significant differences in terms of physiologic parameters after pMCAO, the development of the cerebral vasculature, or the preatherosclerotic lesions in the arterial wall because of hypercholesterolemia (Mori et al., 2003); 2) the results of the present study show no significant differences in the alterations in the regional CBF among 2/2-, 3/3-, or 4/4-KI mice in the periphery or the core of the ischemic area during the initial 60 minutes after pMCAO; 3) as shown in Fig. 4 and Table 2, the distribution and the intensity of apoE immunoreactivity were similar among 2/2-, 3/3-, or 4/4-KI mice in the present study; 4) in a separate experiment using the background strain (C57BL/6N) wild-type mice subjected to pMCAO, the infarct volumes at 1 and 5 days were 13 ± 1 mm3 and 14 ± 2 mm3, respectively (n = 6 for each time-point; data not shown), which were similar to the corresponding infarct volumes in 2/2- or 3/3-KI mice; and 5) in these same animals, GFAP immunoreactive burden at 1 and 5 days after pMCAO was 3 ± 1% and 8 ± 2%, respectively (data not shown), values that are similar to those in 2/2- or 3/3-KI mice. When taken together, the above findings support the view that the observed differences in terms of delayed infarct expansion, peri-infarct reactive astrocytosis, or neurologic deficits among 2/2-, 3/3-, or 4/4-KI mice after pMCAO primarily represent an apoE4 isoform-specific event, precluding the influences of the amount of human apoE expressed or the genetic background of the mice used.
Alterations in the number of TUNEL-positive cells in the peri-infarct area reveal ample evidence to indicate the significant contribution that apoptosis makes to cell death in focal ischemia (Lipton, 1999; MacManus and Linnik, 1997). The apoptotic cells present in the peri-infarct area contribute to expansion of the ischemic lesion (Charriaut-Merlangue et al., 1996; Li et al., 1995; Sharp et al., 2000). Although TUNEL staining is not specific for apoptosis, it is a convenient method to identify DNA fragmentation in dying cells (Ben-Sasson et al., 1995). Therefore, we used the method to estimate the magnitude of cell damage in the peri-infarct area. We have already shown that both the number of TUNEL-positive cells in the peri-infarct area and the infarct volume increased in parallel from 1 to 5 days after pMCAO in the rat (Matsui et al, 2002). This result corroborates the view that some cell-damaging process is in operation for days after pMCAO at the interface between the infarct and the peri-infarct area, leading to DNA fragmentation and delayed infarct expansion. Also, in the present study, the number of TUNEL-positive cells in the peri-infarct area was (similarly) significantly increased from 1 to 5 days after pMCAO in 2/2-, 3/3-, or 4/4-KI mice. The trend was for the number of TUNEL-positive cells in 4/4-KI mice to exceed those in 2/2- or 3/3-KI mice at every time-point examined, although between-mouse groups differences did not reach a statistically significant level. The previously mentioned result may imply that counting the number of TUNEL-positive cells in one coronal section is not sensitive enough to disclose the presumably minute differences in the number of dying cells in the peri-infarct area among the experimental groups of KI mice (Wyllie et al., 1980).
Whereas significant delayed infarct expansion occurred exclusively in 4/4-KI mice, it is hereby important to note that the extent of delayed infarct expansion tends to be variable depending upon the nature of the model used. Our previous study using the rat pMCAO model revealed an increase in the infarct volume of approximately 41% between 1 and 7 days (Matsui et al., 2002). In the present study, however, significant delayed expansion occurred only in 4/4-KI mice, with an increase of approximately 28% between 1 and 5 days. On the other hand, previous studies using the mouse pMCAO model revealed no significant increase in hemispheric infarct volume during the time interval between 24 and 168 hours (Guégan et al., 1998) or between 18 and 96 hours (Hill et al., 1999). Furthermore, the infarct volumes of C57BL/6N mice (the background strain mice in the present study) at 1 and 5 days after pMCAO were similar to the corresponding infarct volumes in 2/2- or 3/3-KI mice but significantly smaller than those in 4/4-KI mice. Thus, the result of the present study reinforces the view that, during the acute as well as the subacute phases, the apoE4 isoform acts to aggravate ischemic brain damage whereas the apoE2 or apoE3 isoform does not.
Whereas peri-infarct reactive astrocytosis (as evidenced by GFAP immunoreactive burden) was enhanced in all of the KI-mice during the initial 3 days, it was significantly more pronounced in 4/4-KI mice than in 2/2- or 3/3-KI mice. During the period from 3 days until 7 days after pMCAO, GFAP immunoreactive burden in 4/4-KI mice continuously increased, whereas that in 2/2-or 3/3-KI mice was not significantly altered (Fig. 6). This correlation between reactive astrocytosis and delayed infarct expansion in 4/4-KI mice is of importance in view of previous evidence implicating glial activation in the pathogenesis of ischemic brain damage. In particular, microglial activation has been shown to precede astrocytic activation in a variety of CNS damage including stroke (Gregersen et al., 2000; Kreutzberg, 1996). Also, it has been suggested that the inflammatory process leading to the progression of the infarct involves activation of both microglia and astrocytes (Benveniste, 1995; Davies et al., 1998). In this regard, apoE was shown to inhibit glial activation in vitro in an isoform-specific fashion, wherein the apoE4 isoform is much less potent than the apoE2 or apoE3 isoform (Barger and Harmon, 1997; Laskowitz et al., 2001). In a study using mixed glial cultures, apoE modulated the endogenous CNS inflammatory response (Lynch et al., 2001). Thus, apoE seems to coordinate the interaction between activated microglia and astrocytes in an isoform-specific fashion. In parallel with the results of the above in vitro studies, the results of the present study point to the possibility that the prolonged enhancement of astroglial activation in the 4/4-KI mice was caused by the presence of apoE4 isoform. Possible mechanisms underlying the correlation between these two events are briefly speculated on in the following sections.
Once activated, astrocytes together with microglia are known to produce a myriad of biologically active inflammatory substance(s), creating a vicious “cytokine cycle” (Eddleston and Mucke, 1993; Griffin et al., 1998; Ridet et al., 1997). Furthermore, recent evidence suggests that reactive astrocytosis may be involved in chronic neuro-degeneration after ischemia. For example, Fujioka and colleagues (2003) suggested that long-lasting glial and inflammatory responses might play an important role in the slowly maturing striatal degeneration after transient MCAO in the rat. Badan and colleagues (2003) have shown that the development of a glial scar after transient focal ischemia in the rat was abnormally accelerated in the aged rat; this coincided with the stagnation of recovery in these animals. In the rat pMCAO model, we have reported that a novel agent (ONO-2506) that suppresses astrocytic S-100β synthesis significantly inhibits reactive astrocytosis while abolishing delayed infarct expansion (Tateishi et al., 2002). The results of the present study showing a significant correlation between peri-infarct reactive astrocytosis and delayed infarct expansion in 4/4-KI mice agree with the previously mentioned reports, lending further support to the view that astrocytosis acts to exacerbate delayed infarct expansion. However, because these data do not exclude the possibility that more severe delayed infarct expansion positively regulates astrocytosis, further investigation into this area is warranted.
CONCLUSION
We have shown that the presence of the apoE4 iso-form aggravates infarct expansion from 1 to 5 days after pMCAO, which is associated with enhanced astroglial activation in the peri-infarct area. Such an association between delayed infarct expansion and peri-infarct reactive astrocytosis, as evidenced in genetically engineered mice, may provide an avenue to explore the putative etiologic contribution of reactive astrocytes to the exacerbation of ischemic brain damage as well as the mechanism underlying the detrimental influence of the apoE4 isoform.
Footnotes
Acknowledgements
The authors thank Mr. N. Koyama and Ms. A. Kudo for expert assistance, as well as Dr. H. Yamaguchi for helpful discussions.