
Abstract
Brain-derived neurotrophic factor (BDNF), acting through the high-affinity receptor tyrosine kinase (TrkB), is widely distributed throughout the central nervous system and displays in vitro trophic effects on a wide range of neuronal cells, including hippocampal, cerebellar, and cortical neurons. In vivo, BDNF rescues motorneurons, hippocampal, and substantia nigral dopaminergic cells from traumatic and toxic brain injury. After transient middle cerebral artery occlusion (MCAO), upregulation of BDNF-mRNA in cortical neurons suggests that BDNF potentially plays a neuroprotective role in focal cerebral ischemia. In the current study, BDNF (2.1 μg/d) in vehicle or vehicle alone (controls) was delivered intraventricularly for 8 days, beginning 24 hours before permanent middle cerebral artery occlusion by intraluminal suture in Wistar rats (n = 13 per group). There were no differences in physiological variables recorded during surgery for the two groups. Neurological deficit (0 to 4 scale), which was assessed on a daily basis, improved in BDNF-treated animals compared with controls (P < 0.05; analysis of variance and Scheffe's test). There were no significant differences in weight in BDNF-treated animals and controls during the experiment. After elective killing on day 7 after MCAO, brains underwent 2,3,5-triphenyltetrazolium chloride staining for calculation of the infarct volume and for histology (hematoxylin and eosin and glial fibrillary acid protein). The mean total infarct volume was 83.1 ± 27.1 mm3 in BDNF-treated animals and 139.2 ± 56.4 mm3 in controls (mean ± SD; P < 0.01, unpaired, two-tailed t-test). The cortical infarct volume was 10.8 ± 7.1 mm3 in BDNF-treated animals and 37.9 ± 19.8 mm3 in controls (mean ± SD; P < 0.05; unpaired, two-tailed t-test), whereas ischemic lesion volume in caudoputaminal infarction was not significantly different. These results show that pretreatment with intraventricular BDNF reduces infarct size after focal cerebral ischemia in rats and support the hypothesis of a neuroprotective role for BDNF in stoke.
Neurotrophic growth factors are polypeptides that promote trophic effects on neuronal cells by acting through specific receptors. Brain-derived neurotrophic factor (BDNF) and the biologically similar nerve growth factor are related members of the neurotrophin family of growth factors. These neurotrophins act on a set of high-affinity receptor kinases to promote survival, differentiation, and neurite extension in many types of mammalian central nervous system neurons (Lamballe et al., 1991; Kaplan et al., 1991). Acting through the receptor tyrosine kinase (TrkB), BDNF is more widely distributed throughout the central nervous system than nerve growth factor and seems to have an additional paracrine or autocrine function (Lindsay, 1995). In vitro BDNF displays trophic effects on a wide range of neuronal cells, including hippocampal, cerebellar, and cortical neurons (Ernfors et al., 1990; Hohn et al., 1990; Hyman et al., 1991; Ebendal, 1992; Ip et al., 1993; Lindholm et al., 1993; Shimohama et al., 1993; Roback et al., 1995). In vivo, BDNF rescues motoneurons and substantia nigral dopaminergic cells from traumatic and toxic brain injury (Oppenheim et al., 1992; Sendtner et al., 1992; Tsukahara et al., 1995). Transient forebrain ischemia induces BDNF mRNA upregulation in the hippocampus (Lindvall et al., 1992; Tsukahara et al., 1994) and correlates with prevention of hippocampal neuronal death in BDNF-treated rats (Tsukahara et al. 1994, Beck et al. 1994). After middle cerebral artery occlusion (MCAO), BDNF mRNA upregulation in the cortex seems to correlate with resistance to injury in this area (Kokaia et al., 1995). Exogeneous administration of BDNF may support this effect and minimize brain damage after ischemic injury.
Because of the reported neuroprotective effects of BDNF in vitro and in vivo and the possible role of BDNF in focal cerebral ischemia, we hypothesize that intraventricular administration of BDNF may reduce infarct volume and improve behavioral outcome in a model of permanent MCAO in rats. In this study, we administered intraventricular BDNF for 8 days, starting 1 day before MCAO and assessed the effects on neurological outcome, infarct volume, and histological findings.
MATERIALS AND METHODS
Male Wistar rats weighing 300–315 g were allowed to have food and water as desired. All animals were randomly assigned before surgery for 2,3,5-triphenyltetrazolium chloride (TTC)-staining or histology in one of the following groups: BDNF-treated group (n = 13), placebo-treated group (n = 13), or sham-operated group (n = 3). The rats were then weighed and anesthetized intraperitoneally with chloral hydrate (400 mg/kg). The left femoral artery was cannulated with PE-50 polyethylene tubing for continuous monitoring of arterial blood pressure and blood sampling for analysis of arterial blood gases. Core temperature was maintained at 36.9 to 37.2°C during surgery using a heating lamp connected to a rectal temperature probe. Cranial temperature was recorded using a small copper-constantan thermocouple (Physitemp Instruments, Clifton, NJ, U.S.A.). Guided by a 23-gauge needle, the thermocouple was inserted between the right temporalis muscle and the right aspect of the skull.
Brain-derived neurotrophic factor was obtained as a concentrated stock solution (15 mg/mL) as a generous gift from Amgen Inc.(Thousand Oaks, CA, U.S.A) and stored at 8°C before use. Fifteen microliters of the BDNF solution (2 mg/mL) were dissolved in 185 μL sterile artificial cerebrospinal fluid. Brain-derived neurotrophic factor was delivered intraventricularly at a rate of 2.1 μg/d with pump rate of 0.5 μL/h (0.087 μg/h). Controls received similar infusions of the vehicle alone. Preparation and implantation of the infusion were performed under sterile conditions.
The rats were placed in a stereotaxic head holder for miniosmotic pump implantation. The dorsal surface was exposed by midline incision, and a bur hole (1 mm) was drilled over the right lateral ventricle (1.6 mm lateral and 0.8 mm posterior to the bregma). A stainless steel cannula was inserted stereotaxically into the ventricle to a depth of 4 mm beneath the surface of the skull, with polyethylene tubing connected to a miniosmotic pump (Alzet 2001 D, 200 μL fill volume, 0.5 μL/h pump rate; Alzet, Palo Alto, CA, U.S.A.) and implanted subcutaneously in the back. A second bur hole (1 mm) was drilled over the left cortex (3 mm lateral and 3 mm posterior to the bregma) and a small machine screw inserted in the skull. Dental cement was used for fixation of the pump on the skull and connected to the screw for stability. Sham-operated animals underwent the same experimental procedures as described above but screws and minipump were not implanted. The wound was closed with a 3-0 silk suture.
After 24 hours, permanent occlusion of the middle cerebral artery (MCA) was induced using the suture occlusion technique (ZeaLonga et al., 1989; Schäbitz et al., 1996). The right common carotid artery and the right external carotid artery were exposed briefly through a midline neck incision. A 4-0 monofilament nylon suture (Ethicon, Germany) coated with silicon (Bayer, Germany) was inserted through an arteriectomy in the common carotid artery, gently advanced into the internal cartotid artery, and positioned approximately 17 mm from the carotid bifurcation. Using this technique, the tip of the suture occludes unilaterally the proximal anterior cerebral artery, the origins of the MCA, and the posterior communicating artery. A large infarct in the territory of the MCA is typically produced (ZeaLonga et al., 1989; Minematsu et al., 1992). Sham-operated animals underwent the same experimental procedures as described above but the nylon filament was just inserted in the common carotid artery and not advanced. After surgery, the catheters were removed, and the animals were allowed to recover from the anesthesia and given food and water as desired.
After ischemia was induced, the animals were weighed and neurologically assessed daily (rating scale: 0 = no deficit to 4 = spontaneous circling) (Menzies et al., 1992). On the seventh day animals were reanesthetized with chloral hydrate (400 mg/kg) and decapitated. The brains were removed and the appropriate location of the intra-arterial suture was confirmed. The brains were then coronally sectioned into five 2-mm coronal slices, incubated for 30 minutes in a 2% solution of TTC at 37°C, and fixed by immersion in a 10% buffered formalin solution. TTC stains viable brain tissue red, while infarcted tissue remains unstained (Bederson et al 1986). TTC-stained brain sections were photographed using a charge coupled device camera (EDC-1000HR Computer Camera, Electrim Corporation, Princetown, NJ, U.S.A; Slices 1 to 5 = bregma coordinates +2.4, +0.4, −1.6, −3.6, and −5.4, respectively). The infarct volumes were calculated blinded to the treatment given (S.S.), and in each of the five slices the infarct size was quantified using an image processing software package (Bio Scan OPTIMAS, Edmonds, WA, U.S.A.). The infarct area was determined separately for cortex and caudoputamen for slices 1, 2, and 3. Then the infarct areas on each slice were summed and multiplied by slice thickness to give the infarct volumes.
For histological study, animals (n = 3 per group) were transcardially perfusion fixed with 4% paraformaldehyde in 0.1 mol/L phosphate buffer. The brains were removed from the skull, allowed to fix overnight in 4% paraformaldehyde at 4°C and then sectioned at the level of the anterior commissure, hippocampus, and substantia nigra. Slices of 10 μm were obtained from paraffin blocks and stained with hematoxylin and eosin. Other sections from these brains were cut at 30 μm and immunostained for glial fibrillary acid protein using the avidin-biotin peroxidase complex method (Garcia et al. 1993). Hematoxylin and eosin stained brain sections and a corresponding TTC-stained brain slice at the level of the anterior commisure were compared for extent of striatal and cortical infarct volume blinded to the treatment given (M.S.). Hematoxylin and eosin- and glial fibrillary acid protein-stained brain sections were then inspected for morphology.
The values presented in this study are mean ± SD. After acquiring all the data, the randomization code was broken. Volumes of infarction between groups were compared by an unpaired, two-tailed t-test. Continuous data were analyzed by analysis of variance and Scheffe's-test.
RESULTS
Permanent intra-arterial suture occlusion of the MCA causes large infarcts of the lateral cortex and the underlying caudoputamen in the ipsilateral hemisphere (Garcia et al., 1993). In this study the mean infarct volume was smaller (ca. 40%) in BDNF-treated animals than in controls (P < 0.01; unpaired, two-tailed t-test) (Table 1; Figs. 1–3). Cortical infarction was reduced (ca. 70%) in BDNF-treated animals versus controls (P < 0.05, unpaired, two-tailed t-test), whereas caudoputaminal infarction was reduced ca. 25% (N.S.), (Table 1).
Infarct volume in mm3 in BDNF-treated animals and controls
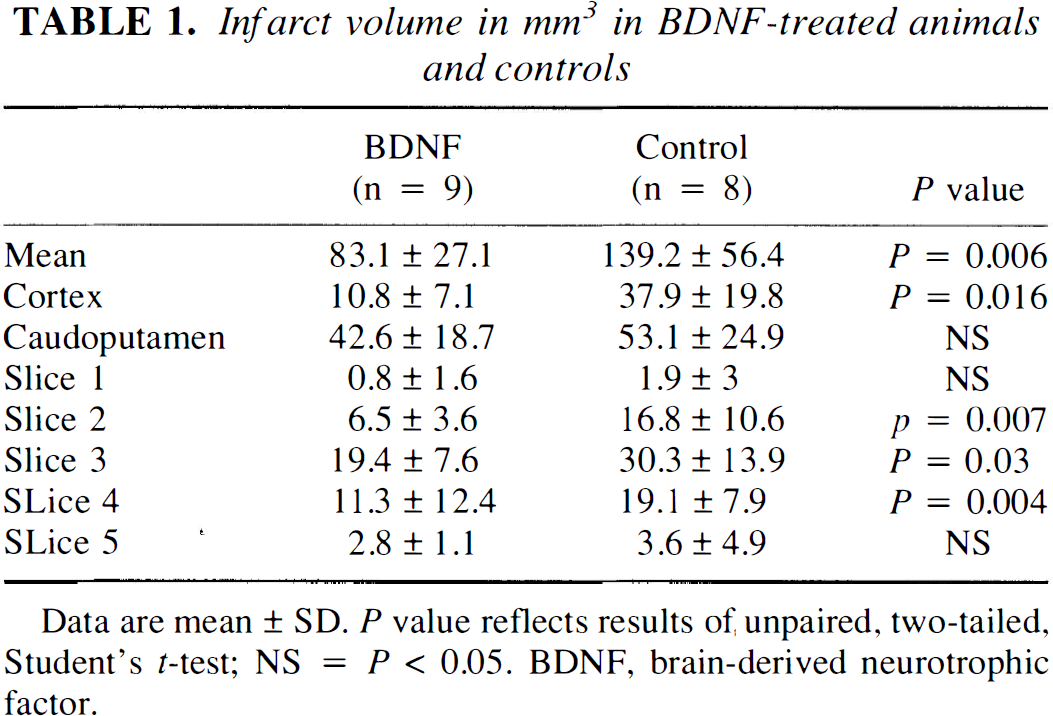
Data are mean ± SD. P value reflects results of unpaired, two-tailed, Student's t-test; NS = P < 0.05. BDNF, brain-derived neurotrophic factor.
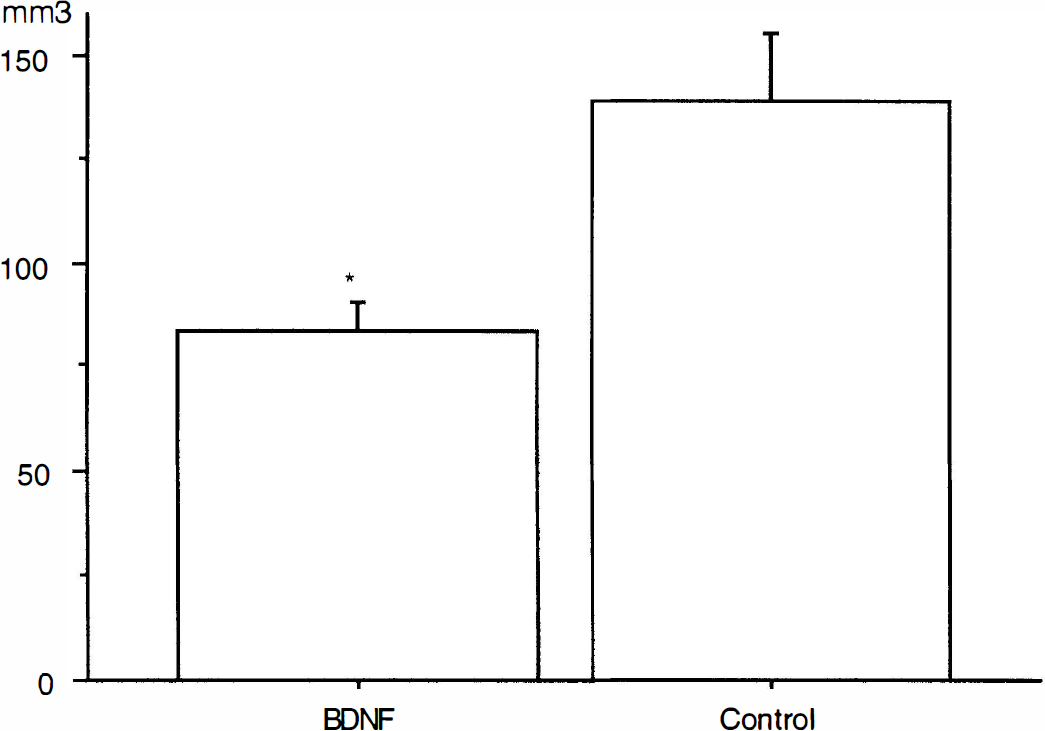
Bar graphs of infarct volume in mm3 in controls and BDNF-treated animals. Bar indicates SD. The infarct volume was significantly smaller in BDNF-treated animals than in controls (*P < 0.01 versus control; unpaired, two-tailed Student's t-test).
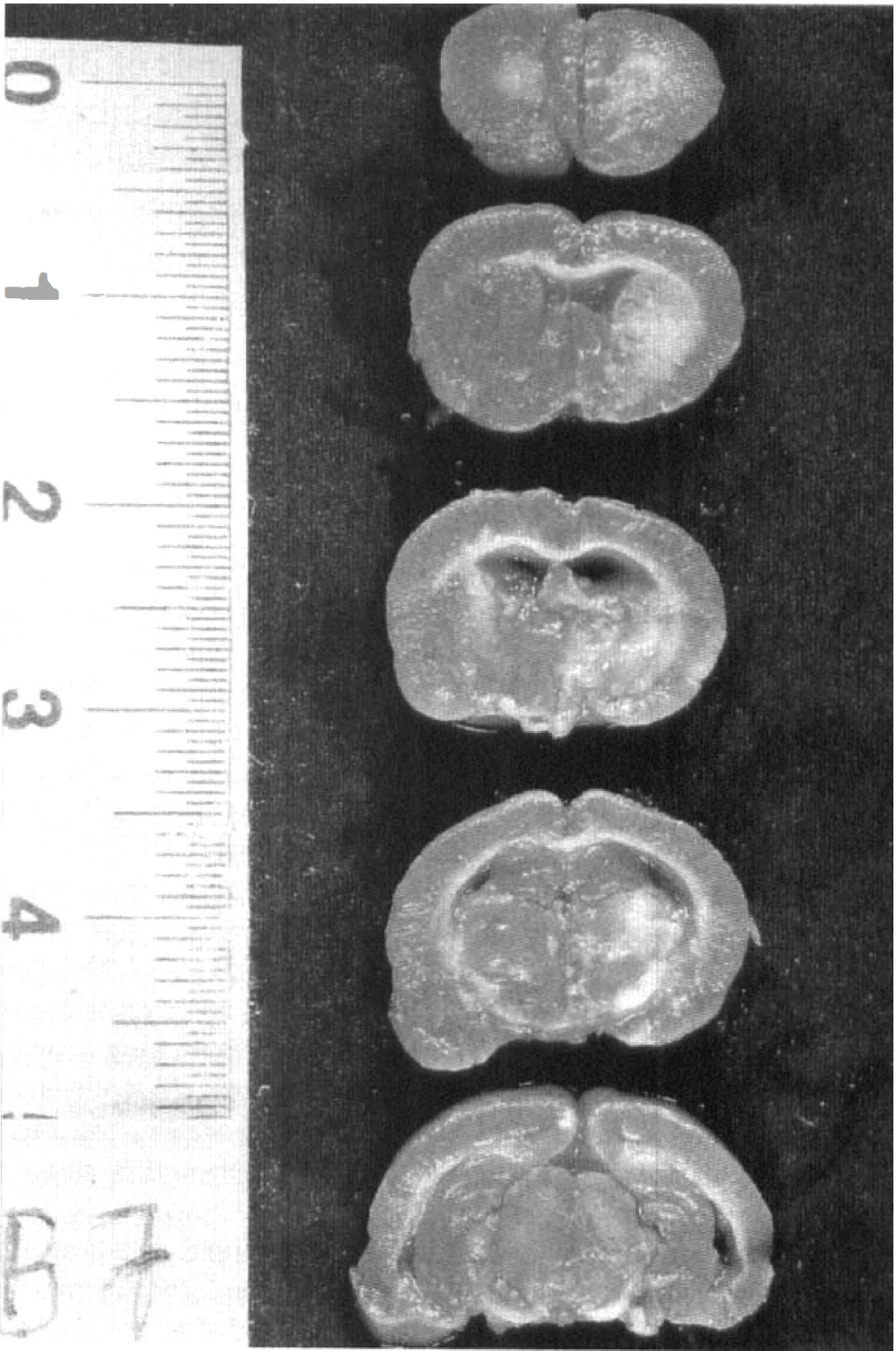
2,3,5-Triphenyltetrazolium chloride-stained brain slices from BDNF-treated animals.
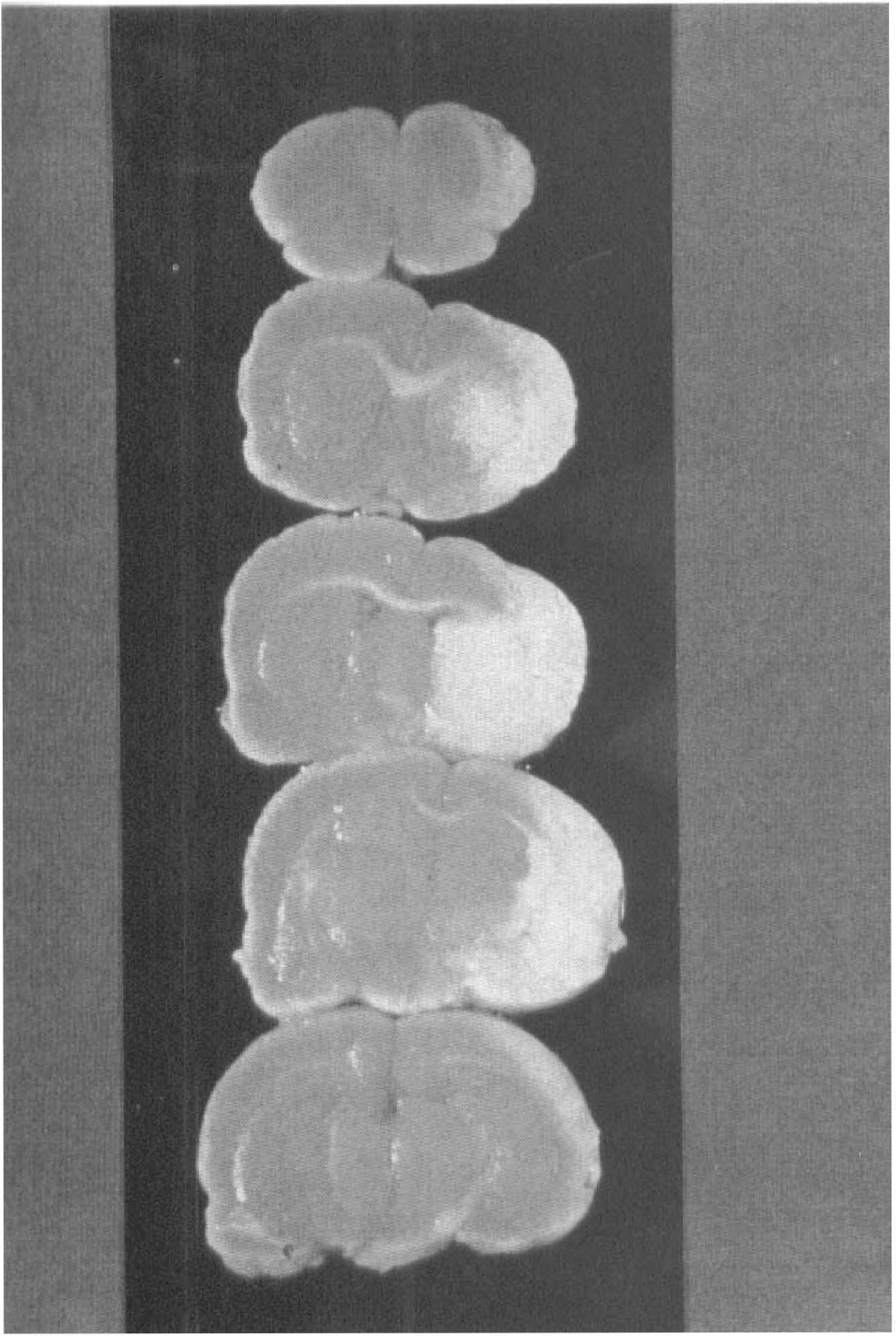
TTC-stained brain slices from control animals. Note the trend toward larger cortical infarctions. Original magnification ×4.
There were no significant differences in the physiological parameters during surgery between BDNF-treated animals and controls (Table 2). There was less neurological deficit after ischemia in the BDNF-treated animals than in the controls (P < 0.05; analysis of variance and Scheffe's test) (Table 3). There were no significant differences in weight loss among the infarcted animals; however weight increased significantly in sham-operated animals as compared with infarcted animals (P < 0.001; analysis of variance and Scheffe's test) (Table 4). One animal in the BDNF-treated group and 2 animals in the control group died 24 to 48 hours after MCAO with signs of massive brain edema and were discarded from the experiment.
Physiological variables in BDNF-treated animals, controls, and sham-operated animals
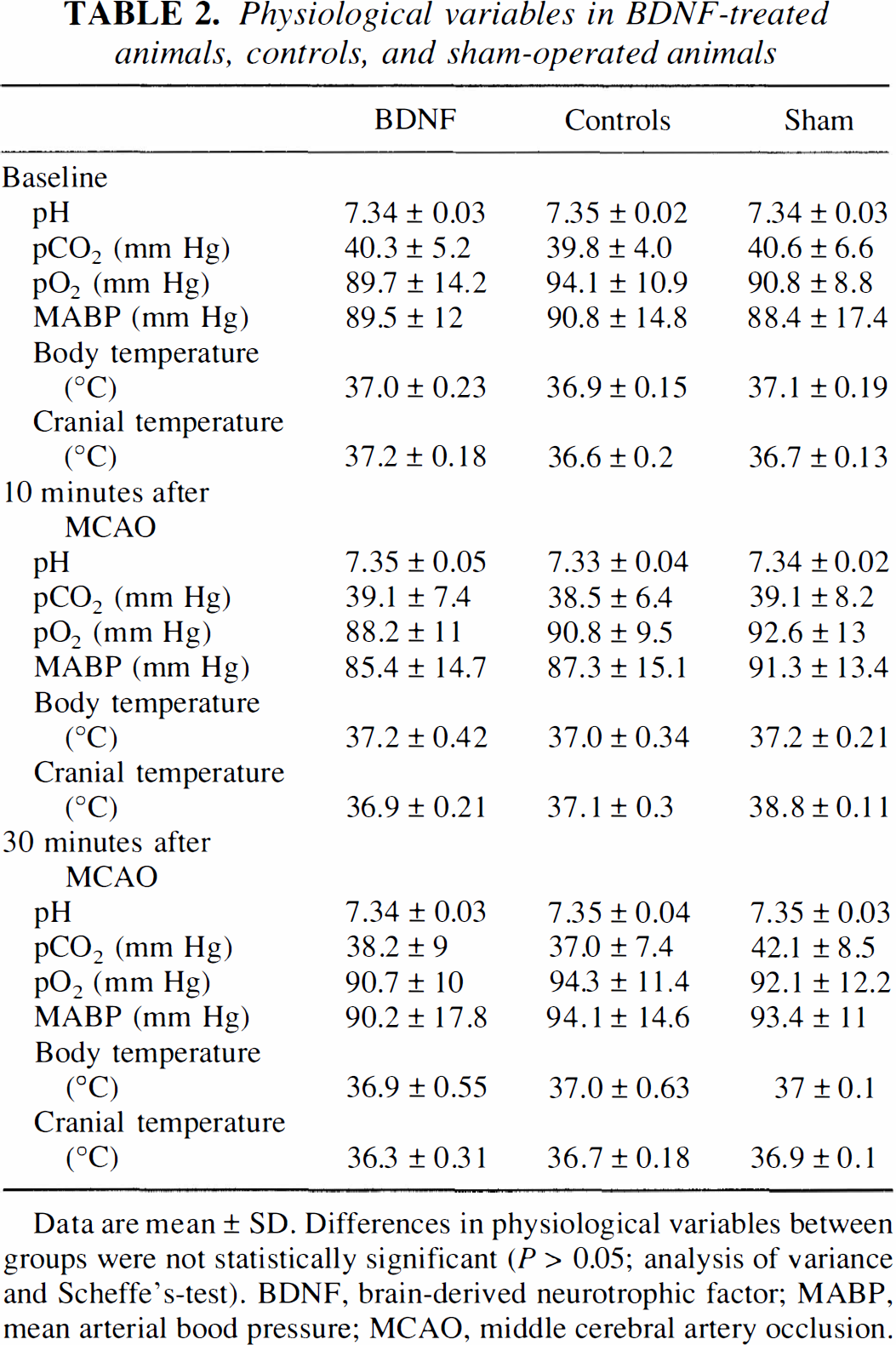
Data are mean ± SD. Differences in physiological variables between groups were not statistically significant (P > 0.05; analysis of variance and Scheffe's-test). BDNF, brain-derived neurotrophic factor; MABP, mean arterial bood pressure; MCAO, middle cerebral artery occlusion.
Neurological score in BDNF-treated animals and controls
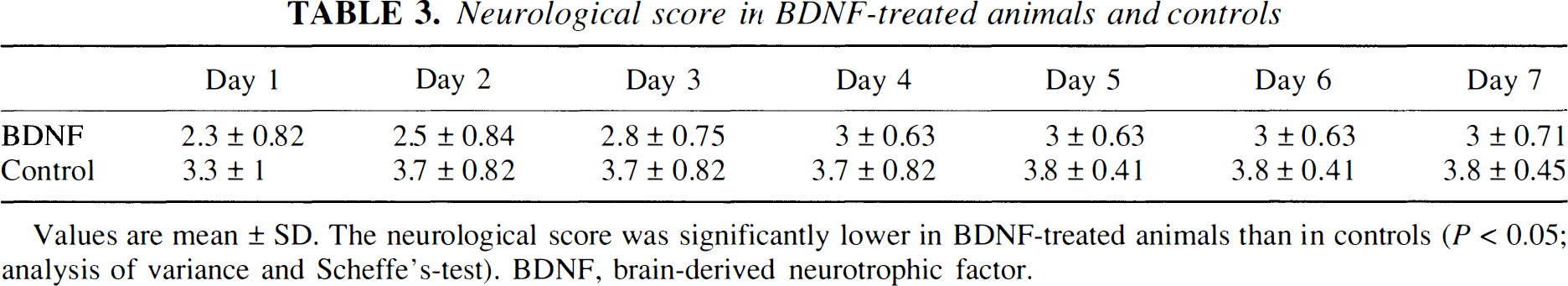
Values are mean ± SD. The neurological score was significantly lower in BDNF-treated animals than in controls (P < 0.05; analysis of variance and Scheffe's-test). BDNF, brain-derived neurotrophic factor.
Weight in % of initial weight in BDNF-treated animals, controls, and sham-operated animals
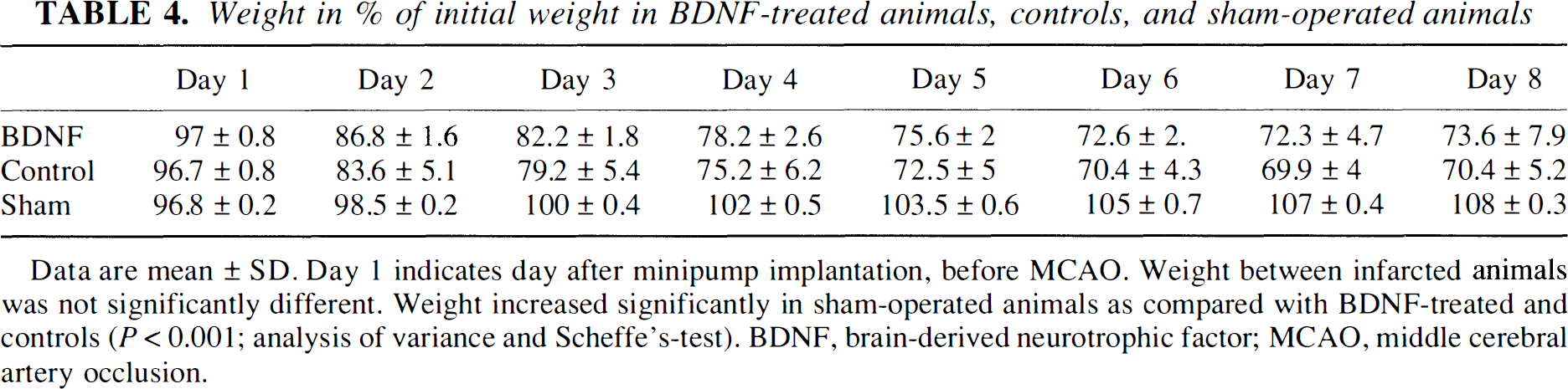
Data are mean ± SD. Day 1 indicates day after minipump implantation, before MCAO. Weight between infarcted animals was not significantly different. Weight increased significantly in sham-operated animals as compared with BDNF-treated and controls (P < 0.001; analysis of variance and Scheffe's-test). BDNF, brain-derived neurotrophic factor; MCAO, middle cerebral artery occlusion.
Extent of striatal and cortical infarction in hematoxylin and eosin-stained brains correlated with corresponding TTC-stained brain sections in both groups. The ischemic lesion, clearly discernible from the surrounding brain, involved the basal ganglia, a portion of the thalamus, and part of the cortex. Pan-necrosis and reabsorption were most prominent in the supraoptic area. Red and ghost neurons, ghost astrocytes, and infiltration by inflammatory cells were found in particular at the margins of the lesion in both groups. Infarcts were, on average, smaller in BDNF-treated animals than in controls. In particular, cortical infarctions appeared visibly smaller in BDNF-treated animals. Moreover, examination of hematoxylin and eosin- and glial fibrillary acid protein-stained brain sections showed no apparent differences in vascular or glial proliferation at the borders of the ischemic lesion in BDNF-treated animals and controls. Examination of hematoxylin and eosin- and glial fibrillary acid protein-stained brain sections in sham-operated animals showed no abnormal histologic findings.
DISCUSSION
The results of this study show for the first time that continuous treatment with intraventricularly administered BDNF for 1 day before and 7 days after ischemia reduced infarct volume and improved neurological outcome in a model of permanent MCAO in rats. The infarct volume was smaller in the cortex than in the caudoputamen.
The dose of BDNF chosen for the current studies (intraventricular administration, 2.1 μg/d) was higher than that used in previous studies of transient forebrain ischemia (Tsukahara et al., 1995; Beck et al., 1994) to maximize any potential infarct-reducing effects. Even with this relatively high dose no significant side effects were observed. In the present study, intraventricular BDNF administration was started 1 day before and continued 7 days after MCAO. Although the rate of BDNF delivery was controlled (0.087 μg/h), we do not know the exact concentration and distribution of BDNF delivered to the ischemic cortex. In healthy brains intraventricular BDNF poorly penetrates the ventricular wall into the surrounding parenchyma (Yan et al., 1994; Dittrich et al., 1996). However, no data are available for distribution of BDNF under traumatical conditions such as MCAO. The suture occlusion of the MCA causes ischemia of the ipsilateral periventricular brain structures (preoptical region, basal ganglia, thalamus) within minutes, whereas infarction of the cortex occurs later (Garcia et al., 1993). Brain-derived neurotrophic factor might penetrate the traumatized periventricular wall and diffuse into the parenchyma. For intrastriatal injection, retrograde transport of BDNF was found within regions known to project to the striatum, such as the frontoparietal cortex, rhomboid, centrolateral, ventrolateral, parafascicular, posterior thalamic areas as well as the pars compacta of the substantia nigra (Mufson et al., 1994).
Protective effects after administration of neurotrophic factors (e.g., BDNF) were observed both in vitro and in vivo after various brain insults. In cell culture, BDNF ameliorates degeneration of hippocampal neurons caused by glutamate (Cheng et al., 1994). Brain-derived neurotrophic factor also protects cultured dentate granule cells and hippocampal, striatal, septal, and cortical neurons against a hypoglycemic insult, evoked by glucose deprivation (Cheng et al., 1991; Cheng et al., 1994; Lindholm et al., 1993; Shimohama et al., 1993; Kokaia et al., 1994; Nakao et al., 1995). Exogeneous administration of BDNF in vivo was shown to ameliorate neuronal necrosis after ischemic insults (Beck et al., 1994; Tsukahara et al., 1994). The mechanisms for the neuroprotective capability of BDNF that cause infarct reduction in focal cerebral ischemia are still unclear.
Glucose deprivation and excitotoxicity with subsequent Ca2+ overload of cells as well as apoptosis and decreased energy reserve in the face of increased requirements (e.g., from spreading depression) are the main causes of, neuronal cell death after ischemia (Choi and Hartley, 1993; Hossmann, 1994; Li et al., 1995). Brain-derived neurotrophic factor protects in vitro neuronal cells against glutamate induced neurotoxicity and the subsequent high intracellular calcium level (Lindholm et al., 1993; Cheng et al., 1994). However, another study showed that treatment by neurotrophins, including BDNF, increased Ca2+ uptake and potentiated the necrotic death of cortical neurons induced by oxygen-glucose deprivation or N-methyl-D-aspartate exposure (Koh et al., 1995). In contrast, in the same study BDNF also attenuates apoptotic cell death induced by serum deprivation or exposure to the calcium-channel antagonist nimodipine. The necrosis- potentiating effect may be caused, at least in part, by increased NMDA receptor-mediated Ca2+ influx. Implying a more sophisticated mechanism Koh et al. (1995) hypothesized that increased concentrations of intracellular free Ca2+ is beneficial to cells undergoing apoptosis, but may also be detrimental to cells already overloaded with Ca2+ because of excitotoxic NMDA receptor overstimulation (Koike et al., 1989; Hartley et al., 1993; Morelli et al., 1986).
In focal cerebral ischemia, critical blood flow rates determine the penumbra, a coronal region intercalated between the necrotic infarct core and the normal brain. This region comprises all tissues that have a cerebral blood flow that is sufficiently reduced so as to put the cells at risk. Penumbral tissues have higher flow rates and can be salvaged by recirculation or by drugs administered either before or during the first hours of MCAO (Memezawa et al., 1992; Hossmann, 1994). Recently, apoptotic cell death after MCAO was defined to the penumbral border of the infarction, where cells are not so rapidly and severely damaged that they can undergo an appropriate cell death rather than necrosis (Li et al., 1995a, b ). Presence and anatomical location of cells showing DNA fragmentation after transient MCAO suggests that apoptosis contributes to the development of the ischemic infarct (Hsu et al., 1993). Brain-derived neurotrophic factor attenuates in vitro apoptosis (Koh et al., 1995) and it may be speculated that a possible prevention of apoptosis by BDNF in vivo contributes to, at least in part, the reduction of infarct size. Moreover, the neuroprotective action of BDNF may depend on neuronal gene transcription and protein synthesis particularly at the margins of focal infarcts. Middle cerebral artery occlusion induces the expression of immediate early genes (c-fos, c-jun) and transcription factors (AP-1) that may activate growth factor gene transcription (Kaku et al., 1993; Kokaia et al., 1995). A BDNF upregulation was found after MCAO in the cortex and the penumbra of the ischemic hemisphere and seems to correlate with resistance to injury in these areas (Kokaia et al., 1995). Exogenous administration of BDNF may support this endogenous effect and reduce infarct size through direct interaction in these mechanisms.
The present study does not include measurements on CBF and we can not exclude that BDNF may have affected CBF. For other growth factors (e.g., basic fibroblast growth factor), an effect on CBF after topical application has been reported. Basic fibroblast growth factor causes a dilation of brain pial arterioles, increases CBF, thereby reduces infarct volume after focal cerebral ischemia. (Rosenblatt et al., 1995; Tanaka et al., 1995).
Another possible mechanism of BDNF-induced reduction in infarct size might be a protection of the degradation of cytoskeletal proteins. Glutamate release and the subsequent Ca2+ overload of neuronal cells caused by ischemia activate calpain-induced proteolysis of the cytoskeletal proteins neurofilament and microtubule-associated protein, the major component of microtubules (Tanaka et al., 1994; Black et al., 1986). Neurotrophins, and particularly nerve growth factor, promote stability and formation of microtubules and inhibit degradation of microtubules through activation of microtubule-associated-protein (Hofer and Barde 1988). Although not completely understood, BDNF might have a function similar to that of nerve growth factor in this system (Hofer and Barde, 1988; Ebendal, 1992).
No changes in physiological parameters such as MABP, blood gases, and body and cranial temperature were seen between treated groups during surgery, although we cannot exclude such changes during preoperative or postoperative periods. No differences in weight were seen between BDNF-treated animals or controls during this relatively short experiment. However, BDNF-treated animals with reduced brain damage should suffer from less loss of weight after ischemia than controls. This possible effect may be hidden by BDNF-induced weight loss after central infusion (Lapchak et al., 1992; Pelleymounter et al., 1994; Dittrich et al., 1996). Weight loss during central infusion of BDNF occurred in brains that were not pathologically altered and may reflect a central appetite suppression perhaps through a hypothalamic seretonergic mechanism (Lapchak et al., 1992; Pelleymounter et al., 1994). Unfortunately, weight behavior during pathophysiological (e.g., ischemic) conditions was not reported (Beck et al., 1994; Tsukahara et al., 1994).
Clearly, further studies are necessary to clarify the phenomenon and mechanisms of reduction in infarct size by BDNF. Potential clinical utility of BDNF for stroke treatment requires demonstration of effectiveness when given intravenously and after cerebral infarction. Nonetheless, our present findings suggest that administration of BDNF may represent a new approach to the treatment of focal cerebral ischemia.
Footnotes
Acknowledgment:
Brain-derived neurotrophic factor was provided by Amgen Inc., Thousand Oaks, CA, U.S.A.