
Abstract
After focal cerebral ischemia, tumor necrosis factor-α deteriorates cerebral edema and survival rate. Therefore, tumor necrosis factor-α neutralization could reduce cerebral microvascular permeability in acute cerebral ischemia. Left middle cerebral artery occlusion for 120 mins followed by reperfusion was performed with the thread method under halothane anesthesia in Sprague-Dawley rats. Antirat tumor necrosis factor-α neutralizing monoclonal antibody with a rat IgG Fc portion (15 mg/kg) was infused intravenously right after reperfusion. Stroke index score, infarct volume, cerebral specific gravity, and the endogenous expression of tumor necrosis factor-α, matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in the brain tissue were quantified in the ischemic and matched contralateral nonischemic hemisphere. In the antitumor necrosis factor-α neutralizing antibody-treated rats, infarct volume was significantly reduced (P = 0.014, n = 7; respectively), and cerebral specific gravity was dramatically increased in the cortex and caudate putamen (P<0.001, n = 7; respectively) in association with a reduction in MMP-9 and membrane type 1-MMP upregulation. Tumor necrosis factor-α in the brain tissue was significantly elevated in the ischemic hemisphere 6 h after reperfusion in the nonspecific IgG-treated rats (P = 0.021, n = 7) and was decreased in the antitumor necrosis factor-α neutralizing antibody-treated rats (P = 0.001, n = 7). Postreperfusion treatment with antirat tumor necrosis factor-α neutralizing antibody reduced brain infarct volume and cerebral edema, which is likely mediated by a reduction in MMP upregulation.
Introduction
Microvascular permeability as a result of extracellular matrix degradation plays a critical role in cerebral edema. Focal cerebral ischemia is responsible for the loss in microvascular integrity, which accompanies major alterations in endothelia cell permeability and the loss of basal lamina matrix ligands. The major basal lamina constituents, laminin, collagen IV, and fibronectin, decrease roughly in parallel during experimental middle cerebral artery occlusion (MCAO), followed by reperfusion (MCAO/R) (Hamann et al, 1995). Those major basal lamina constituents are degraded by matrix metalloproteinases (MMPs). The disruption of normal cell-matrix adhesion has been associated with cell death, edema, and loss of cell viability in several central nervous system injuries (Frisch and Francis, 1994; Re et al, 1994; Rosenberg and Navratil, 1997). Some or all of these MMP-dependent effects could contribute to central nervous system injury.
In conjunction with the increases of MMPs, early inflammatory molecules are also increased in the ischemically injured brain parenchyma. Among these inflammatory molecules, tumor necrosis factor (TNF)-α is reported to stimulate the expression of proteases on several types of cells in vitro. Tumor necrosis factor-α is a potent stimulator of MMP secretion and expression in several types of cultured cells (Brenner et al, 1989; Hanemaaijer et al, 1993). Tumor necrosis factor-α mRNA and protein are increased in cerebral ischemic area (Liu et al, 1994). Furthermore, TNF-α inhibition reduced the total volume of cerebral ischemia (Dawson et al, 1996; Barone et al, 1997; Nawashiro et al, 1997a). Moreover, TNF-α might be involved in blood-brain barrier disruption and the initiation of inflammation in the brain (Gong et al, 1998; Yang et al, 1999). Conversely, a significant reduction in infarct size was noted in mice pretreated with intracisternal delivery of TNF-α (Nawashiro et al, 1997b). Hence, it was suggested that TNF-α played a neuroprotective role after acute brain insults (Gary et al, 1998). Therefore, in focal cerebral ischemia, the role of TNF-α in cerebral vascular and neuronal injury and recovery processes is still controversial.
In the brain, experimental occlusion of MCA is associated with a time-dependent series of events affecting both the microvasculature and the surrounding parenchyma. Endogenous TNF-α and MMP-9 were increased after cerebral ischemia (Rosenberg et al, 1996; Gong et al, 1998). It is known that transcription of MMP-9 is stimulated in the microvascular endothelial cells and smooth muscle cells in vitro by TNF-α (Cho et al, 2000; Hummel et al, 2001). These findings are consistent with the degradation of collagen IV in the extracellular matrix, and the appearance of TNF-α during focal ischemia reperfusion. Therefore, we hypothesized that TNF-α neutralization with an anti-rat TNF-α monoclonal antibody (mAb) would result in MMP-9 reduction and a concomitant amelioration of cerebral edema. The major objectives of the present study are (i) to quantitate temporal expression of TNF-α, MMP-2, MMP-9, and membrane type (MT) 1-MMP in the ischemic parenchyma; (ii) to understand the role of TNF-α in cerebral ischemia by neutralizing TNF-α with a specific anti-rat TNF-α mAb; and (iii) to define the roles of MMP-2, MMP-9, and MT1-MMP in cerebral ischemia.
MATERIALS AND METHODS
Male SD rats (5 weeks old) were obtained from Clea Japan, Inc. (Tokyo, Japan). Animals were kept under controlled conditions with respect to temperature and humidity and were housed on a 12-h light/12-h dark cycle with free access to food and water. No animal should have evidence of disease during a mandated standard quarantine period before entry into this study. All animals must be clinically free of infection or apparent inflammation, and have normal neurologic function. At 6 to 7 weeks of age, the experiments were conducted and the animals weighed between 180 and 250 g. The experimental procedures were approved by the Institutional Animal Care and Use Committee.
Focal cerebral ischemia and reperfusion in the SD rat was induced according to procedures described by Zea-Longa et al (1989). Briefly, each animal was anesthetized with 4% halothane and spontaneously respired with 1% halothane in a 2:1 N2O/O2 mixture, with the use of a face-mask. Under the operating microscope, the left common carotid artery was exposed through a midline incision. The external carotid artery and the occipital artery were ligated, and the internal carotid artery was isolated from the adjacent vagus nerve. Further dissection identified the origin of the pterygopalatine artery, which was not ligated. A microvascular clip was placed across the common carotid artery. A 30-mm segment of 4-0 ethylon monofilament, its tip rounded by heating, was introduced into the external carotid artery at the origin of the occipital artery. The external carotid artery was tied around the intraluminal nylon monofilament to prevent hemorrhage, and the common carotid artery clip was removed. The nylon monofilament was gently advanced from the external carotid artery into the internal carotid artery lumen until a slight resistance was felt and the skin incision closed. Animals were returned to their cages. The rats were reanesthetized and the filament was withdrawn at 2 h after induction of ischemia. Animals were then returned to their cages again and closely monitored until they recovered from anesthesia. Rats were killed at different time points after transient ischemia for cerebral specific gravity and infarct volume, or for measurement of TNF-α, MMP-2, MMP-9, and MT1-MMP. Rectal temperature was monitored continuously and maintained between 36.5 and 37.5°C throughout the anesthesia via a feedback-regulated heating pad placed underneath the rat.
Anti-rat TNF-α neutralizing mAb with a rat IgG Fc portion (dosed at 15 mg/kg based on previous experience (Duan et al, 2002), and kindly provided by Centocor, Inc., Malvern, PA, USA) or nonspecific rat IgG (15 mg/kg; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was administered with intravenous injection at the onset of reperfusion after MCAO. Stroke index score was evaluated 7 days after MCAO/R (n = 13, respectively). For infarct volume measurement, the experiments were terminated 7 days after MCAO/R with decapitation under halothane anesthesia (n = 7, respectively), and for specific gravity measurement, the experiments were terminated at 6 h, 24 h, and 7 days after MCAO/R (n = 7, respectively). During the entire experimental period (7 days), mean blood pressure was monitored with the tail-cuff method. For brain tissue TNF-α, MMP-2, MMP-9, and MT1-MMP measurement, the experiments were terminated at 6, 24 h, and 7 days after MCAO/R under halothane anesthesia by left ventricular transcardiac perfusion of the cranial structures with chilled (4°C) perfusate containing heparin (200 IU/L), nitroprusside (1 mg/L), and bovine serum albumin (50 g/L). The brain tissue perfusion was regarded as complete by the visual absence of blood, and the absence of blood elements on microscopic inspection. Brains were then removed and quickly frozen in 2-methylbutane/dry ice for brain tissue TNF-α, MMP-2, MMP-9, and MT1-MMP measurement. Brain tissue TNF-α, MMP-2, MMP-9, and MT1-MMP were quantified in the same subjects.
Stroke Index Score
To determine the neurologic deficit, neurologic examination was performed at 7 days after reperfusion. The animals were evaluated for a decrease in alertness and movement, ptosis, cocked head, circling behavior, splayed or rotated limbs, seizures, and tremor. Each symptom was assigned a numerical weight, which was used in calculating the stroke index (McGraw, 1977). The maximum score was 34 in dead subjects.
Infarct Volume
The brain was cut into 2-mm-thick coronal slices with a rodent brain matrix. The extent of ischemic infarction was revealed by reaction with a 2% 2,3,5-triphenyltetrazolium chloride solution (Sigma-Aldrich, Inc., St Louis, MO, USA) for 20 to 30 mins. The border between infarct and noninfarct tissue was outlined with an image analysis system, and the area of infarction was measured by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral side. The volume of infarction was calculated by integration of the lesion areas. The traditional direct measurement of infarct volume was associated with an overestimation of infarct volume during the development of brain edema after ischemia (Lin et al, 1993). This artifact can be reduced with indirect measurement, which is based on noninfarcted cortex volume.
Brain Specific Gravity
The specific gravity of the cortex and caudate putamen was determined by microgravimetry using seven samples from each animal. The samples were obtained from the ischemic cortex and caudate putamen and identical contralateral brain using a 1.0-mm punch needle and placed in a bromobenzene-kerosene density gradient column that was calibrated daily with potassium sulfate standards (Hosomi et al, 1996). The position of the sample was determined 2 mins after insertion. The specific gravity of each sample was determined by linear regression analysis. Only columns with correlation coefficients >0.995 were used for analysis. The highest and lowest values were excluded and the mean specific gravity was calculated based on the values in the remaining five samples.
Brain Tissue Tumor Necrosis Factor-α, Matrix Metalloproteinase-2, Matrix Metalloproteinase-9, and membrane type 1-Matrix Metalloproteinase
The brain was minced and homogenized in lysis buffer (50 mmol/L Tris-HCl (pH 7.6), 1.5 mmol/L NaCl, 0.5 mmol/L CaCl2, 1 μmol/L ZnCl2, 1.0% Triton X-100, 0.01% Brij 35, and 0.25% Triton X-100) on ice and centrifuged at 4°C for 20 mins at 9000g. Supernatants were divided into aliquots and stored at –80°C. Preliminary experiments determined the optimal conditions for homogenization and activity extraction. Tumor necrosis factor-α, MMP-2, MMP-9, and MT1-MMP in brain tissue extract were determined by commercially available ELISA (TNF-α) or activity assay (MMP-2, MMP-9, and MT1-MMP) kits (TNF-α from BioSource International, Camarillo, CA, USA; MMP-2, MMP-9, and MT1-MMP from Amersham Biosciences UK Limited, Buckinghamshire, UK). Assays were performed according to the manufacturer's instructions. The sensitive linearity ranges were from 4 to 1000 pg/mL for TNF-α, 0.19 to 3 ng/mL for MMP-2, 0.125 to 4 ng/mL for MMP-9, and 0.125 to 4 ng/mL for MT1-MMP. Concentrations were calculated in picograms per milligram protein for TNF-α. In the TNF-α assay, free form was detected. Concentrations were calculated in nanograms per milligram protein for MMP-2, MMP-9, and MT1-MMP. In these MMP assays, both latent and active forms were detected. These assays cross over to the different species, but not to the other MMPs, thus retaining the specificity for each molecule tested. The crossreactivities of these MMP assays were less than 1% as evaluated with recombinant human MMP-2 and/or MMP-9 (Genzyme; Cambridge, MA, USA), since those assays used the specific antibody to catch the selected MMP.
Statistical Analysis
Differences in stroke index score between groups were examined using the Mann-Whitney U-test. Differences in changes on blood pressure among groups were examined using repeated-measure ANOVA. Differences in infarct volume and specific gravity among groups were examined using one-way ANOVA. Differences in brain tissue TNF-α, MMP-2, MMP-9, and MT1-MMP among groups were examined using two-way ANOVA. When the P-value was <0.05, the Bonferroni's correction for multiple comparisons was used to evaluate the significance of the difference between groups. Differences between the ischemic hemisphere and the matched nonischemic contralateral hemisphere were examined using the paired t-test. The statistical significance was considered when the P-value was <0.05. All results are expressed as the means±s.d.
RESULTS
There was no difference in mean blood pressure between the anti-TNF-α neutralizing mAb-treated rats and the nonspecific IgG-treated rats through the 7-day experimental period (data not shown). Stroke index score was significantly reduced with anti-TNF-α neutralizing mAb treatment 7 days after MCAO/R (P<0.001, n = 13; Figure 1).
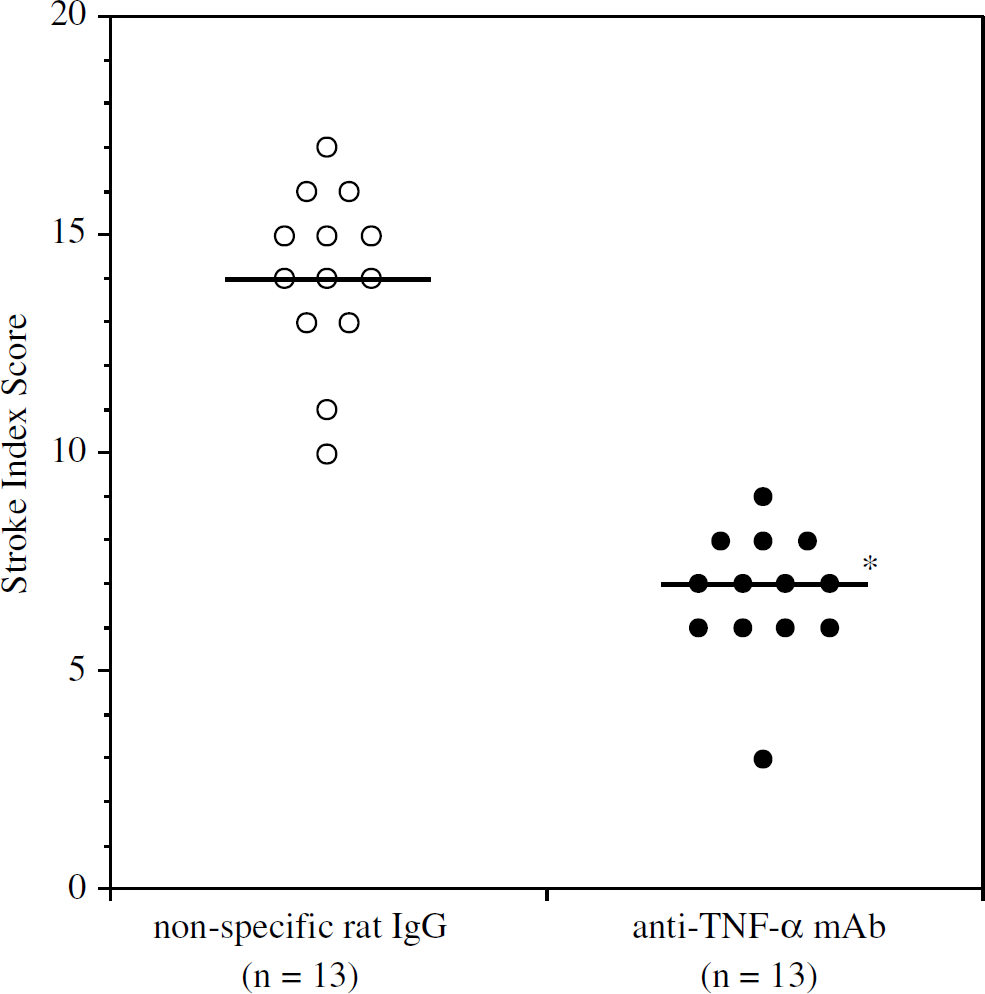
Stroke index score of the nonspecific IgG and anti-TNF-α neutralizing mAb treatment 7 days after MCAO/R (n = 13 per cohort). The bar showed the median value. mAb: monoclonal antibody. *P<0.001 to the nonspecific IgG rats with the Mann-Whitney U-test.
Infarct volume was significantly reduced in anti-TNF-α mAb-treated rats compared with the nonspecific IgG-treated rats 7 days after MCAO/R (F1,12 = 8.228, P = 0.014, n = 7 per cohort; Figure 2). Cerebral specific gravity, presuming cerebral edema, was significantly decreased at the cortex and caudate putamen in the infarct area compared with those in the matched nonischemic contralateral hemisphere (P<0.001, n = 7; respectively; Figure 3). In the anti-TNF-α mAb-treated rats, cerebral specific gravity was significantly higher than that in the nonspecific IgG-treated rats at the cortex and caudate putamen in the infarct area (F1,36 = 19.343, P<0.001 at the cortex; F1,36 = 46.279, P<0.001 at caudate putamen, n = 7 per cohort at each time point; Figure 3).
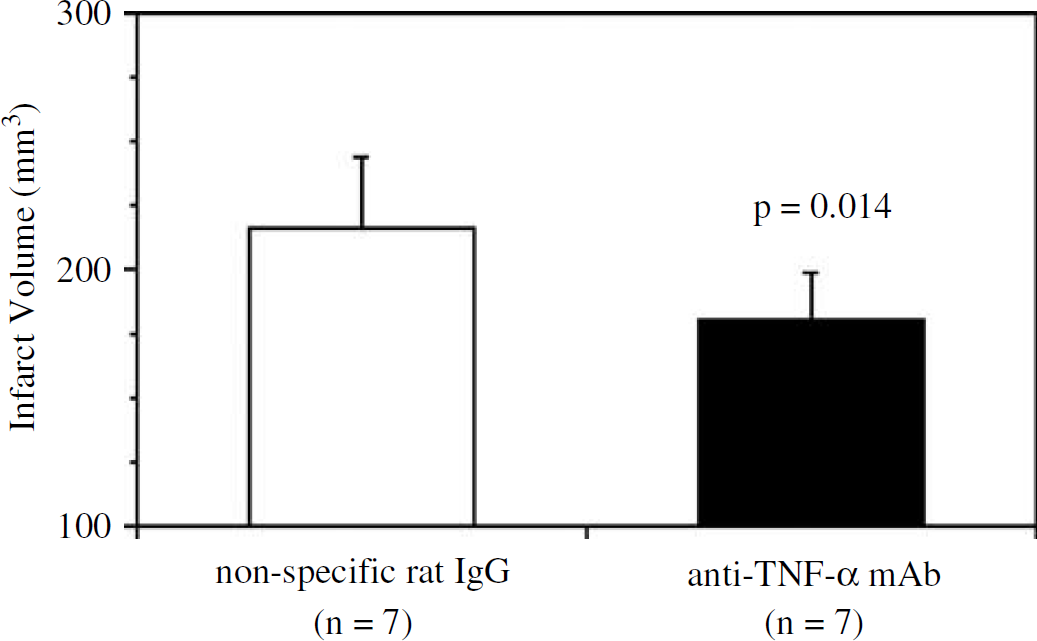
Difference in infarct volume defined 7 days after MCAO/R. Infarct volume was decreased with anti-TNF-α neutralizing mAb treatment (n = 7). mAb: monoclonal antibody.
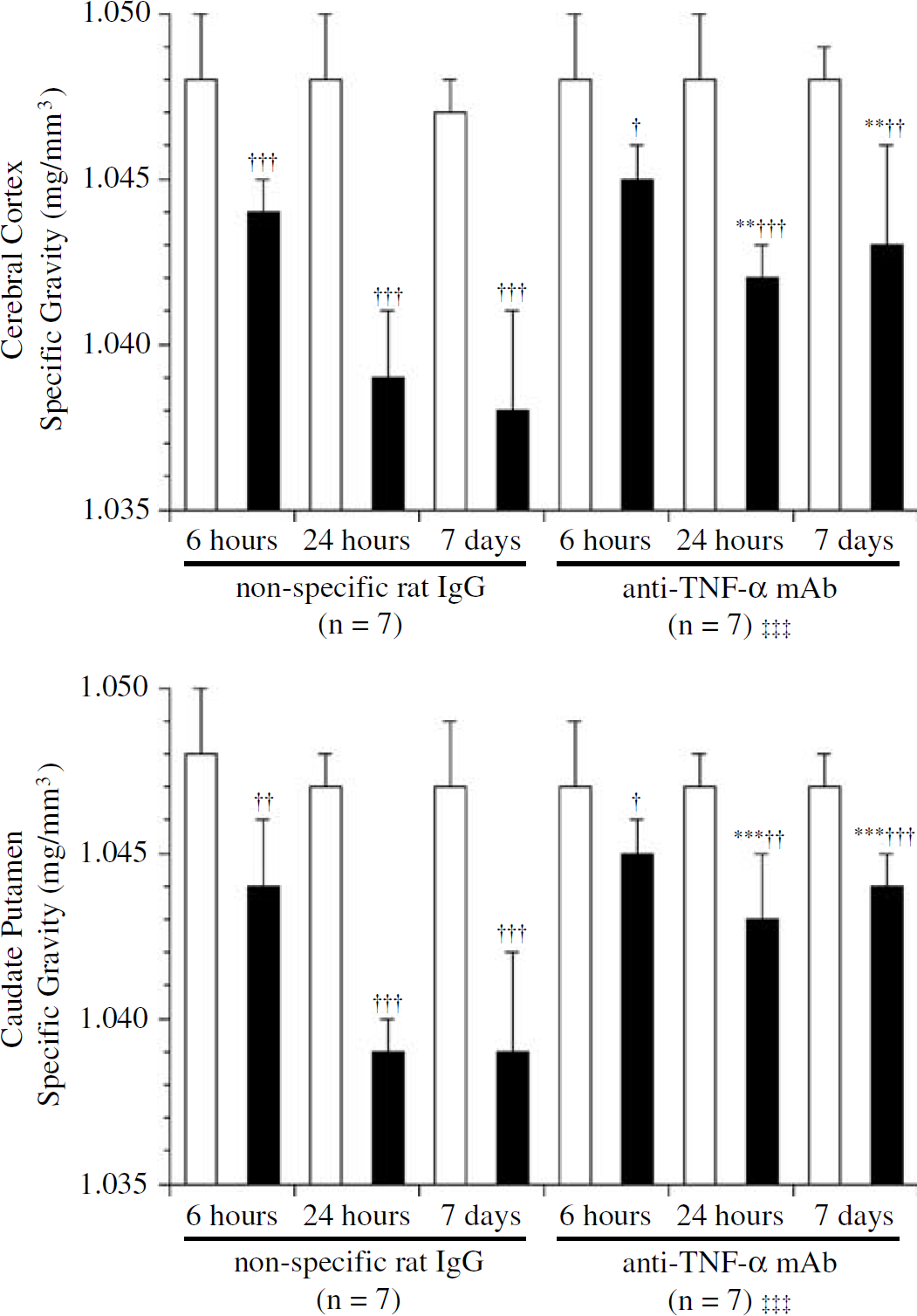
Difference in cerebral specific gravity presuming cerebral edema in the cortex and caudate putamen defined at 6, 24 h, and 7 days after MCAO/R. Cerebral specific gravity was increased with anti-TNF-α neutralizing mAb treatment (n = 7). mAb: monoclonal antibody;white square: the nonischemic hemisphere; black square: the ischemic hemisphere. **P<0.01, ***P<0.001 compared with the nonspecific rat-IgG-treated rats at each time point with one-way ANOVA. †P<0.05, ††P<0.01, †††P<0.001 compared with the matched nonischemic hemisphere with paired t-test. †††P<0.001 compared with the nonspecific rat-IgG-treated rats with two-way ANOVA.
To assess the effects of anti-TNF-α mAb treatment on TNF-α level in the brain tissue, brain tissue was evaluated at 6, 24 h, and 7 days after MCAO/R. In the nonspecific IgG-treated rats, brain tissue TNF-α increased 6 h after MCAO/R in the ischemic hemisphere compared with the matched nonischemic contralateral hemisphere (P = 0.021, n = 7; Figure 4). More importantly, this elevated TNF-α expression in the brain tissue of the ischemic hemisphere was markedly reduced by anti-TNF-α mAb treatment at 6 and 24 h after MCAO/R (F1,12 = 12.952, P = 0.004, n = 7 per cohort and F1,12 = 24.295, P<0.001, n = 7 per cohort; respectively; Figure 4). The effect of anti-TNF-α mAb on serum TNF-α level was also evaluated 24 h after MCAO/R. Serum TNF-α level was 74.5±7.8 pg/mL with the nonspecific rat IgG, treatment and was 17.7±3.6 pg/mL with anti-rat TNF-α mAb treatment (n = 5, P<0.001). Therefore, serum TNF-α level was also significantly reduced with antirat TNF-α mAb treatment.
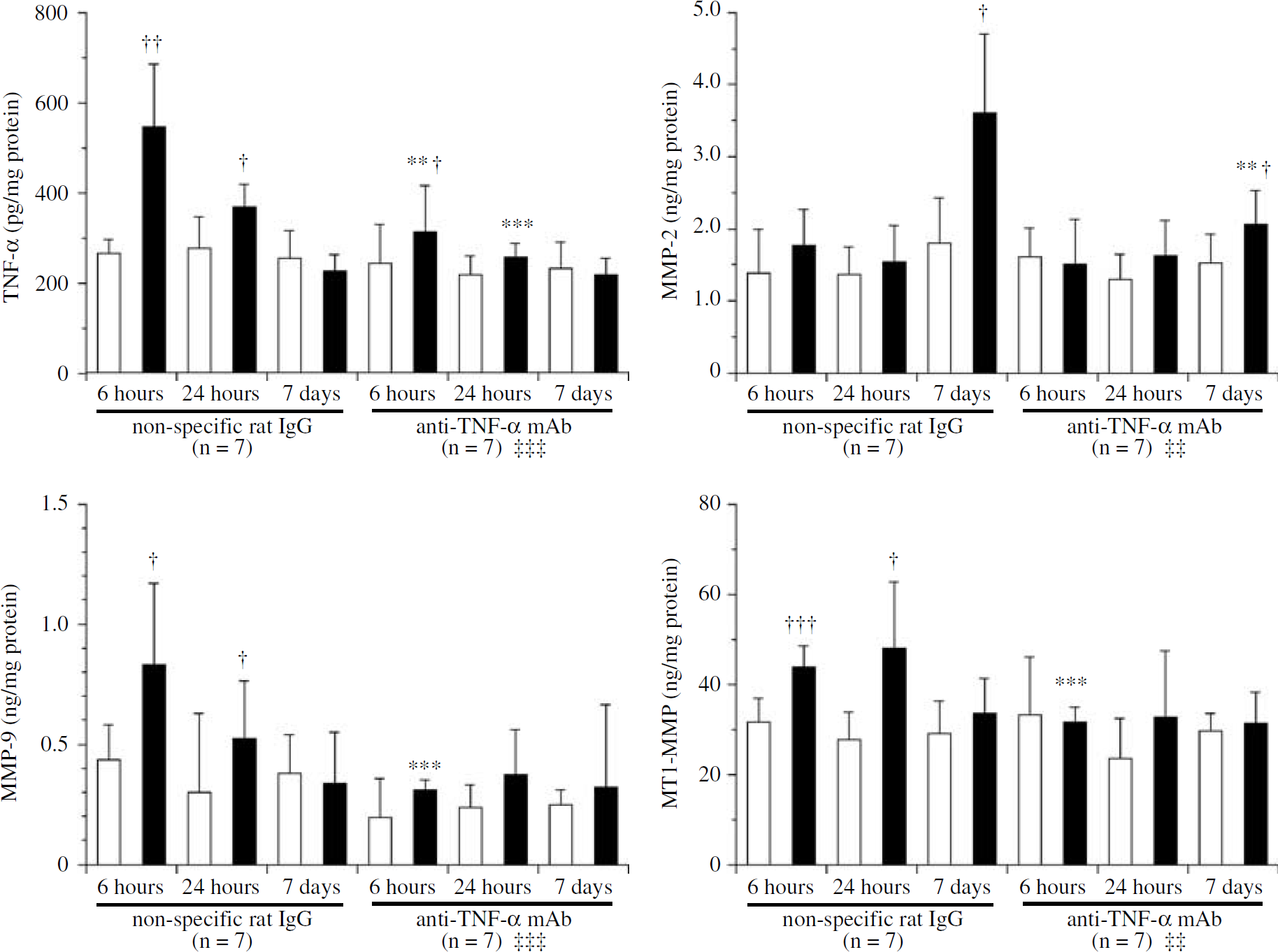
Brain tissue TNF-α, MMP-2, MMP-9, and MT1-MMP defined 6, 12 h, and 7 days after MCAO/R (n = 7 at each time point; respectively). Reactive TNF-α, MMP-2, MMP-9, and MT1-MMP upregulations were reduced with anti-TNF-α neutralizing mAb treatment in the ischemic hemisphere. mAb: monoclonal antibody; white square: the nonischemic hemisphere; black square: the ischemic hemisphere. **P<0.01, ***P<0.001 compared with the nonspecific IgG-treated rats at each time point with one-way ANOVA. †P<0.05, ††P<0.01, †††P<0.001 compared with the matched nonischemic hemisphere with paired t-test. ††P<0.01, †††P<0.001 in the ischemic hemisphere compared with the nonspecific IgG-treated rats with two-way ANOVA.
To understand the mechanism used by TNF-α in inducing cerebral edema, we hypothesized that TNF-α may stimulate the production of MMPs, which subsequently lead to the increased basal lamina cleavage and impact on the cerebral microvascular permeability after cerebral ischemia (Galis et al, 1994; Rosenberg et al, 1995). Matrix metalloproteinase-2, MMP-9, and MT1-MMP in the brain tissue were thus evaluated at 6, 24 h, and 7 days after MCAO/R to define the role of these MMPs in the cerebral edema reduction on anti-TNF-α mAb treatment. Matrix metalloproteinase-2 in the ischemic hemisphere was significantly increased 7 days after MCAO/R as compared with the matched nonischemic contralateral hemisphere (P = 0.011, n = 7; Figure 4). Anti-TNF-α neutralizing mAb treatment reduced reactive MMP-2 upregulation in the ischemic hemisphere 7 days after MCAO/R (F1,12 = 11.773, P = 0.005, n = 7 per cohort), but did not affect MMP-2 production in the nonischemic hemisphere (Figure 4). Similarly, MMP-9 in the ischemic hemisphere was significantly increased at 6 and 24 h after MCAO/R when compared with the matched nonischemic contralateral hemisphere (P = 0.023 and 0.027, n = 7 at each time point; Figure 4). Anti-TNF-α neutralizing mAb treatment reduced reactive MMP-9 upregulation in the ischemic hemisphere 6 h after MCAO/R (F1,12 = 23.255, P<0.001, n = 7 per cohort), but did not affect MMP-9 production in the nonischemic hemisphere (Figure 4). Finally, MT1-MMP in the non-ischemic hemisphere was significantly increased at 6 and 24 h after MCAO/R as compared with the matched nonischemic contralateral hemisphere (P< 0.001 and = 0.012, n = 7 at each time point; Figure 4). Anti-TNF-α neutralizing mAb treatment reduced reactive MT1-MMP upregulation in the ischemic hemisphere 6 h after MCAO/R (F1,12 = 29.740, P<0.001, n = 7 per cohort), but did not affect MT1-MMP production in the nonischemic hemisphere (Figure 4).
To define the relationship between the reduced levels of TNF-α and MMP-2, MMP-9, or MT1-MMP, regression analysis was performed. There was no significant linear association between TNF-α and MMP-2 (n = 84, P = 0.249, r2 = 0.016; Figure 5). Conversely, there were significant linear associations between TNF-α and MMP-9 or MT1-MMP at 6 h after MCAO/R (n = 84, P<0.001, r2 = 0.322; n = 84, P<0.001, r2 = 0.161, respectively; Figure 5).
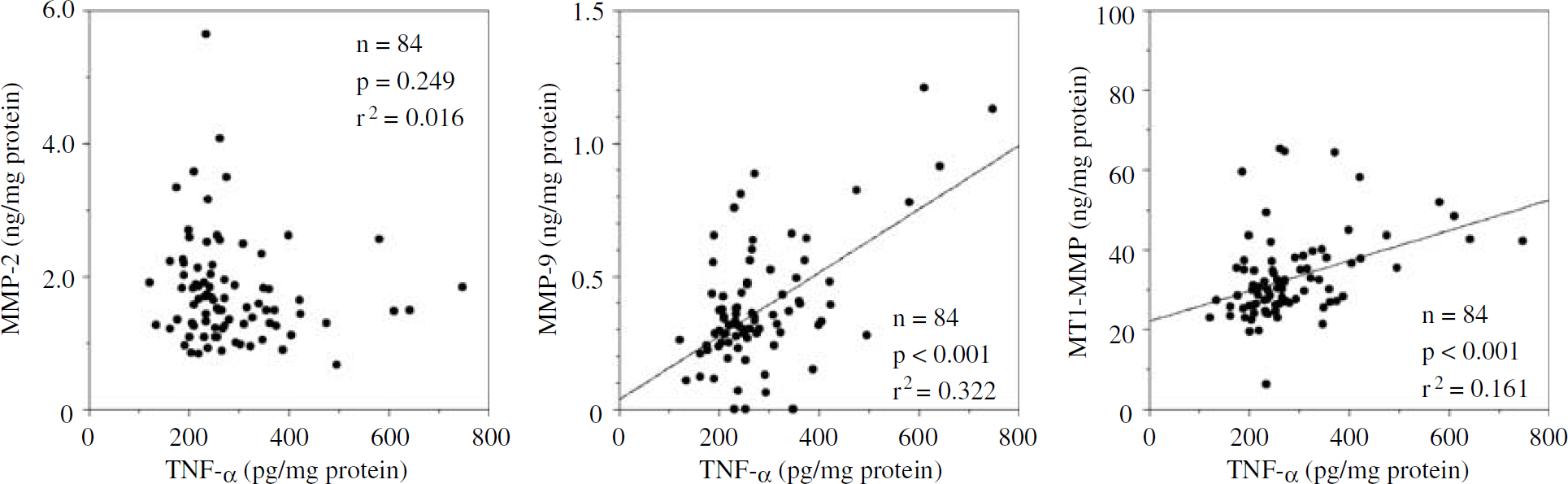
Univariate regression analysis between TNF-α and MMP-2, MMP-9, or MT1-MMP (n = 84). A significant linear relationship was observed between TNF-α and MMP-9 or MT1-MMP.
DISCUSSION
Anti-TNF-α neutralizing mAb treatment on acute cerebral ischemia reduced infarct volume and cerebral edema. In parallel with these favorable effects on acute cerebral ischemia, anti-TNF-α neutralizing mAb treatment decreased reactive TNF-α, MMP-2, MMP-9, and MT1-MMP upregulation in cerebral ischemic area, but did not affect the production of these proteins in the nonischemic hemisphere. From the present study, anti-TNF-α mAb treatment reduced the infarct size by 16.6%. In addition, TNF-α was reduced by 43.0%, MMP-2 by 42.6%, MMP-9 by 60.9%, and MT1-MMP by 32.0% as measured when these molecules were produced at the peak level. The reduction ratios in TNF-α, MMP-2, MMP-9, and MT1-MMP were more than twice greater than that in infarct size reduction. Therefore, the reduction in TNF-α, MMP-2, MMP-9, and MT1-MMP were caused not only by the reduction in infarct size but also by the direct effect of neutralizing TNF-α with anti-TNF-α mAb treatment. From the present study, it was suggested that MMP-9 and MT1-MMP might be involved in the mechanisms of anti-TNF-α neutralizing mAb treatment on cerebral edema reduction after MCAO/R.
Under normal conditions, very little immunoglobulins can cross the intact blood-brain barrier. However, the blood-brain barrier was disturbed after cerebral ischemia, and the extravasation of serum proteins, albumin, leukocyte common antigen, and immunoglobulins was observed in the ischemic core (Nishino et al, 1994; Remmers et al, 1999). Therefore, in the present study, the TNF-α antibody could transudate through the compromised blood-brain barrier after cerebral ischemia and therefore neutralize the brain TNF-α. Tumor necrosis factor-α is one of the interesting cytokines that has many different actions. From the previous and present studies, TNF-α was related to the cerebral ischemic injury (Gong et al, 1998; Yang et al, 1999) and the cerebral ischemic injury was reduced with TNF-α neutralizing treatment after MCAO/R (Dawson et al, 1996; Barone et al, 1997; Nawashiro et al, 1997a). Tumor necrosis factor-α is involved in stroke initiation, progressive brain damage in acute stroke, and also in the development of tolerance to ischemia (Nawashiro et al, 1997b; Liu et al, 2000). The regulation of the neuronal damage by TNF-α after focal cerebral ischemia was mediated by TNF-α receptor-1 (Gary et al, 1998). Upregulation of TNF-α receptor-1 immunoreactivity was detectable within 4 to 6 h of ischemia onset in these same cell types. Receptor-mediated TNF-α signaling can lead to apoptosis (Hsu et al, 1996) and interference with survival peptides (Venters et al, 1999). When TNF-α pretreatment was made, the subjects showed the ischemic tolerance against cerebral ischemia (Nawashiro et al, 1997b). Furthermore, in the studies evaluating cerebral ischemia with TNF receptor knockout mice, cerebral infarct volume and ischemic injury was increased in the knockout mice compared with the wild-type mice (Bruce et al, 1996; Gary et al, 1998). These results suggested that a low amount of TNF-α might be neuroprotective, whereas the presence of excessive amount of TNF-α could be detrimental. Further dose-ranging studies with either TNF-α and/or anti-TNF-α will be helpful to understand the functions of TNF-α in cerebral ischemia.
In preclinical rodent models of brain ischemia, TNF-α mRNA increases within 1 h in the ischemic injury zone, followed by expression of immunoreactive protein within 2 to 6 h of the onset of ischemia (Liu et al, 1994). Tumor necrosis factor-α immunoreactivity appears in neurons, astrocytes, microglia, perivascular cells, and endothelium. Microglia is the major source of TNF-α in acute focal brain ischemia in mice (Gregersen et al, 2000). Tumor necrosis factor-α stimulates expression of tissue factor and adhesion molecules for leukocytes, enhances the release of interleukin-1, nitric oxide, factor VIII/von Willebrand factor, platelet-activating factor and endothelin, suppresses thrombomodulinprotein C-protein S system, reduces tissue-plasminogen activator, and releases plasminogen activator inhibitor-1. Tumor necrosis factor-α also activates MMPs that contribute to increased vascular permeability and hemorrhage (Hummel et al, 2001). Matrix metalloproteinases have been found to contribute both to the progression of blood-brain barrier damage in brain ischemia (Rosenberg et al, 2001) and to TNF-α related neurodegeneration (Schlomann et al, 2000).
Matrix metalloproteinase-2 and MMP-9 are secreted by neurons, astrocytes, microglia, oligodendroglia, Schwann cells, endothelial cells, and smooth muscle cells, and can be regulated by various cytokines in vitro. Nonstimulated vein endothelial cells expressed the mRNAs for MMP-1, MMP-2, TIMP-1, and TIMP-2. Tumor necrosis factor-α induces/enhances the production of several MMPs including MMP-2 and MMP-9, in human endothelial cells (Hanemaaijer et al, 1993). Tumor necrosis factor-α is a potent stimulator of MMP secretion and expression in several types of cultured cells (Brenner et al, 1989; Hanemaaijer et al, 1993). Therefore, the reduction in MMP activity is highly likely a result of TNF-α neutralization. Smooth muscle cells constitutively produce MMP-2, tissue inhibitors of MMP (TIMP)-1, and TIMP-2. On stimulation with interleukin-1 or TNF-α, smooth muscle cells synthesized in addition to MMP-9, stromelysin, and interstitial collagenase, MMPs that together can degrade all of the extracellular matrix components (Galis et al, 1994). Matrix metalloproteinase-9 induced by TNF-α can contribute to the increased vascular endothelial permeability through the degradation of specific extracellular matrix components. Rosenberg et al has shown that MMP-9 is the intermediate substance linking TNF-α to modulation of capillary permeability in the brain (Rosenberg et al, 1995). In the present study, cerebral specific gravities began to decrease 6-h after MCAO/R and almost reached the maximum reduction in 24 h after MCAO/R in the cortex and caudate putamen. The development of cerebral edema and reactive increases in MMP-9 occurred concomitantly. Confirmatory to these reports, inhibition in reactive increases in MMP-9 with anti-TNF-α neutralizing mAb was also associated with a reduction in cerebral edema after MCAO/R.
In the present study, not only MMP-2 and MMP-9, which degrade the basal lamina, but also MT1-MMP, which is a potent activator of MMP-2, was expressed after cerebral ischemia in parallel with the cerebral edema formation. Membrane type 1-MMP is secreted by astrocytes, endothelial cells, smooth muscle cells, and other cell types in vitro and is expressed at low levels in the mouse central nervous system (Pagenstecher et al, 1998). Membrane type 1-MMP mRNA and protein were increased with TNF-α exposure via activation of NF-κB on smooth muscle cells, monocytes, endothelial cells, and astrocytes in vitro (Rathke-Hartlieb et al, 2000; Han et al, 2001). Moreover, pro-MMP-9 is expressed in the hippocampus, and MT1-MMP in the cortex and hypothalamus after central nervous system viral infection (Khuth et al, 2001). Chang et al (2003). have recently reported that the expressions of MT1-MMP were increased 1 h after MCAO in the ischemic core in nonhuman primates. Therefore, MT1-MMP likely promotes matrix degradation by activating MMP-2 in the ischemic core in rat cerebral ischemic model. In the present study, anti-TNF-α neutralizing mAb treatment reduced MT1-MMP in the brain tissue and inhibited its reactive overexpression after cerebral ischemia.
Anti-TNF-α therapy has proven beneficial in the long-term treatment of rheumatoid arthritis (Feldmann, 2002). In addition, infliximab (anti-human TNF-α neutralizing antibody with human Fc fraction) is now approved for the treatment of Crohn's disease. In the clinic, anti-TNF-α antibody has shown long-lasting efficacy for more than 3 months. The known adverse effects of infliximab are respiratory infection, reactivation of latent tuberculosis, infusion reactions, delayed hypersensitivity reaction, and lupus-like symptoms. Although measurements of TNF-α levels in blood and cerebrospinal fluid of stroke patients have supported a role for TNF-α in stroke pathology (Sairanen et al, 2001; Zaremba and Losy, 2001) and the potential benefit of blocking TNF-α has been proposed (Clark and Lutsep, 2001), anti-TNF-α strategies have not yet been applied to treat stroke in the clinic. Further translational studies will be necessary to find the therapeutic time window and the optimal dose of TNF-α neutralizing agent in cerebral ischemia and to establish whether TNF-α neutralization has any clinically significant antiischemic effect in humans.
Footnotes
Acknowledgements
Anti-rat TNF-α neutralizing antibody with rat IgG Fc portion was kindly supplied by Centocor, Inc., Malvern, PA, USA.