
Abstract
Preischemic hyperglycemia or superimposed hypercapnia exaggerates brain damage caused by transient forebrain ischemia. Because high regional levels of brain-derived neurotrophic factor (BDNF) protein correlate with resistance to ischemic damage, we studied the expression of BDNF mRNA using in situ hybridization in rats subjected to 10 minutes of forebrain ischemia under normoglycemic, hyperglycemic, or hypercapnic conditions. Compared with normoglycemic animals, the increase of BDNF mRNA in dentate granule cells was attenuated and that in CA3 pyramidal neurons completely prevented in hyperglycemic rats. No ischemia-induced increases of BDNF mRNA levels in the hippocampal formation were detected in hypercapnic animals. Hyperglycemic and hypercapnic rats showed transiently decreased expression of BDNF mRNA levels in the cingulate cortex, which was not observed in normoglycemic animals. The results suggest that suppression of the BDNF gene might contribute to the increased vulnerability of the CA3 region and cingulate cortex in hyperglycemic and hypercapnic animals.
Brain-derived neurotrophic factor (BDNF) is a member of a family of neurotrophic molecules called the neurotrophins (Lindsay et al., 1994). It is widely but unevenly distributed in the brain, with the highest mRNA levels in the hippocampal formation, particularly in the dentate gyrus and CA3 region (Ernfors et al., 1990; Hofer et al., 1990). Although its role in the mature brain is poorly understood, BDNF may have a neuroprotective effect. This contention is supported by the fact that various brain insults lead to an upregulation of the BDNF gene. These encompass epileptic seizures (Ernfors et al., 1991; Isackson et al., 1991), hypoglycemic coma (Lindvall et al., 1992), cortical spreading depression (Kokaia et al., 1993b), traumatic brain injury (Ballarín et al., 1991; Hughes et al., 1993; Mudó et al., 1993), and global as well as focal brain ischemia (Lindvall et al., 1992; Comelli et al., 1993; Takeda et al., 1993; Kokaia et al., 1995). Furthermore, BDNF protects several types of cultured neurons against hypoglycemic damage in vitro (Cheng and Mattson, 1994; Kokaia et al., 1994; Nakao et al., 1995b), and intraventricular injection of BDNF ameliorates neuronal necrosis in the hippocampal CA1 region after transient forebrain ischemia in the rat (Beck et al., 1994).
A recent study from our laboratories casts additional light on the concept of a neuroprotective role for BDNF (Kokaia et al., 1996). In these experiments, the relationship between selective neuronal necrosis and the regulation of regional BDNF mRNA and protein levels was explored after 10 minutes of transient forebrain ischemia in the rat. In support of the concept were results demonstrating elevated BDNF gene expression in the dentate gyrus and CA3 sector, two structures that are relatively resistant to ischemic insults under normoglycemic conditions, with a transient increase in BDNF protein. Two vulnerable regions (the CA1 region and parietal cortex) failed to show an increased gene expression, and the protein levels actually decreased. Furthermore, the highest basal BDNF protein concentrations were found in the dentate gyrus and CA3 region, whereas the levels in the CA1 region and parietal cortex were markedly lower.
The objective of the present study was to explore the possibility that the severity of the insult influences BDNF gene expression after transient forebrain ischemia. We used a similar experimental protocol as in our previous study (Kokaia et al., 1996), but the insult was aggravated by preischemic hyperglycemia or superimposed hypercapnia. Preischemic hyperglycemia is known to increase brain damage caused by transient forebrain ischemia (for recent reviews, see Siesjö, 1988; Siesjö et al., 1996). Critical plasma glucose concentrations are 10 to 12 mmol/L; above these values damage is incurred, not only by vulnerable regions such as the CA1 region and the neocortex, but also by normally resistant structures such as the CA3 region and the cingulate cortex (Li et al., 1994; Li et al., 1995b). Recent results indicate that most of the aggravating effect of hyperglycemia is caused by an exaggerated intra- and extracellular acidosis. In support, if a comparable exaggeration of the intraischemic acidosis is induced in normoglycemic animals by superimposed hypercapnia, the ischemic damage is aggravated (Katsura et al., 1994). Furthermore, hypercapnia also aggravates brain damage caused by hypoglycemic coma (Kristián et al., 1995). For these reasons, we included normoglycemic groups with superimposed hypercapnia.
MATERIALS AND METHODS
Animals and study design
Male Wistar rats (Møllegaard's Breeding Centre, Copenhagen, Denmark) weighing 295 to 325 g were used. The rats were housed under 12-hours light/12-hours dark conditions with ad libitum access to food and water. Before the ischemic insult or sham procedure, the animals were fasted overnight. Brain-derived neurotrophic factor mRNA expression was analyzed using in situ hybridization in three groups of animals (n = 16 in each group), which had been subjected to 10 minutes of normoglycemic–normocapnic (“normoglycemic”), hyperglycemic–normocapnic (“hyperglycemic”), or normoglycemic–hypercapnic (“hypercapnic”) forebrain ischemia and sacrificed after 1, 2, 4, or 18 hours of recirculation (n = 4 at each time point). Four control rats subjected to a sham procedure were included in each group for each time point.
Procedure for cerebral ischemia
Global forebrain ischemia was induced by bilateral common carotid artery occlusion with simultaneous hypotension as described previously (Smith et al., 1984). Briefly, animals were anesthetized with 3.5% halothane in N2O/O2 (70:30), intubated, and then artificially ventilated with the halothane concentration lowered to 1.0% to 1.5%. The common carotid arteries were isolated, and loose ligatures were placed around them. The tail artery was cannulated to monitor physiologic variables, and stainless steel electrodes were inserted in skull muscles for EEG recording. The body temperature was continuously measured and animals were maintained normothermic in all experimental conditions. Hyperglycemia (plasma glucose 20 to 25 mmol/L) was induced by intravenous infusion of a 25% glucose solution during the preischemic period. Normoglycemic animals were given the same volume of saline. For induction of hypercapnia, the animals were ventilated with 50% CO2, starting 10 to 15 minutes before ischemia and continuing throughout the ischemic period (Katsura et al., 1994). After bilateral occlusion of the common carotid arteries for 10 minutes, combined with hypotension (mean arterial blood pressure = 50 mm Hg) by exsanguination, circulation was restored by removal of the occluding clamps and reinfusion of blood. Control rats were subjected to a sham procedure including induction of hyperglycemia or hypercapnia, but the common carotid arteries were not occluded and blood pressure was only briefly reduced to 80 mm Hg.
In situ hybridization and image analysis
The animals were deeply anesthetized and decapitated, and the brains were immediately removed and frozen in powdered dry ice. Coronal cryostat sections (14 μm), taken through the dorsal hippocampus, were fixed with 4% paraformaldehyde and hybridized overnight at 42°C (for details on procedure see Kokaia et al., 1993a). Hybridization buffer (50% deionized formamide, 0.3 mol/L NaCl, 20 mmol/L Tris-HCl, 0.5 mg/mL yeast tRNA, 1× Denhardt's solution, 0.1 mg/mL poly A, 1 mmol/L EDTA, 0.1 mol/L dithiotreitol, and 10% dextran sulfate) contained 2.5 × 106 cpm/mL 35S-labeled BDNF cRNA probe (Kokaia et al., 1993a). After hybridization, sections were washed and treated with RNase A. Sections were opposed to β-max X-ray film for 10 to 14 days. To visualize radioactive grains, sections were dipped in Ilford K5 emulsion for 4 to 6 weeks and, after being developed, counterstained with hematoxylin and eosin. Quantification of hybridization signals was performed by computerized image analysis using Image 1.57 software (Wayne Rasband, National Institutes of Health, Bethesda, MD) as described before (Kokaia et al., 1995). Gray levels from standards were used in a third-degree polynomial calibration to obtain equivalent values of tissue radioactivity (nCi/g).
All values are presented as mean ± SD. Evaluation of differences in BDNF mRNA expression between animal groups was performed using one-way analysis of variance followed by Bonferroni/Dunn post-hoc test with significance set at P < 0.05.
RESULTS
Before the ischemia, the hyperglycemic animals had a mean plasma glucose value of 21.7 ± 0.6 mmol/L, compared with 5.1 ± 0.4 mmol/L in the normoglycemic group. In the hypercapnic animals, P
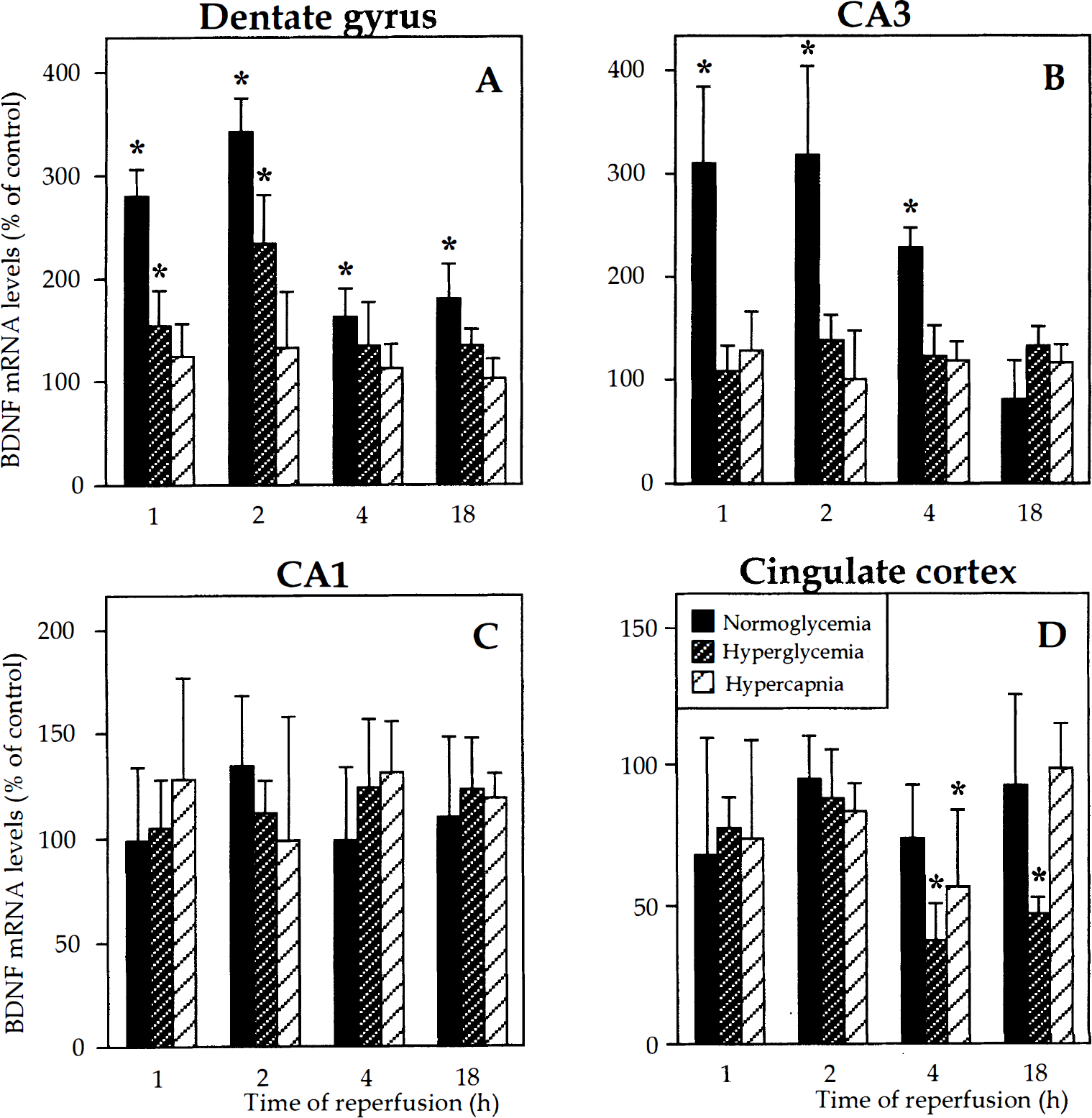
Ten minutes of global forebrain ischemia led to elevated BDNF mRNA expression in the dentate granule cells of normo- and hyperglycemic animals (Figs. 1A and 2G–J). In both groups, significantly increased levels of BDNF mRNA were detected at 1 hour of reperfusion, and at 2 hours had reached maximum values (310% and 233% of control in normo- and hyperglycemic animals, respectively). After 4 and 18 hours of reperfusion, BDNF mRNA expression in dentate granule cells of normoglycemic animals had decreased, but was still significantly higher compared with that in controls. In contrast, BDNF mRNA levels in dentate granule cells of hyperglycemic animals tapered off more rapidly and reached control values by 4 and 18 hours postischemia. No changes of BDNF mRNA expression were detected in dentate granule cells of animals subjected to hypercapnia.
Levels of brain-derived neurotrophic factor (BDNF) mRNA in the dentate granule cell layer (
Dark-field photomicrographs of autoradiograms (
In CA3 pyramidal neurons, BDNF mRNA expression was significantly increased only in normoglycemic animals (Figs. 1B and 2G). Maximum levels occurred at 1 to 2 hours postischemia, and expression then gradually decreased and had returned to control values by 18 hours. Irrespective of whether the ischemic insult was delivered under normoglycemic, hyperglycemic, or hypercapnic conditions, no changes of BDNF mRNA levels were detected in the CA1 pyramidal layer at any time point examined (Figs. 1C and 2G, 2I, and 2K).
Under normoglycemic conditions, the forebrain ischemia did not cause any alteration of BDNF mRNA expression in the cingulate cortex (Fig. 1D). However, when the ischemia was combined with hyperglycemia, the levels of BDNF mRNA in this cortical region at 4 hours of reperfusion were markedly reduced compared with control (37% of control) and remained significantly decreased also at 18 hours (47% of control). A transient reduction of BDNF mRNA expression was also detected in the cingulate cortex of hypercapnic animals (57% of control) after 4 hours of reperfusion (Fig. 1D). Hyperglycemia and hypercapnia did not lead to significant changes of BDNF mRNA expression in any examined structure of the control animals not subjected to ischemia.
DISCUSSION
The expression of the BDNF gene after transient forebrain ischemia was clearly influenced by the characteristics of the insult. The normoglycemic animals exhibited a more long-lasting and marked increase of BDNF mRNA levels in dentate granule cells than hyperglycemic and, in particular, hypercapnic animals, which showed no upregulation of BDNF gene expression after ischemia. In the CA3 region, BDNF gene activation was observed only under normoglycemic conditions. In contrast to normoglycemic animals, hyperglycemic and hypercapnic rats showed decreased expression of BDNF mRNA levels in the cingulate cortex.
Recent experimental data have indicated that BDNF can enhance excitatory synaptic transmission under normal, physiologic conditions (Knipper et al., 1993; Lohof et al., 1993; Leßmann et al., 1994; Kang and Schuman, 1995). Hypothetically, high levels of BDNF could therefore enhance excitotoxic damage after brain insults. However, intraventricular injection of BDNF significantly increases the survival of CA1 cells after forebrain ischemia (Beck et al., 1994), and BDNF protects hippocampal, cortical, and striatal neurons in vitro against excitotoxic lesions (Cheng and Mattson, 1994; Nakao et al., 1995a), arguing in favor of a neuroprotective role. Furthermore, in our previous study, we demonstrated that high BDNF protein levels in the dentate gyrus and CA3 region under basal conditions and after normoglycemic ischemia correlated with resistance to ischemic brain damage. In contrast, the vulnerable CA1 region and parietal cortex had low basal BDNF protein levels and showed a further reduction after ischemia (Kokaia et al., 1996). In relation to the possible neuroprotective role of BDNF, it is of clear interest that both hyperglycemia and hypercapnia attenuated the elevated BDNF mRNA expression in the CA3 region observed after normoglycemic ischemia and led to decreased BDNF mRNA levels in the cingulate cortex. Although these regions are usually spared in normoglycemic animals, they are frequently recruited in the damage process in hyperglycemic and hypercapnic animals (Katsura et al., 1994; Li et al., 1994; Li et al., 1995b). Thus, the suppressant action of hyperglycemia and hypercapnia on BDNF gene expression in CA3 and cingulate cortex correlated to the vulnerability of these regions to the ischemic damage. Recent studies have shown that changes of BDNF mRNA levels induced by seizure activity or transient forebrain ischemia in most cases give rise to the presumed alterations of BDNF protein levels (Nawa et al., 1995; Kokaia et al., 1996; Elmér et al., 1997). It is conceivable, therefore, that hyperglycemia and hypercapnia influence not only BDNF gene expression but also protein levels after the ischemia. However, despite attenuation or lack of increase of BDNF mRNA upregulation in the dentate granule cells of hyperglycemic and hypercapnic animals, respectively, there is no cell death in this layer (Katsura et al., 1994; Li et al., 1996). This may, at least partly, be explained by an already high BDNF protein level in the dentate gyrus under basal conditions.
As remarked, the aggravation of ischemic brain damage by hyperglycemia and hypercapnia is probably related to reduction of extra- and intracellular pH (Siesjö et al., 1993; Katsura et al., 1994). It should be recalled, though, that superimposed hypercapnia does not duplicate all the effects of hyperglycemia. For example, hypercapnic animals fail to develop postischemic seizures and do not show damage to the substantia nigra pars reticulata or the cingulate cortex (Katsura et al., 1994; Li et al., 1996). It seems justified, therefore, to discuss separately mechanisms that are common to hyperglycemia and hypercapnia, and those which could explain differences in effects.
The question arises whether acidosis could suppress BDNF gene expression by influencing calcium influx into cells. Entry of calcium ions has been shown to activate BDNF gene transcription in hippocampal and cortical neurons (Zafra et al., 1990; Zafra et al., 1991; Zafra et al., 1992; Ghosh et al., 1994), but the intracellular mechanisms mediating this action of the Ca2+ signal are not known in detail (Ghosh and Greenberg, 1995). The mode of entry of Ca2+ influences the induction of BDNF mRNA expression. In cultured cortical neurons, activation of N-methyl-
Although both preischemic hyperglycemia and superimposed hypercapnia suppressed the BDNF mRNA levels observed after normoglycemic ischemia, there were some differences. (1) Hypercapnia totally prevented the ischemia-induced increase of BDNF mRNA expression in dentate granule cells. In contrast, ischemia under hyperglycemic conditions still led to activation of the BDNF gene in these cells, although less pronounced and for a shorter time compared with that in the normoglycemic animals. (2) The reduction of BDNF mRNA levels in the cingulate cortex was more marked and long-lasting in hyperglycemic as compared with hypercapnic animals. At least in the parietal cortex, extracellular pH is reduced to similar levels during ischemia in hyperglycemic and hypercapnic animals (Katsura et al., 1994). Therefore, we need to involve other factors to explain differences between these conditions in terms of BDNF gene regulation. One possible explanation is that in hypercapnic animals, pH is reduced before the ischemia is induced, whereas in hyperglycemic rats the lowering of pH is caused by the ischemia. However, it is also possible that the differences in results are related to corresponding differences in molecular mechanisms, notably in the effect of acidosis on free radical production. At least in vitro, a lowering of pH to 6.1 to 6.2 by addition of lactic acid gives a higher rate of iron-catalyzed free radical production than if pH is lowered by hypercapnia (Rehncrona et al., 1989; Siesjö et al., 1996).
In conclusion, when hyperglycemia or hypercapnia is added to the ischemic insult, which aggravates the brain damage, the expression of the BDNF gene is suppressed in vulnerable areas. Our study provides further support for the hypothesis that endogenous BDNF might have a neuroprotective effect after brain insults.