
Abstract
Programmed cell death occurs in ischemia when cell surface death receptors (DRs) are stimulated by death-inducing ligands (DILs). Matrix metalloproteinases are extracellular matrix-degrading enzymes involved in the shedding of DRs and DILs from the cell surface. Tissue inhibitor of metalloproteinase-3 (TIMP-3), which is bound to the extracellular matrix, has been shown to promote apoptosis in cancer cell lines by inhibiting cell surface sheddases. Since apoptosis is an important mechanism of cell death in ischemia, the authors hypothesized that TIMP-3 would be expressed in ischemic neurons that are undergoing programmed cell death. Spontaneously hypertensive rats had a 90-minute middle cerebral artery occlusion with reperfusion. Transcription of TIMP-3 mRNA was measured by quantitative reverse transcription–polymerase chain reaction at 2, 6, 24 and 48 hours after reperfusion. Western blots were used to measure TIMP-3 protein expression. Spatial distribution and production of TIMP-3 was studied by immunohistochemistry at 3, 24, and 48 hours, 5 days, and 3 weeks. DNA fragmentation in cells dying by necrosis and apoptosis was identified with terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL). After 2 hours of reperfusion, TIMP-3 mRNA increased significantly in both ischemic and nonischemic hemispheres. Western blot analysis confirmed the identity of the TIMP-3, which appeared to be increased on the ischemic side. After 3 hours of reperfusion, TIMP-3 immunostaining was increased in neurons on the ischemic side, and by 24 hours the majority of the ischemic neurons were TIMP-3–positive. Dual-fluorescence staining for TUNEL and TIMP-3 showed that they were coexpressed in many neurons. The results suggest that ischemic neurons express TIMP-3, which may be inhibiting sheddases. The authors propose that TIMP-3 facilitates cell death in ischemic neurons. Further studies are needed to identify the sheddases inhibited by the TIMP-3, and on the effect of inhibition of matrix metalloproteinases on cell death mechanisms.
Keywords
Ischemia induces molecular cascades that lead to death of neurons by necrosis and programmed cell death (PCD) (Chopp et al., 1996; Endres et al., 1998; Dirnagl et al., 1999; Graham and Chen, 2001). Cell death occurs when cell surface death receptors (DRs) are engaged by death-inducing ligands (DILs) with the activation of the caspase cascade (Ashkenazi and Dixit, 1998). Ischemia induces the DRs, such as Fas (CD95/APO-1) and tumor necrosis factor-α (TNF-α) receptor (Matsuyama et al., 1994; Martin-Villalba et al., 1999; Rosenbaum et al., 2000). Metalloproteinases (MPs), which include the matrix metalloproteinases (MMPs) and ADAMs (a disintegrin and metalloproteinase domain) protease gene families, act as sheddases in the removal of cell surface DRs and DILs (Schlondorff and Blobel, 1999). The MMPs that act as sheddases include stromelysin-1 (MMP-3) and matrilysin (MMP-7), while the ADAMs that have a sheddase function include ADAM-17 or TNF-α–converting enzyme (TACE) and ADAM-10 (Schlondorff and Blobel, 1999; Powell et al., 1999). Four endogenous tissue inhibitors of metalloproteinases (TIMPs) regulate the activity of the MPs (Brew et al., 2000). TIMP-3, which is bound to the extracellular matrix, has additional functions, including the facilitation of PCD (Leco et al., 1994; Brew et al. 2000). Overexpression of TIMP-3 in colon cancer cells caused apoptosis (Baker et al., 1999). TIMP-3 has been shown to facilitate cell death by the inhibition of TNF-α–receptor cleavage (Smith et al., 1997). Two MMPs have been found to have a sheddase function, namely, stromelysin-1 (MMP-3), which removed the Fas ligand from synovial cells in patients with rheumatic arthritis, and matrilysin (MMP-7), which removed the Fas ligand in prostate cancer cells (Powell et al. 1999; Matsuno et al., 2001). These studies suggested the hypothesis that TIMP-3 affects apoptosis in the brain by blocking the action of the sheddases. In an earlier study, we observed MMP-3 expression in ischemic neurons and in activated microglia, whereas MMP-7 was not detected (Rosenberg et al., 2001). Sheddases, DRs, and DILs, therefore, are present in the central nervous system, but no information is available on the role of TIMP-3 in cerebral ischemia. To test the hypothesis, we exposed adult spontaneously hypertensive rats (SHR) to 90 minutes of transient ischemia with reperfusion for various times. We measured tissue levels of TIMP-3 mRNA by quantitative reverse transcription–polymerase chain reaction (RT–PCR), and TIMP-3 protein expression by Western immunoblot analysis. Colabeling with fluorescent antibodies specific for neurons and astrocytes identified cells expressing TIMP-3 in the ischemic regions. To determine the cells that were dying by either apoptosis or necrosis, we colabeled cells for TIMP-3 and terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL).
MATERIALS AND METHODS
Transient middle cerebral artery occlusion with reperfusion
The study was approved by the University of New Mexico Animal Care Committee and conformed to the NIH Guidelines for use of animals in research. Spontaneously hypertensive rats (SHR), weighing 280 to 320 g, were anesthetized with 1.5% halothane in 70% nitrous oxide and 30% oxygen. Temporary middle cerebral artery occlusion (MCAO) was done by insertion of an intraluminal nylon suture with a bulb on the end (Longa et al., 1989). Neck vessels were exposed through a midline incision, and branches of the right external carotid were isolated and ligated. A 6-0 silk suture was loosely tied around the external carotid artery stump. A 4-0-monofilament nylon suture was introduced into the external carotid and advanced to occlude the middle cerebral artery. Reperfusion was done by slowly pulling the thread back. Reperfusion times were varied for the different experimental measurements.
Real-time quantitative reverse transcription–polymerase chain reaction for tissue inhibitor of metalloproteinase-3
Comparison of the time course of mRNA for TIMP-3 was done on SHR rats exposed to 90 minutes of ischemia and periods of reperfusion of 2, 6, 24, and 48 hours. Four rats were studied at each time point for comparison with sham-operated animals. Quantitative real-time RT–PCR with the GeneAmp 5700 (PerkinElmer Life Sciences, Boston, MA, U.S.A.) was used to measure mRNA for TIMP-3. Tissue from the caudate and cortex was homogenized in Trizol reagent (Invitrogen, Carlsbad, CA, U.S.A.) for extraction of total RNA, according to manufacturer's protocol. First-strand cDNA was generated from RNA, according to the manufacturer's instructions for the TaqMan Reverse Transcriptase Kit (PerkinElmer Applied Biosystems). In a 40-μL reaction, 0.5 μg RNA was used with 1 x TaqMan RT buffer, 5.5 mmol/L MgCl2, 500 μmol/L of each dNTP, 2.5 μmol/L Random Hexamers, 0.4U/L of RNAse inhibitor, 1.25U/L of reverse transcriptase and diethylpyrocarbonate water. Thermal cycling parameters were 10 minutes at 25°C, 30 minutes at 48°C, and 5 minutes at 95°C. Primers and probe for TIMP-3 were designed using Primer Express software (PerkinElmer) from a rat TIMP-3 sequence (Gene Bank U27201). Each sample was also run with primers and probe for a “housekeeping gene,” ribosomal protein rpl-32, based on published sequences (Wang et al., 2000). Primers (Integrated DNA Technologies, Inc., Coralville, Iowa, U.S.A.) and probes (PerkinElmer) were synthesized. Probes had a reporter dye, 6-carboxyfluorescein (FAM), at the 5′ end, and a quencher dye, 6-carboxy-tetramethyl-rhodamine (TAMRA), at the modified thymine at position 651 of the 3′ end. The primers and probe for rat TIMP-3 are as follows: TIMP-3 Forward Primer (613-632): 5′ AGC ATC AGC AAT GCC ACA GA-3′, TIMP-3 Reverse Primer (679-663): 5′ CAG CGG GAT GGG AAG GA-3′, TaqMan Probe (634-658): 5′ CCC TGA ACC CAG ACC TGT CCC ACC T-3′.
The concentrations of primers and probes in the PCR reaction mix for both TIMP-3 and rpl-32 were optimized before running all samples. Ten samples were run as controls of the reverse transcription reaction, with template RNA but without enzyme, to determine whether amplification occurred from any contaminating DNA. Plasmids for both TIMP-3 and rpl-32 were created by inserting the purified amplicon into a pGEM T-easy TA cloning vector (Promega, Madison, WI, U.S.A.) to permit quantification and comparisons across reaction plates. The region of the plasmid containing the TIMP-3 PCR fragment was then sequenced. Final PCR comparison reactions were run in a 96-well plate that included reactions containing 10-fold dilutions using the TIMP-3 plasmid and the rpl-32 plasmid as templates. This allowed for the creation of a standard quantitative curve. The standard curve for the TIMP-3 amplicon ranged from 6.11 fg to 61.1 pg, or from 3.26 to 7.26 log molecules. The standard curve for the rpl-32 plasmid ranged from 36.2 fg to 362 pg, or 4.03 to 8.03 log molecules. Each plate included control with no template, to check for contamination, and the unknown samples that were split and then run with primers and probe for the gene of interest (TIMP-3) and with primers and probes for the endogenous control (rpl-32). In a 50-μL reaction, 2 μL of sample first-strand reaction, or the equivalent of 25 ng of the original RNA, was used. All samples, controls, and standard curve points were run in triplicate. TIMP-3 primers (both forward and reverse) were used at a final concentration of 300 nmol/L, and the probe at 100 nmol/L. The rpl-32 primers were used at a final concentration of 600 nmol/L, and the probe at 200 nmol/L. The PCR incubation and cycling parameters were 50°C for 2 minutes, 10 minutes at 95°C, then 40 cycles of 15 seconds at 95°C, and 1 minute at 60°C.
Data was expressed as copy number based on the plasmid standard curves. Dividing the copy number of the TIMP-3 reaction by the copy number of the equivalent rpl-32 endogenous control normalized sample data. Data was analyzed statistically, using Prism (GraphPad Software, San Diego, CA, U.S.A.).
For immunohistochemical staining, air-dried and blocked (PBS with 0.1% Tween, 1% bovine serum albumin and 5% normal goat serum) slides were incubated overnight at 4°C with the primary antibodies. Diaminobenzidine (DAB; DAKO Corporation, Kyoto, Japan) staining was done by incubating the sections for 90 minutes with either biotinylated goat antirabbit or goat antimouse immunoglobulin G (Jackson Immuno-Research Laboratories, Inc.). Sections were rinsed, incubated with 0.3% hydrogen peroxide–methanol to quench endogenous tissue peroxidases, and incubated with avidin and biotinylated horseradish peroxidase (ABC Elite Kit; Vector Laboratories, Burlingame, CA, U.S.A.) for 30 minutes at room temperature. Immunostaining was visualized with DAB and sections counter-stained with hematoxylin (Sigma). Slides were dehydrated through alcohols and xylene and mounted in Permount (Sigma-Aldrich Corp., St. Louis, MO, U.S.A.).
Primary antibodies tested were rabbit antihuman TIMP-3 (Chemicon). Astrocytes were identified with either a polyclonal rabbit antibovine or a monoclonal mouse glial fibrillary acidic protein (Accurate Chemical & Scientific Corp. [Westbury, NY, U.S.A.] and Sigma, respectively). Microglial cells were identified, using a monoclonal mouse antirat Ox-42 antibody (Harlan Sera-Lab, Crawley Down, Sussex, U.K.) and neurons identified with mouse antineuronal nuclear antigen, NeuN (Chemicon). Hoechst 33342 (Sigma) was used as a nucleic acid marker on some tissue sections at 1 μg/mL for 2 minutes at room temperature after the incubation of secondary antibody. Fluorescent staining of tissue was done in sections that were incubated with secondary antibodies, goat antirabbit or goat antimouse immunoglobulin G labeled with fluorescein isothiocyanate conjugate (FITC) or cyanine (CY3; JacksonImmunoresearch), rinsed and mounted in Prolong Antifade (Molecular Probes, Eugene, OR, U.S.A.).
Four animals from the 24-hour reperfusion time point were immunostained for TIMP-3 in combination with either TUNEL (NeuroTacsII; Trevigen, Gaithersburg, MD, U.S.A.). Secondary antibodies conjugated with either FITC or CY3 were used to colocalize the substrates of interest. Immunostained slides were photographed using a SPOT digital camera (on an Olympus BX40 microscope; Olympus America Inc., Melville, NY, U.S.A.) or with a fluorescent microscope (Nikon Labophot; Nikon USA, Melville, NY, U.S.A.). Composite pictures were made with Adobe Photoshop 6.0.
RESULTS
Time course of real-time quantitative reverse transcription–polymerase chain reaction
The mRNA expression was linear in the controls and the plasmid studies, confirming that the mRNA was from TIMP-3 (data not shown). Figure 1 shows the levels of TIMP-3 mRNA in the tissues as measured by quantitative RT–PCR. After 2 hours of reperfusion TIMP-3 mRNA significantly increased in both ischemic and nonischemic hemispheres. A second increase in mRNA was seen at 48 hours in the nonischemic side, and no further increases were seen in the ischemic side. Values of mRNA in the caudate and the cortex were similar in the ischemic hemisphere, but the rise at 48 hours in the nonischemic side was significant only in the cortex.
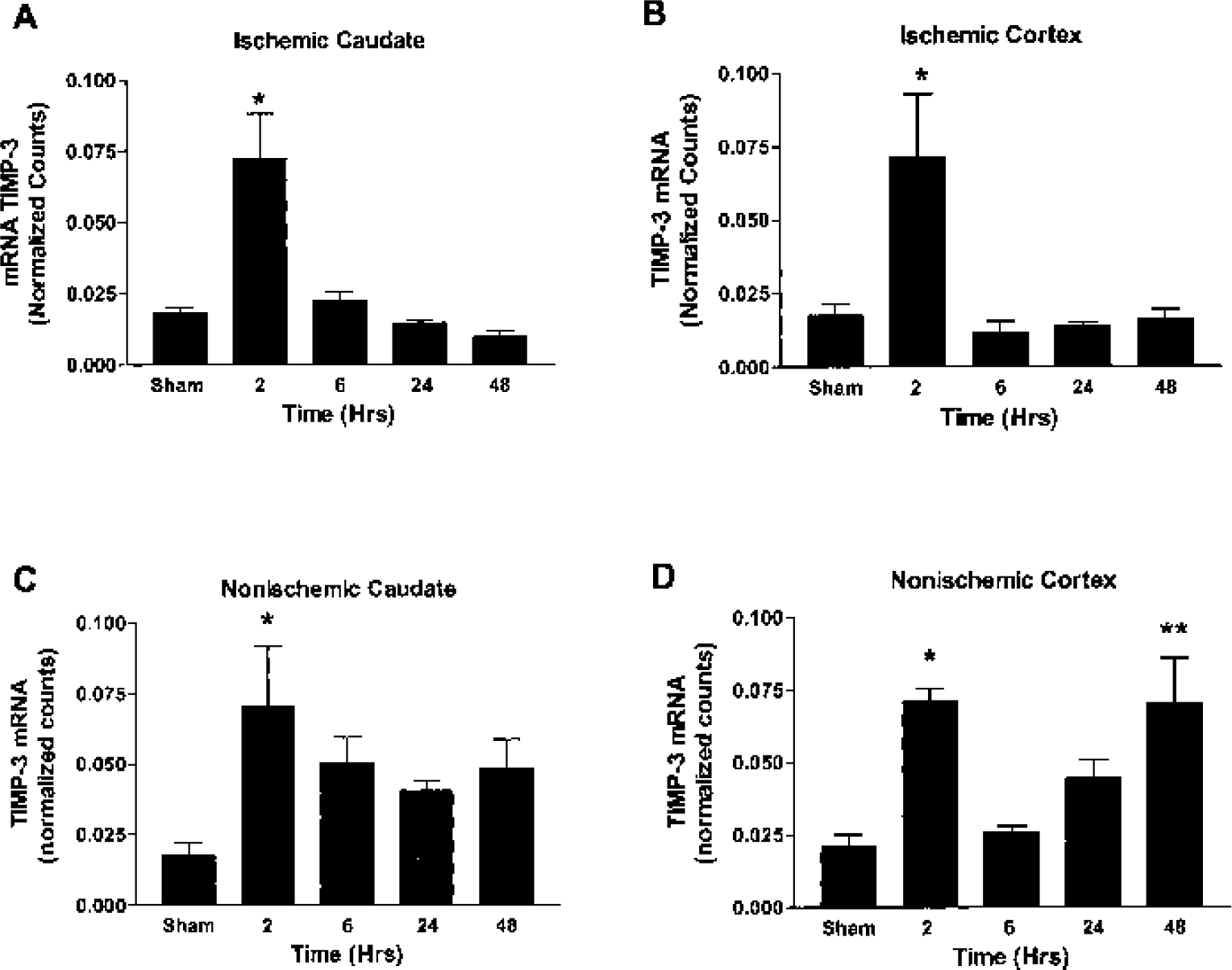
Real-time quantitative measurements of tissue inhibitor of metalloproteinase-3 (TIMP-3) mRNA in rats with 90-minute middle cerebral artery occlusion and reperfusion times as shown on the x-axis.
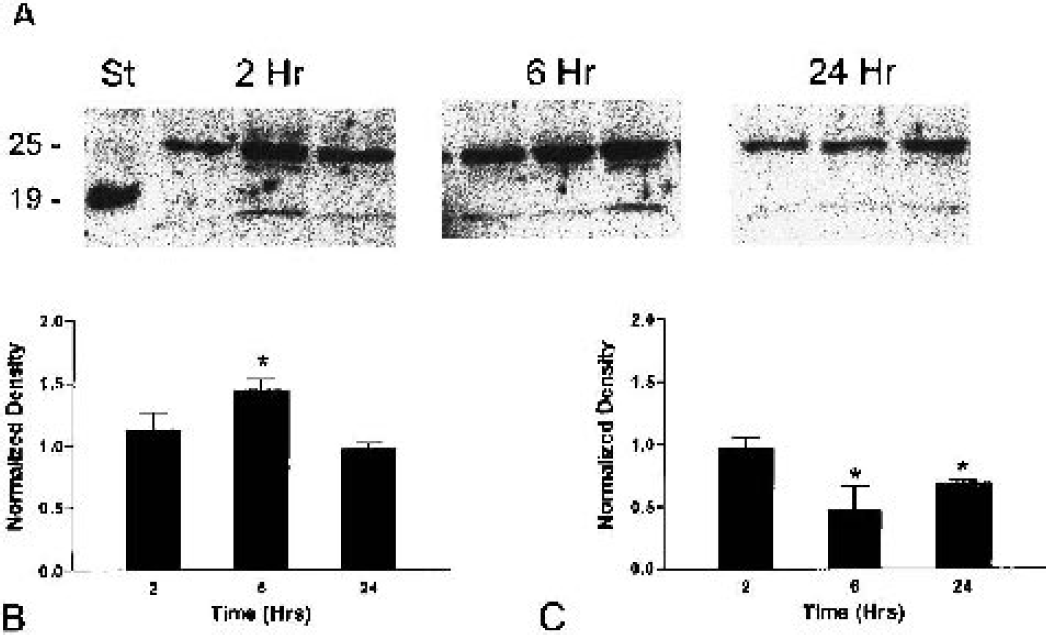
Western blots of the ischemic and nonischemic cortex.
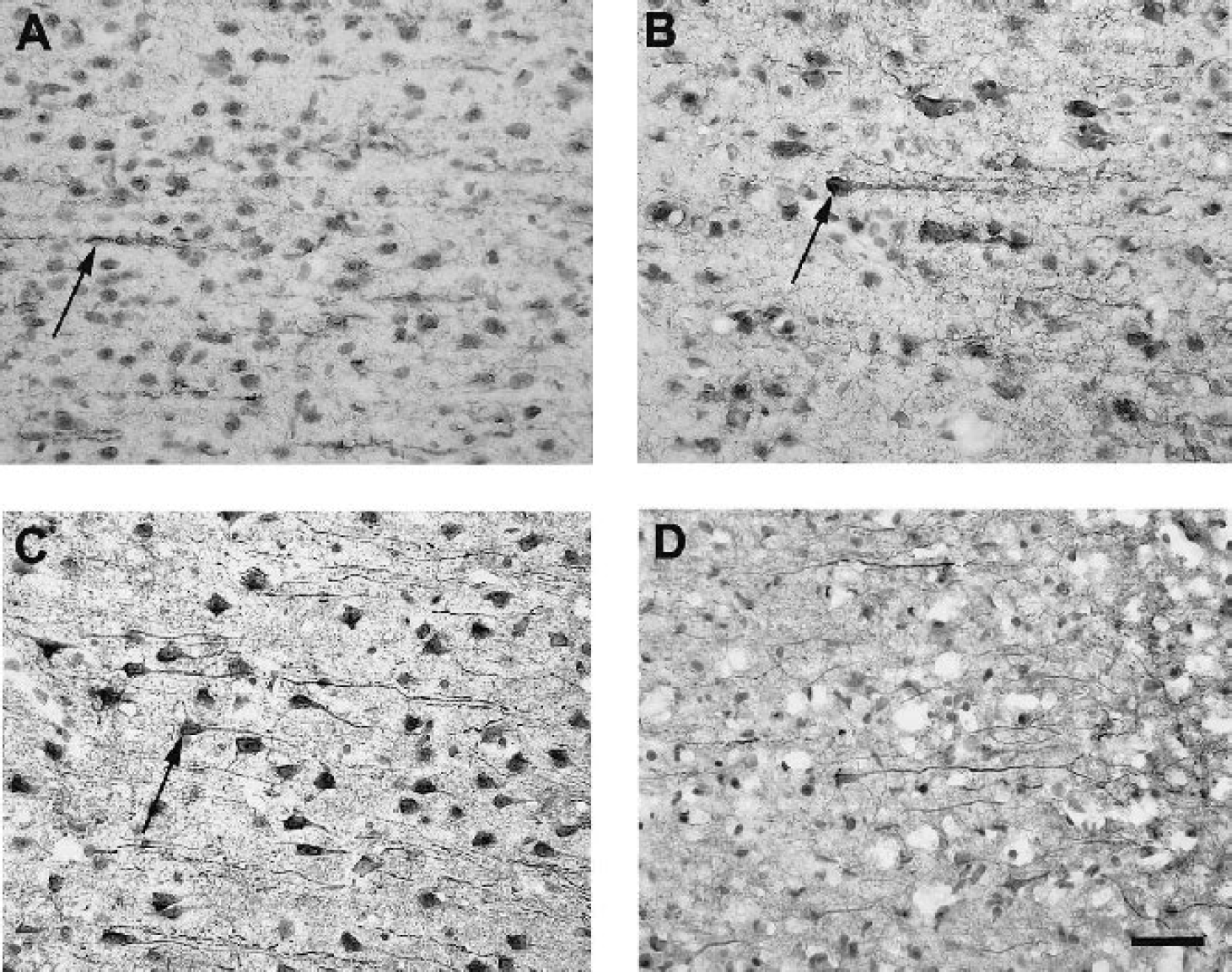
Tissue inhibitor of metalloproteinase-3 (TIMP-3) immunostaining in the cerebral cortex at various intervals of reperfusion after transient middle cerebral artery occlusion.
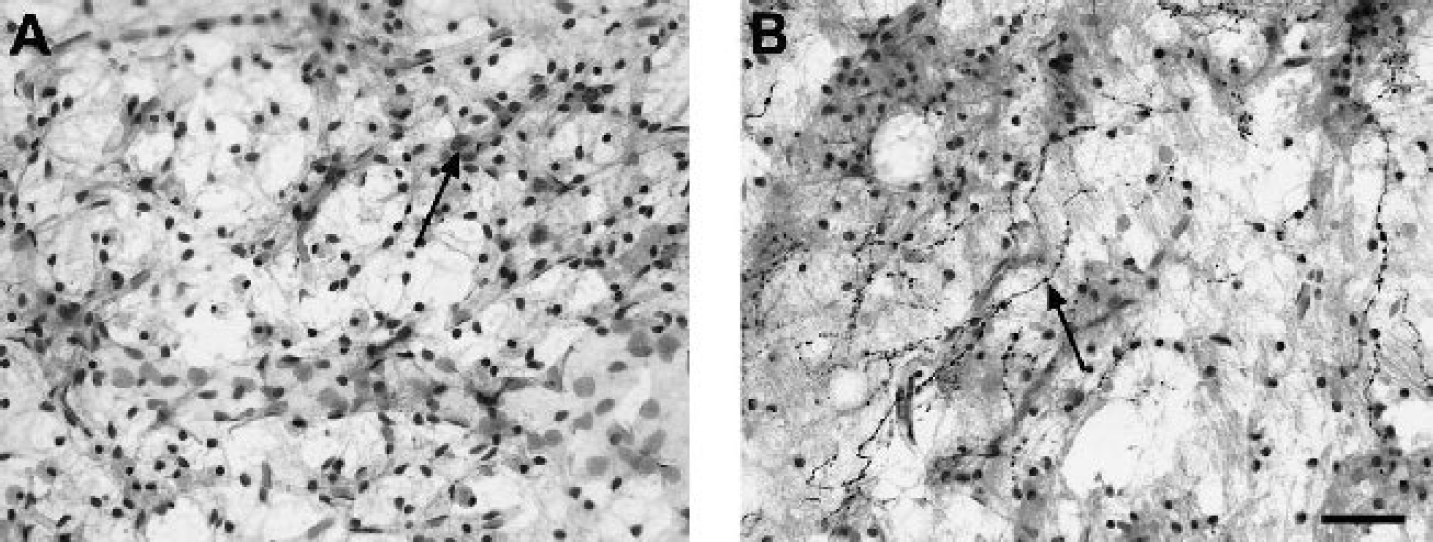
Immunostaining for TIMP-3 was also observed in cells in the caudate nucleus. In the nonischemic side, no staining for TIMP-3 was found in the caudate after 3 hours of reperfusion (Fig. 4A). By comparison, the caudate in the ischemic side showed many cells that had TIMP-3 immunostaining interspersed between white matter bundles, which were unstained (Fig. 4B). By 24 hours, darkly stained TIMP-3-containing cells and processes were seen throughout the ischemic caudate (Fig. 4C). By 5 days of reperfusion, however, few TIMP-3–immunostained elements appeared in the caudate (Fig. 4D).
Immunostaining for tissue inhibitor of metalloproteinase-3 (TIMP-3) in the caudate nucleus at varying periods of reperfusion after transient middle cerebral artery occlusion. (A) No staining for TIMP-3 is seen in the caudate of the nonischemic hemisphere of an experimental animal at 3 hours of reperfusion.
At 3 weeks, the immunostaining for TIMP-3 in both the cortex and caudate was minimal (Fig. 5A). Degenerating regions of the caudate, however, showed numerous darkly stained TIMP-3–positive processes that appeared to be axons with beaded varicosities that emerged from the margins of the intact caudate, and looked like they were entering and growing into degenerating areas of the remaining caudate (Fig. 5B).
Immunostaining for tissue inhibitor of metalloproteinase-3 (TIMP-3) in the cortex and caudate of animals at 3 weeks of reperfusion after transient middle cerebral artery occlusion.
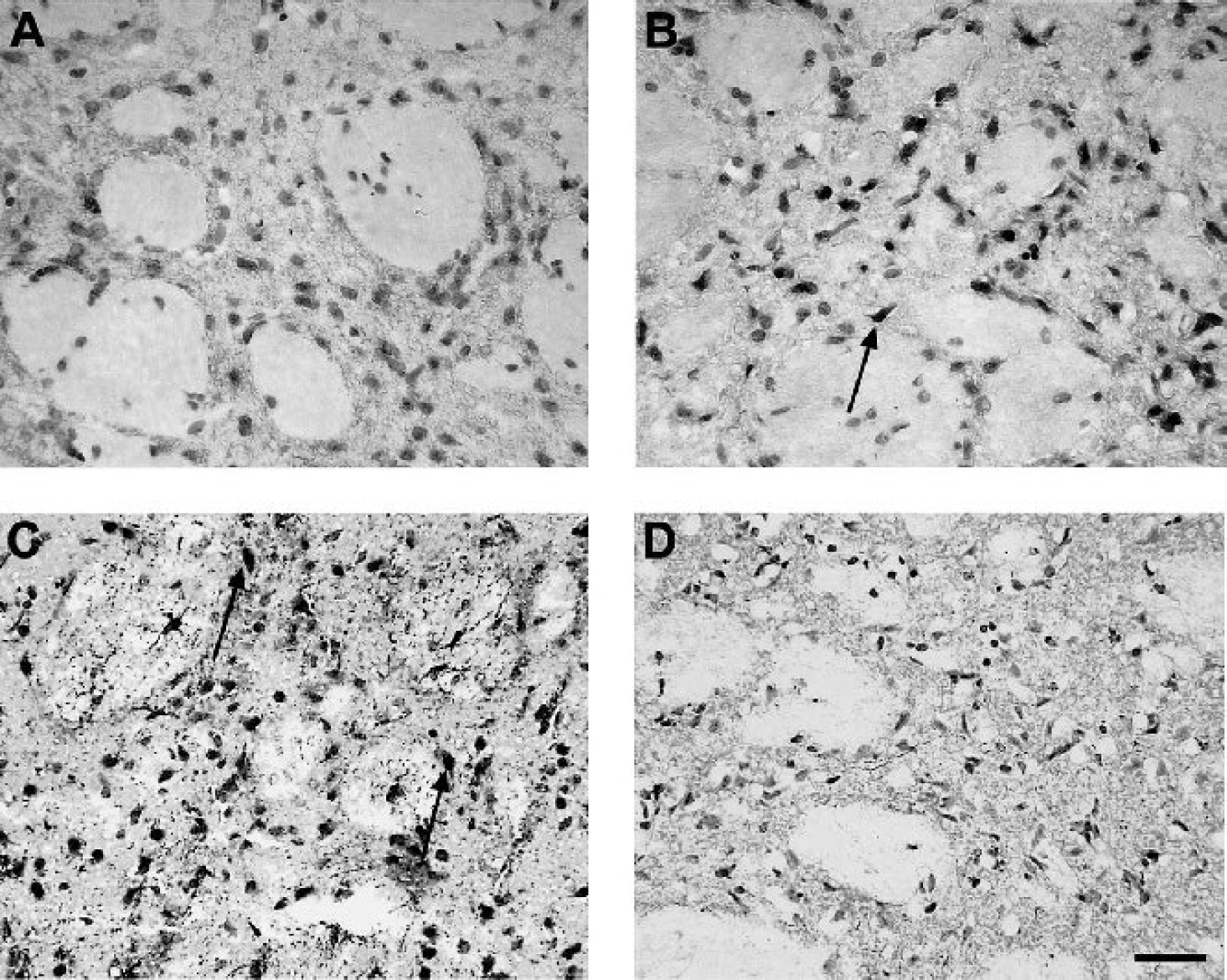
Dual-labeling immunofluorescence was done with TIMP-3 and a neuronal marker, NeuN, in the cortex and caudate at 24 hours after reperfusion. In the nonischemic hemisphere, NeuN-positive neurons appear throughout the cortical tissue, while only punctuate dots and occasional TIMP-3–stained processes were found (Fig. 6A). In the ischemic cortex, TIMP-3 immunostaining was dramatically increased, showing a dense network of TIMP-3–stained dendritic processes along with many NeuN, dual-labeled, pyramidal neurons (Fig. 6B). In the nonischemic caudate widespread NeuN-positive cells were observed with infrequent dots of TIMP-3 staining (Fig. 6C). In contrast, the majority of NeuN-positive cells in the ischemic caudate also contained TIMP-3, as shown by their dual labeling (Fig. 6D). An increase in TIMP-3 in the ischemic caudate is also apparent by the numerous and widely distributed punctate TIMP-3–positive staining.
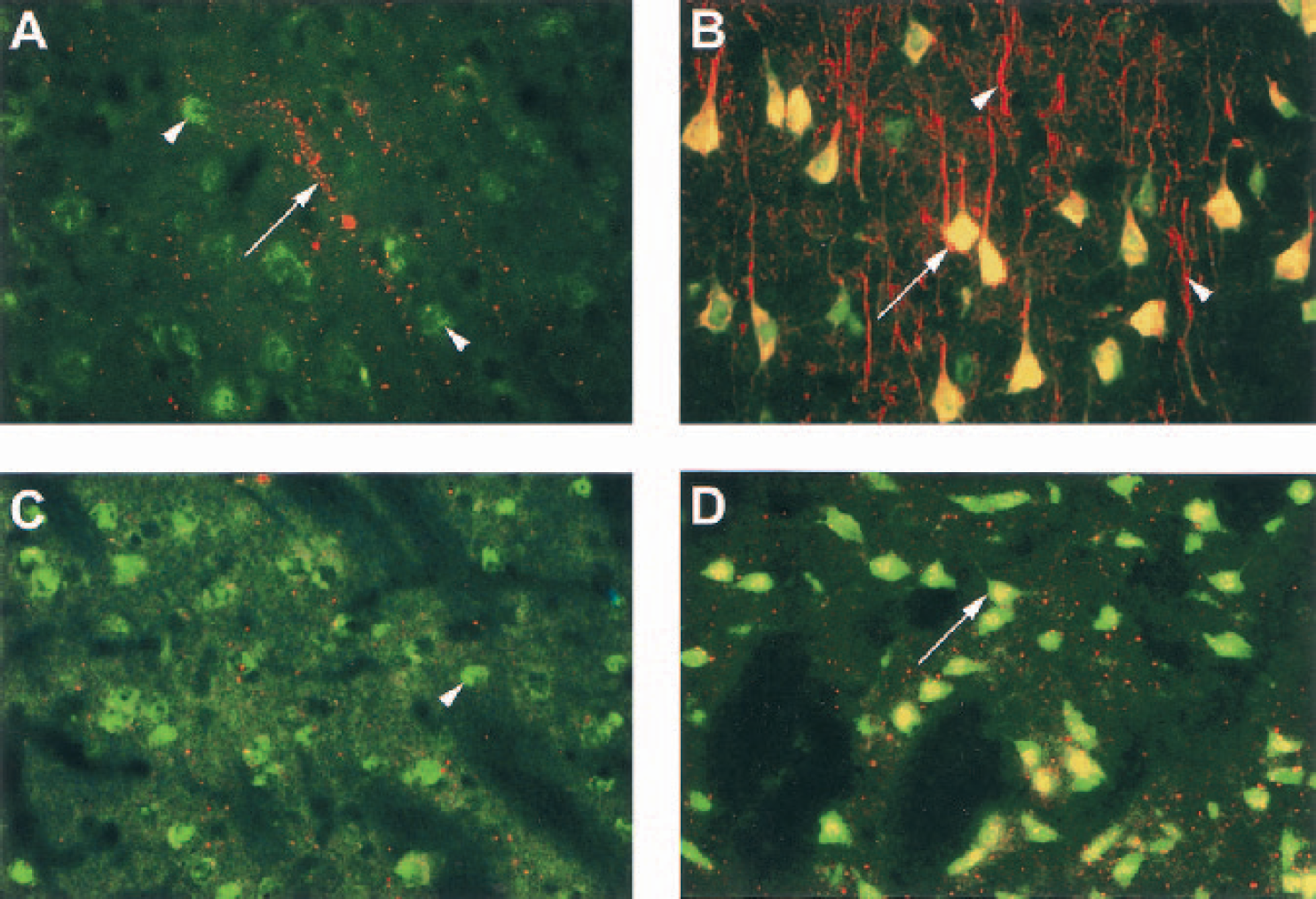
Dual-labeling immunofluorescence with tissue inhibitor of metalloproteinase-3 (TIMP-3) and a neuronal marker, NeuN postive, in the cortex and caudate of an animal at 24 hours of reperfusion after middle cerebral artery occlusion.
Fluorescent dual labeling for TIMP-3 and TUNEL showed no apoptotic cells in the nonischemic hemisphere and only faint TIMP-3 immunoreactivity. In the ischemic cortex, however, numerous TUNEL-stained cells were seen, in addition to a marked increase in TIMP-3–containing cells and processes (Fig. 7A). At higher magnification, TIMP-3 was detected in numerous damaged neurons, as demonstrated by colocalization of TIMP-3 and TUNEL (Fig. 7B). In some regions, such as the piriform cortex and in the core of the caudate, almost all TUNEL-stained neurons appeared to also contain TIMP-3 at 24 hours of reperfusion. Although the degree of overlap in TUNEL and TIMP-3 staining varied between regions in the ischemic hemisphere, the majority of TUNEL-positive cells contained TIMP-3 immunoreactivity.
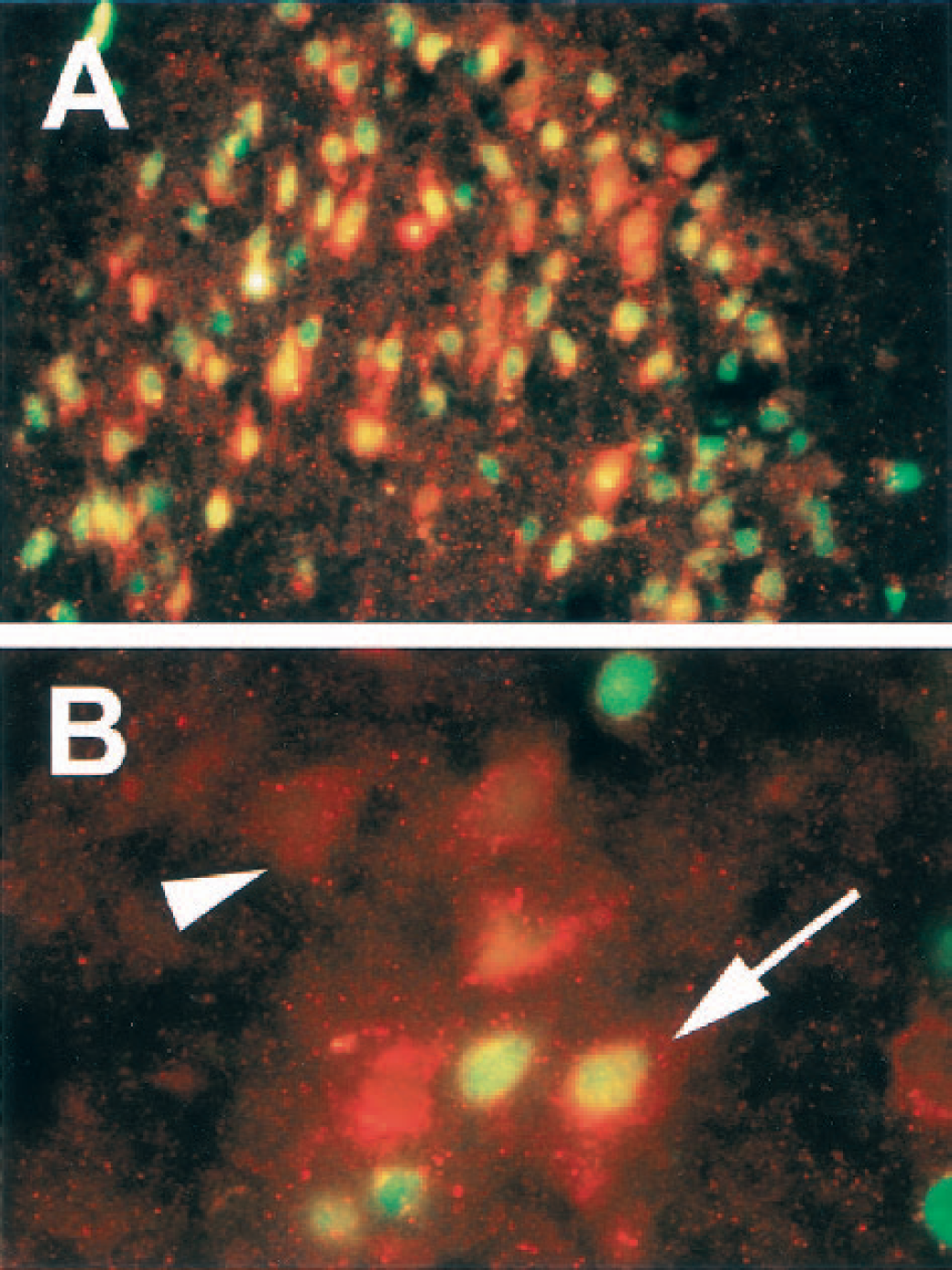
Dual-labeled immunofluorescence of ischemic neurons in the rat piriform cortex 24 hours after a 90-minute middle cerebral artery occlusion.
DISCUSSION
We found marked TIMP-3 expression in neurons in the cortex and caudate of the ischemic hemisphere, and colocalization of TIMP-3 with TUNEL immunostaining. TIMP-3 alone appears insufficient to initiate cell death because it was seen in neuronal process in normal tissues. TIMP-3 gene expression was found at 2 hours after reperfusion in the ischemic and nonischemic regions, and the identity of the protein was confirmed by Western immunoblot analysis. Immunohistochemistry of TIMP-3 showed expression as early as 3 hours after initiation of reperfusion, and maximal expression appeared at 24 hours. The TIMP-3 immunoreactivity was seen predominantly in neurons, where it was colocalized with the TUNEL staining. Our finding of the colocalization of TIMP-3 and TUNEL in hypoxic-ischemic neurons suggests a role of TIMP-3 in cell death in the brain.
Tissue inhibitor of metalloproteinase-3 mRNA appeared rapidly and was translated into TIMP-3 protein, as shown by the immunoreactivity at 3 hours and Western blots at 6 hours after the onset of reperfusion. Expression of the TIMP-3 mRNA was seen in both hemispheres, while the Western blots and immunohistochemistry showed an increase in protein only on the ischemic side. Complex posttranslational effects are seen in a number of proteins, and it is possible that ubiquitination may have been accelerated, accounting for the reduced protein expression in Western blots on the non-ischemic side. The increase in TIMP-3 protein by Western blots at 6 hours was consistent with induction. The reason for the smaller band at 25 kd at 24 hours is less clear. One possibility is that the TIMP-3 was bound to the MMPs that are produced by 12 and 24 hours, resulting in a higher molecular-weight complex. Nevertheless, bound TIMP-3 would be detectable by immunohistochemistry, explaining the increase in TIMP-3 immunostaining seen at 24 hours. Although further studies will be necessary to resolve the effect on the nonischemic side, the results of the mRNA, Western blot, and immunohistochemistry provide convincing support for an increase in TIMP-3 on the ischemic side. This is consistent with reports of TIMP-3 gene expression in brain injury and in Wobbler mice (Jaworski, 2000; Rathke-Hartlieb et al., 2000).
Tissue inhibitor of metalloproteinase-3 was found in normal tissues, suggesting that other factors are needed along with TIMP-3 to promote cell death. Programmed cell death occurs by release of caspase-activating factors from mitochondria or by the stimulation of cell surface receptors that may or may not act through mitochondrial activation (Ashkenazi and Dixit, 1998). These are referred to as the intrinsic and extrinsic pathways (Graham and Chen, 2001). Previous studies have suggested that TIMP-3 acts through cell surface receptors. The mechanism proposed is that TIMP-3 inhibits MMPs and ADAMs that are involved in the shedding of the DRs and DILs from the cell surface (Amour et al., 1998, 2000).
Proposed sheddases include MMP-3, MMP-7, ADAM-10, and ADAM-17. Sheddases release either the DRs or the DILs from the cell surface, and, depending on the cell types and the substrate released, the sheddases can play a protective or damaging role (Powell et al., 1999; Matsuno et al., 2001). Death receptors include the members of the TNF superfamily, such as Fas. One study showed that inhibition of the sheddases by TIMP-3 stabilized TNF receptors on the cell surface, promoting cell death (Smith et al., 1997).
Regulatory elements in the TIMP-3 gene promoter region respond to inflammatory stimuli. The TIMP-3 gene has a p53 consensus–like sequence (Onishi et al., 2001). This could be important since cerebral ischemia leads to p53 expression with peak levels of p53-immunoreactive protein found after 24 hours of reperfusion in rodent stroke models (Chopp et al., 1992; Li et al., 1994). Other factors, including hypoxia-inducing factor-1α and nitric oxide, impact the expression of p53 and could be influential in TIMP-3 gene expression (Halterman and Federoff, 1999; Brune et al., 2001). Mutation of the p53 gene interferes with its normal function as a tumor suppressor gene that regulates other genes required in cell cycle arrest or apoptosis. Mutants of the p53 gene repress TIMP-3 gene transcription by greater than 10-fold (Loging and Reisman, 1999). Although a number of studies support the concept that expression of p53 and TIMP-3 in ischemia could contribute to cell death, a recent study of a p53-knockout mouse showed that the lack of the gene failed to affect apoptosis, and, in fact, benefited other metabolic measures (Maeda et al., 2001). Further studies in conditional knockouts, therefore, would be useful since the effect on other genes in the p53 knockout was not studied.
Fas ligand and Fas receptors are associated with cell death in ischemic neurons. Mice lacking the Fas ligand gene have smaller strokes. In the same study (Martin-Villalba et al. 1999), it was shown that the anticancer drug, doxorubicin, induces the Fas ligand in cultured neurons and leads to apoptosis of the cells. We have studied the role of TIMP-3 in embryonic neuronal cell cultures, using doxorubicin as a stimulus for apoptosis. We found that cultured neurons normally express TIMP-3 and that doxorubicin leads to apoptosis. More important, an antibody to TIMP-3 prevented doxorubicin-induced apoptosis in the cultured neurons. The effect of doxorubicin was reduced by the addition of the sheddase, MMP-3, to the medium, which was not seen with MMP-7 (Wetzel et al., unpublished data, 2002). The in vitro neuronal culture studies thus corroborate the in vivo studies, supporting the facilitation of neuronal cell death by TIMP-3.
Our findings of TIMP-3 gene expression and TIMP-3 protein immunoreactivity in ischemic neurons suggest that it may have a role in apoptosis. Previous studies by us showing the expression of MMP-3 in ischemic neurons and by others showing the expression of Fas in ischemic neurons, combined with in vitro cell culture results, strongly support a novel role for TIMP-3 in the facilitation of the extrinsic pathway of PCD. The ischemic injury caused by 90-minute MCAO is extensive. Since TUNEL immunoreactivity is found in either apoptosis or necrosis, the type of cell death involved is uncertain (Colbourne et al., 1999). Shorter periods of ischemia, which favor apoptosis over necrosis, will need to be studied. We propose that TIMP-3 facilitates cell death in neurons. Although much remains to be learned about the regulation and actions of TIMP-3 in the brain, these studies raise the potential for agents that interfere with the action of TIMP-3, to act as novel treatments for the reduction of PCD. Since apoptosis is critical in many chronic neurodegenerative diseases, TIMP-3 may also play a role in those conditions.