
Abstract
Intracerebral hemorrhage (ICH) initiates an inflammatory response with secondary growth of hemorrhage and cell death. Matrix metalloproteinase (MMP) gelatinolytic activity is increased in ICH, and synthetic inhibitors to MMPs reduce edema and hemorrhage size. Recently, we found that tissue inhibitor of metalloproteinase-3 (TIMP-3) is elevated after ischemia and colocalizes with TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end-labeled)-labeled cells. Tissue inhibitor of metalloproteinase-3 promotes neuronal apoptosis in vitro by blocking the shedding of the tumor necrosis factor (TNF) superfamily of death receptors/ligands by stromelysin-1 (MMP-3). However, the effect of TIMP-3 and synthetic MMP inhibitors on cell death in ICH is unclear. Therefore, we used the collagenase-induced intracerebral hemorrhage (CIH) model in Timp-3 knockout and C57Bl/6 wild-type mice to study MMP expression, hemorrhage volume, and cell death. Real-time PCR showed an increase in Mmp-3 mRNA in CIH, but similar Mmp-2 and -9 mRNA expression levels in CIH and saline-injected mice. Protein levels of pro and cleaved MMP-3 were increased in CIH, and zymographic gelatinolytic activity of MMP-9 was elevated after CIH at 72 h, suggesting an exogenous source. Apoptosis was shown by increased caspase-3 levels at 2 and 72 h, and active caspase-8 by 2 and 24 h. The Timp-3 null mouse and wild types had similar hemorrhage sizes and TUNEL-labeled cells. Unexpectedly, the broad-spectrum MMP inhibitor BB-94 increased hemorrhage size and TUNEL-labeled cells. Our results fail to implicate TIMP-3 in apoptosis in CIH, but show that BB-94 increased apoptosis in CIH, possibly by blocking shedding of TNF death receptors and/or their ligands.
Keywords
Introduction
Intracerebral hemorrhage (ICH) is the most debilitating form of cerebrovascular disease (Qureshi et al, 2001). The ability to observe the evolution of the mass lesion by computed tomography and magnetic resonance imaging has shown growth of the hemorrhagic lesion in approximately 30% of patients in the first 24 h (Brott et al, 1997). Matrix metalloproteinase (MMP) levels are increased in ICH in animal models and in humans, and MMP levels in blood have been associated with the extent of hemorrhagic transformation (Rosenberg and Navratil, 1997; Montaner et al, 2001; Rosell et al, 2006; Tejima et al, 2007). Matrix metalloproteinases contribute to hemorrhagic transformation after cerebral ischemia by degrading the basal lamina around blood vessels and disrupting tight junction proteins in endothelial cells (Yang et al, 2007). Synthetic inhibitors to metalloproteinases have been shown to reduce edema and hemorrhage size in animal models of ICH (Power et al, 2003; Wang and Tsirka, 2005). Additionally, MMPs contribute to cell death (Gu et al, 2002). In cerebral ischemia, stromelysin-1 (MMP-3) protects neurons from death by releasing death receptors and ligands from the cell surface, whereas tissue inhibitor of metalloproteinase-3 (TIMP-3) promotes apoptosis by inhibiting MMP-3, thus preventing death receptor/ligand shedding (Cunningham et al, 2005). In bacterial collagenase-induced hemorrhage (CIH), metalloelastase (MMP-12) was shown to be elevated in mice and the antiinflammatory agent that suppresses MMPs, minocycline, both reduced the injury and lowered levels of MMP-12 (Power et al, 2003). In their study, mRNA for MMP-3 was increased but levels of protein were not measured.
Evidence for apoptotic cell death appears in experimental hemorrhage models including microballoon inflation, autologous blood injection, and bacterial collagenase (BC) injection (Hickenbottom et al, 1999; Nakashima et al, 1999; Matsushita et al, 2000). Apoptotic cells were found to be distributed both within the matrix of the hematoma and in the perihematomal region in these animal models. Additionally, apoptosis has been observed in perihematomal human tissue after ICH (Qureshi et al, 2003). Furthermore, TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end-labeled)-labeled cells were found to be mainly neurons and astrocytes within and around the hemorrhage in the CIH model (Matsushita et al, 2000). Autologous blood injection resulted in greater than 95% neuronal TUNEL-labeled cells around the clot (Gong et al, 2001). After injection of autologous blood, there is activation of MMP-9 by thrombin, and the injury is reduced in MMP-9 knockout mice (Xue et al, 2006).
Stimulation of neuronal cultures with Doxorubicin (Dox) induced Fas-mediated apoptosis, and inhibition of endogenous MMP sheddase activity on death receptors/ligands by TIMP-3 enhanced apoptosis and in the absence of TIMP-3, the synthetic inhibitor BB-94 promoted cell death (Wetzel et al, 2003). Apoptotic cells, especially those of the perihematomal region, may represent a potentially salvageable group of cells, and because cell death is delayed, these cells may be amenable to therapeutic interventions (Qureshi et al, 2003). From the evidence that inhibition of MMPs by endogenous and synthetic MMP inhibitors enhances apoptosis in an in vitro model of neuronal apoptosis and that Timp-3 mRNA was elevated and the protein localized with TUNEL-labeled cells in an in vivo cerebral ischemic model (Wallace et al, 2002; Wetzel et al, 2003), we investigated the role of TIMP-3 and a synthetic MMP inhibitor in the CIH model. We measured MMPs and caspases in CIH in mouse and tested the effect of the broad-spectrum MMP inhibitor BB-94 (Batimastat) and Timp-3 gene deletion on cell death and hemorrhage volume.
Materials and methods
Stereotactic Collagenase-Induced Intracerebral Hemorrhage
Timp-3 knockout and wild-type mice on the C57Bl/6 background were a generous gift from Dr Rama Khokha. All animal studies were approved by the University of New Mexico Health Science Center's Animal Care and Use Committee and conformed to the National Institutes of Health standards for use of animals in research.
Male mice weighing 24 to 30 g were anesthetized with 2.5% halothane delivered in a 70% nitrous oxide/30% oxygen mixture and maintained with 1.75% halothane. Mice were injected subcutaneously with 0.1 mL of 0.027 mg/mL buprenorphine and placed in a Kopf (Kopf Instruments, Tujunga, CA, USA) stereotaxic head holder. A midline incision was cut and a burr hole was made 2.5 mm lateral from bregma over the left hemisphere. A 32-gauge needle was inserted 3.0 mm ventral from dura into the region of the caudoputamen. Bacterial collagenase (0.5 μL of 0.0375 U Type VII-S; Sigma-Aldrich Corp, St Louis, MO, USA) or sterile normal saline (Sigma-Aldrich Corp) was injected for 2 mins; the needle was left in place for another 2 mins. The burr hole was sealed with bone wax and the skin incision was closed. For studies using MMP inhibition, C57Bl/6 mice were administered a suspension of 30 mg/kg BB-94 (British Biotech, Oxford, UK) in sterile normal saline by intraperitoneal injection starting at the time of BC injection twice a day for 3 days.
Tissue Collection and Processing for Zymography
After euthanasia, brains were immediately removed and frozen in −80°C cooled 2-methylbutane (Fisher Scientific Company, Pittsburgh, PA, USA) and stored at −80°C for later analysis. Each brain was sectioned into a 4 mm coronal section of tissue encompassing the hemorrhage: 2 mm rostral and 2 mm caudal of the injection site. Tissue was collected into 200 μL ice-cold 100 mmol/L phosphate buffer, pH 7.2 with 0.2% Triton X-100 (Bio-Rad Laboratories Inc., Hercules, CA, USA), and homogenized. After centrifugation of each homogenized sample, supernatant was collected and a separate aliquot was saved for bicinchoninic (BCA) protein content analysis performed in triplicate (Pierce Biotechnology Inc., Rockford, IL, USA). Supernatants were incubated with gelatin-coated Sepharose 4B beads (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) to concentrate the gelatinases (Zhang and Gottschall, 1997).
Gelatin-Substrate Zymography
Samples were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels containing 1% gelatin (Sigma-Aldrich Corp). Standards included a protein ladder (Bio-Rad Laboratories Inc.), 187.5 μU BC (Sigma-Aldrich Corp), conditioned media from HT1080 human fibrosarcoma cells, which constitutively express MMP-9 (92 kDa) and MMP-2 (72 kDa), and activated murine MMP-9 (Chemicon, Temecula, CA, USA). After electrophoresis, the gels were washed in 2.5% Triton X-100 (Sigma-Aldrich Corp) to remove the sodium dodecyl sulfate (Fisher Scientific Company). After incubation, gels were stained with Coomassie R-250 dye (Sigma-Aldrich Corp) and proteolytic bands were visualized as clear regions of lysis after subsequent differentiation in 10% acetic acid. Gels were scanned with an AGFA Duoscan scanner (AGFA Corporation, Ridgefield Park, NJ, USA) and analyzed using AlphaEase (Alpha Innotech Corporation, San Leandro, CA, USA). The bands measured included the pro-MMP-9 band of 98 kDa and the pro-MMP-2 band of 72 kDa. Sample load was corrected for total protein content, as determined by the Micro BCA assay (Pierce Biotechnology Inc.). The organomercurial compound 4-aminophenylmercuric acetate (APMA; Sigma-Aldrich Corp) was used to activate MMPs.
Real-Time PCR
Comparison of the time course of mRNA in mice after 12, 24, 48, and 72 h of CIH or saline injection was performed for Mmp-2, -3, -9, and Timp-3. Rpl-32 was used to normalize the mRNA. Primers were designed using Primer Express software (Applied Biosystems, Foster City, CA, USA) and constructed by Integrated DNA Technologies Inc. (Coralville, IA, USA). Primer and probe sequences are listed in Table 1. Optimal concentrations used for SYBR Green PCR (Invitrogen Corp., Carlsbad, CA, USA) were rpl-32 (1,000 nmol/L), MMP-2 (1,000 nmol/L), MMP-3 (1,000 nmol/L), and MMP-9 (500 nmol/L). Optimal primer and probe concentrations for TaqMan PCR (Applied Biosystems) were rpl-32 and TIMP-3 primers (500 and 750 nmol/L, respectively), and 4 μmol/L for the probe. PCR amplification for Timp-3, Mmp-2, -3, -9, and Rpl-32 was performed using the following cycling parameters: 2 mins hold at 50°C, 10 mins hold at 95°C, followed by 40 cycles of 15 secs at 95°C and 1 min at 60°C.
: Murine primers for RT-PCR

MMP, matrix metalloproteinase; RT-PCR, real-time PCR; TIMP-3, tissue inhibitor of metalloproteinase-3.
The Timp-3 mRNA levels were quantitated using the TaqMan real-time PCR assay (Applied Biosystems) as described previously (Medhurst et al, 2000; Wallace et al, 2002). Timp-3 and rpl-32 mRNA levels in the reaction mixtures were calculated from a standard curve generated by amplification of known concentrations of TIMP-3 and rpl-32 plasmids. The plasmids for TIMP-3 and rpl-32 were created using pGEM-T Easy Vector Systems II (Promega, Madison, WI, USA). Data are expressed as copy number based on the plasmid standard curves and normalized by dividing the copy number for each sample by the copy number of the corresponding Rpl-32 housekeeping gene. All samples and controls were run in triplicate.
Western Blots for Matrix Metalloproteinase-3 and the Caspases
For each separate animal, a sample was collected by pooling 1-mm diameter tissue punches from slide mounted 400-μm-thick sections collected from the center of the rostral—caudal extent of the striatum; both the injected ipsilateral and noninjected contralateral hemispheres were separately sampled.
The tissue was collected and dissociated in a buffered protease inhibitor cocktail containing 50 mmol/L Tris-HCl, pH 7.4, 0.5% sodium dodecyl sulfate, 150 mmol/L NaCl, 1 mmol/L NaVO3, 50 mmol/L NaF, 5 mmol/L sodium pyrophosphate tetrabasic decahydrate, and a 1 × stock of protease inhibitor (Applied Biosystems). Sample protein was loaded on acrylamide gels for MMP-3 and caspase Western blots (Bio-Rad Laboratories Inc.). Gels were run with prestained and biotinylated molecular weight ladders (Bio-Rad Laboratories Inc., and Cell Signaling Technology Inc., Danvers, MA, USA, respectively). Positive controls included APMA-activated murine MMP-3 (R&D Systems, Minneapolis, MN, USA), mouse thymus tissue lysate (Axxora, San Diego, CA, USA) for intact caspase-8, and mouse L929 cell lysates treated with or without staurosporine (Cell Signaling Technology Inc.) for caspase-3, caspase-9, and their cleavage products.
The caspase primary antibodies (Cell Signaling Technology Inc.) were all used at a 1:1,000 dilution and included monoclonal rabbit anti-caspase-3 and polyclonal rabbit anti-caspase-8. The secondary antibodies included horseradish peroxidase anti-rabbit and horseradish peroxidase anti-biotin (1:2000 dilution; Cell Signaling Technology Inc.). Matrix metalloproteinase-3 analysis used goat polyclonal anti-MMP-3 (2 μg/mL; Abcam Inc., Cambridge, MA, USA) in 1% bovine serum albumin (Sigma-Aldrich Corp) and horseradish peroxidase anti-goat (1:2,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Chemiluminescent detection of the samples used the West Pico kit (Pierce Biotechnology Inc.) for MMP-3 and Chemicon's ChemiLucent kit for the caspases. The films were densitometrically scanned for semiquantitative analysis and were normalized to the saline uninjected contralateral striata for the corresponding time points. Blots were probed with rabbit anti-actin (Sigma-Aldrich Corp) or were stained with Amido Black (Sigma-Aldrich Corp) to confirm equal loading and transfer.
Stereological Analysis and Immunohistochemistry
At 72 h, the mice were gas anesthetized and injected with 50 mg/kg of Nembutal. The mice were perfused transcardially with 2% paraformaldehyde solution containing lysine and sodium periodate (PLP) (Sigma-Aldrich Corp). The brains were then postfixed overnight in PLP at 4°C, cryoprotected in a solution of 30% sucrose (Sigma-Aldrich Corp) at 4°C, and frozen sections were cut on a cryostat for measurements of hemorrhage volume and TUNEL-labeled cells.
Counting of TUNEL-labeled cells and measurement of hemorrhage volume were performed stereologically on an Olympus fluorescent scope using the optical fractionator method and StereoInvestigator v.6.0 (MicroBrightField Inc., Williston, VT, USA). The hemorrhage lesion size was circumscribed in each coronal section undergoing analysis by optical dissection through the extent of the region defined rostral—caudally as the genu of the corpus callosum. The software used for analysis determines the estimated volume of the hemorrhage based on the given block advance, the actual mounted tissue thickness, and the planimetry measured. The region of interest for all sections was delimited by normal brain parenchyma and the border of the hemorrhage as seen by endogenous autofluorescence as a result of blood components (Figure 7A).
Immunohistochemical analysis of TUNEL-labeled cells was performed on slide-mounted sections (20 μm) using NeuroTACS II (Trevigen Inc., Gaithersburg, MD, USA) with every 10th coronal section collected from the same rostral—caudal extent as above, resulting in 20 sections of each brain for stereological analysis.
The cell types expressing TUNEL were determined in animals treated with BB-94 by dual-labeling of TUNEL with NeuN (1:400; Chemicon) for neurons, glial fibrillary acidic protein (1:400; Sigma-Aldrich Corp) for astrocytes, myeloperoxidase (MPO) (1:500; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for neutrophils, von Willibrand Factor (5 μg/mL; Chemicon) for endothelial cells, and Iba1 (1.25 μg/mL; Wako Chemicals USA Inc., Richmond, VA, USA) for microglia and macrophages. After blocking with goat serum, slides were incubated overnight in primary antibody at 4°C. The tissue was incubated with secondary antibodies labeled with indocarbocyanine (Cy3), and then rinsed and mounted with GelMount (Sigma-Aldrich Corp). Sections incubated in the absence of primary antibody were not immunofluorescent.
Edema Measurement
The effect of BB-94 on edema was determined in adult male C57Bl/6 mice (Harlan, Indianapolis, IN, USA) by calculating percent water content after drying tissues for 24 h at 100°C. Twenty-four hours after treatment, the mice were killed and brains were sectioned coronally into two quadrants each of the left and right hemispheres. Coronal section 1 contained regions from bregma +1.0 to −1.0 for samples L-1 and R-1, whereas coronal section 2 contained regions from approximately bregma −1.0 to −4.5. Water content was expressed as the percentage change between wet weight (WW) and dry weight (DW): [(WW−DW)/WW] × 100. Five of the animals were treated with BB-94 (30 mg/kg, intraperitoneally) 10 mins before receiving their intracerebral injection.
Statistical Analyses
Statistical analyses were performed using the software package, GraphPad Prism (v. 4.03, San Diego, CA, USA). Where required, a two-way analysis of variance was performed with a Bonferroni posttest. For stereological analysis, a two-tailed, nonparametric Mann—Whitney t-test with a 95% confidence level was used. Significant differences between means were determined using one-way analysis of variance with Dunnett's multiple comparison post hoc analysis. Unpaired data were compared using the Student's t-test for parametric data. Data are expressed as means±s.e.m. Significance was set at P<0.05.
Results
Matrix metalloproteinase-9 Protein Increased in Collagenase-Induced Intracerebral Hemorrhage Without mRNA Production
We measured Mmp-2 and -9 mRNA levels at 12, 24, 48, and 72 h after either saline or BC injection (Figures 1A to 1D). Levels of Mmp-9 mRNA increased in both hemispheres; however, there were no statistical differences between saline and BC (Figures 1A and 1B). Mmp-2 mRNA levels were significantly increased in the saline-injected side at 72 h (Figure 1C); no contralateral effect was seen (Figure 1D).
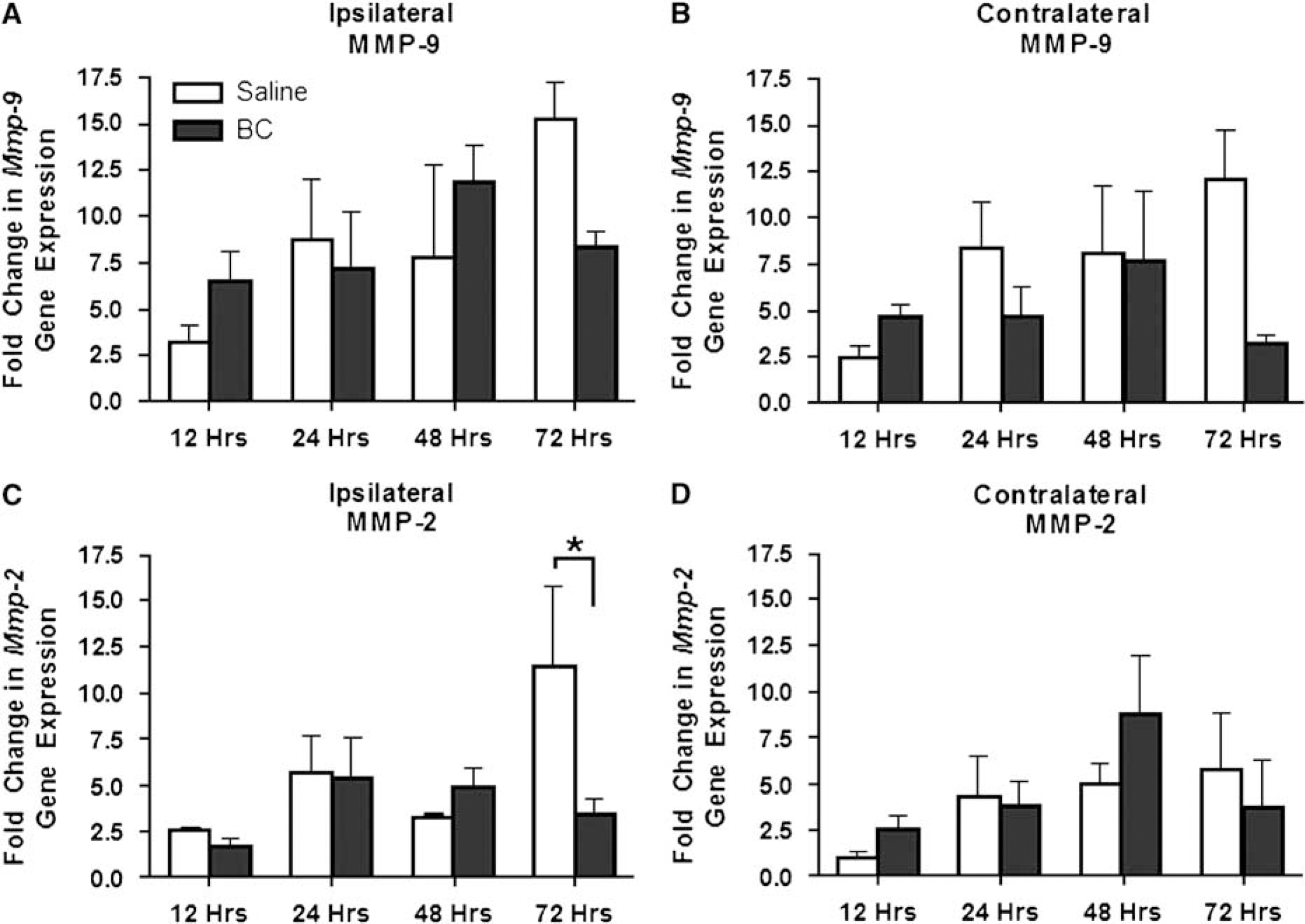
Semiquantitative real-time PCR for Mmp-2 and -9 mRNA after saline injection or BC-induced injury. (
Gelatin zymography showed endogenous production of MMP-2 and -9 after BC injection (Figure 2A) with minimal contralateral effect. Matrix metalloproteinase-9 gelatinolytic activity was significantly increased over saline injection at 72 h and 3 weeks (Figure 2B), but no change was seen after saline injection (Figures 2B and 2C). At 72 h, MMP-2 activity in BC-treated mice was significantly increased over saline-treated mice (Figure 2C). No statistical contralateral effect was observed for MMP-2 and -9 (data not shown).
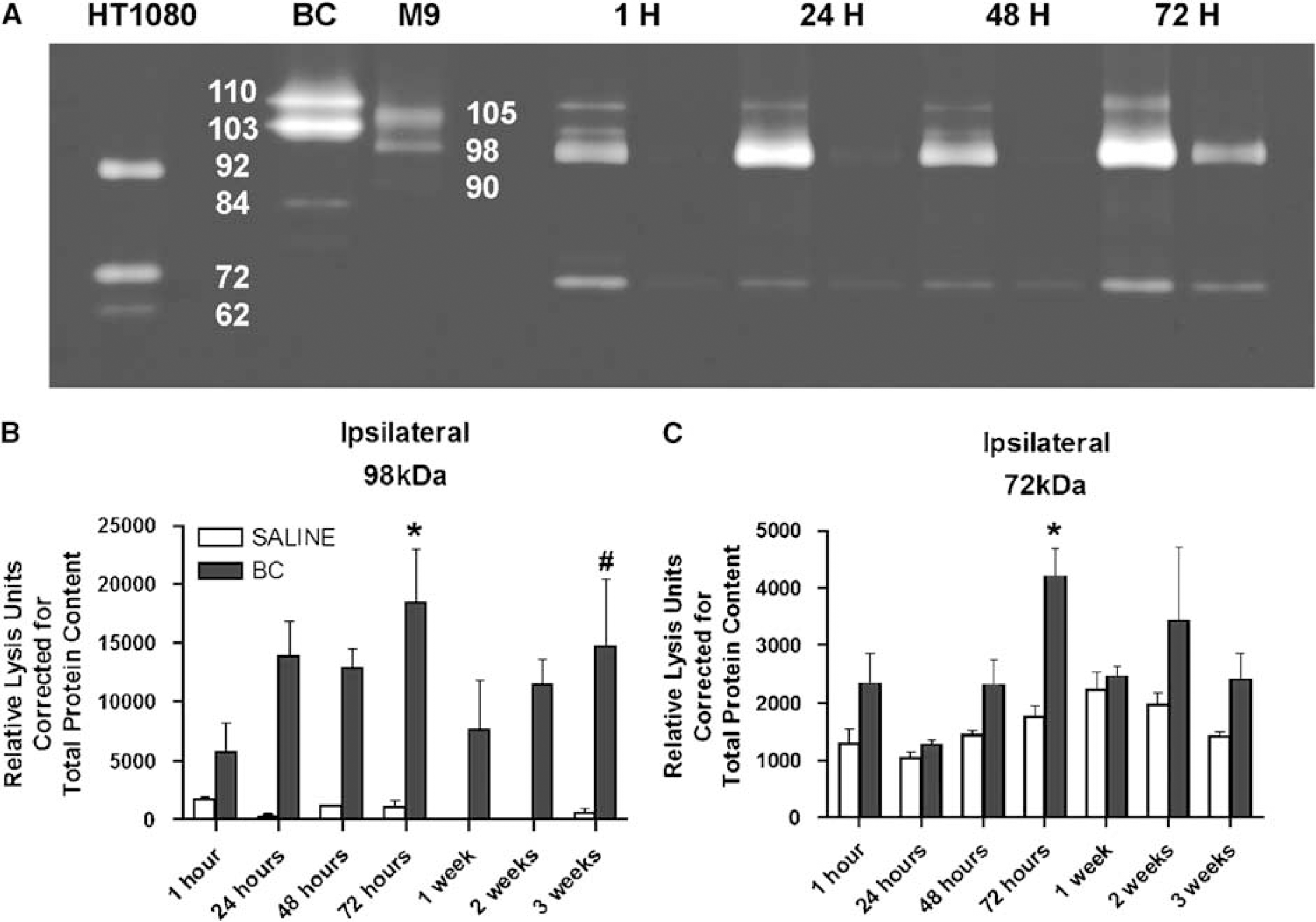
Densitometric analysis of zymograms from BC or sterile normal saline-injected ipsilateral (left) striatum versus noninjected contralateral (right) striatum. (
Bacterial Collagenase and Endogenous Promurine Matrix Metalloproteinase-9 (98 kDa) Molecular Weights
When run separately, BC had two predominant bands with apparent molecular weights of 110 and 103 kDa with lower molecular species found at 93, 84, and 76 kDa by zymography. One hour after BC was injected into the brain, bands were seen at 110, 103, and 84 kDa with a new band at the molecular weight of 98 kDa, which corresponded to the murine MMP-9 standard (Figure 2A). The proform of the MMP-9 standard had a molecular weight of 105 kDa, which could be separated on the zymograms from BC. However, the main species of MMP-9 that was detected after injection was seen at 98 kDa. The higher 105 kDa form, which could be glycosylated, may undergo posttranslational deglycosylation to give rise to the lower-molecular-weight proform observed (Opdenakker et al, 2001b). An active 90 kDa band was observed in the APMA-activated murine MMP-9 standard. Additionally, MMP-9 activation with APMA of a CIH brain sample resulted in a new, lower molecular weight active form present at 90 kDa that corresponded to the murine MMP-9 standard; pro-MMP-2 (72 kDa) converted to its active band at 62 kDa (data not shown). 4-Aminophenylmercuric acetate (APMA) activation of BC did not result in the formation of lower-molecular-weight species (data not shown).
Stromelysin-1 mRNA Induction and Protein Expression in Collagenase-Induced Intracerebral Hemorrhage
Mmp-3 mRNA was significantly increased by approximately 20-fold at 12 and 24 h compared with saline (Figure 3A). Bacterial collagenase-induced mRNA levels decreased by 48 and 72 h; no statistical contralateral effect was observed (data not shown). At 72 h, pro-MMP-3 was detected in its glycosylated form at approximately 60 kDa (Figure 3B). Samples were normalized to the contralateral hemisphere of the saline-injected brain. The proform was statistically elevated in the BC-injected hemisphere at 72 h compared to the saline-injected brain (Figure 3C). More importantly, active MMP-3 (45 kDa), which was detectable only with BC injection, was significantly increased compared to saline at 72 h (Figure 3D).
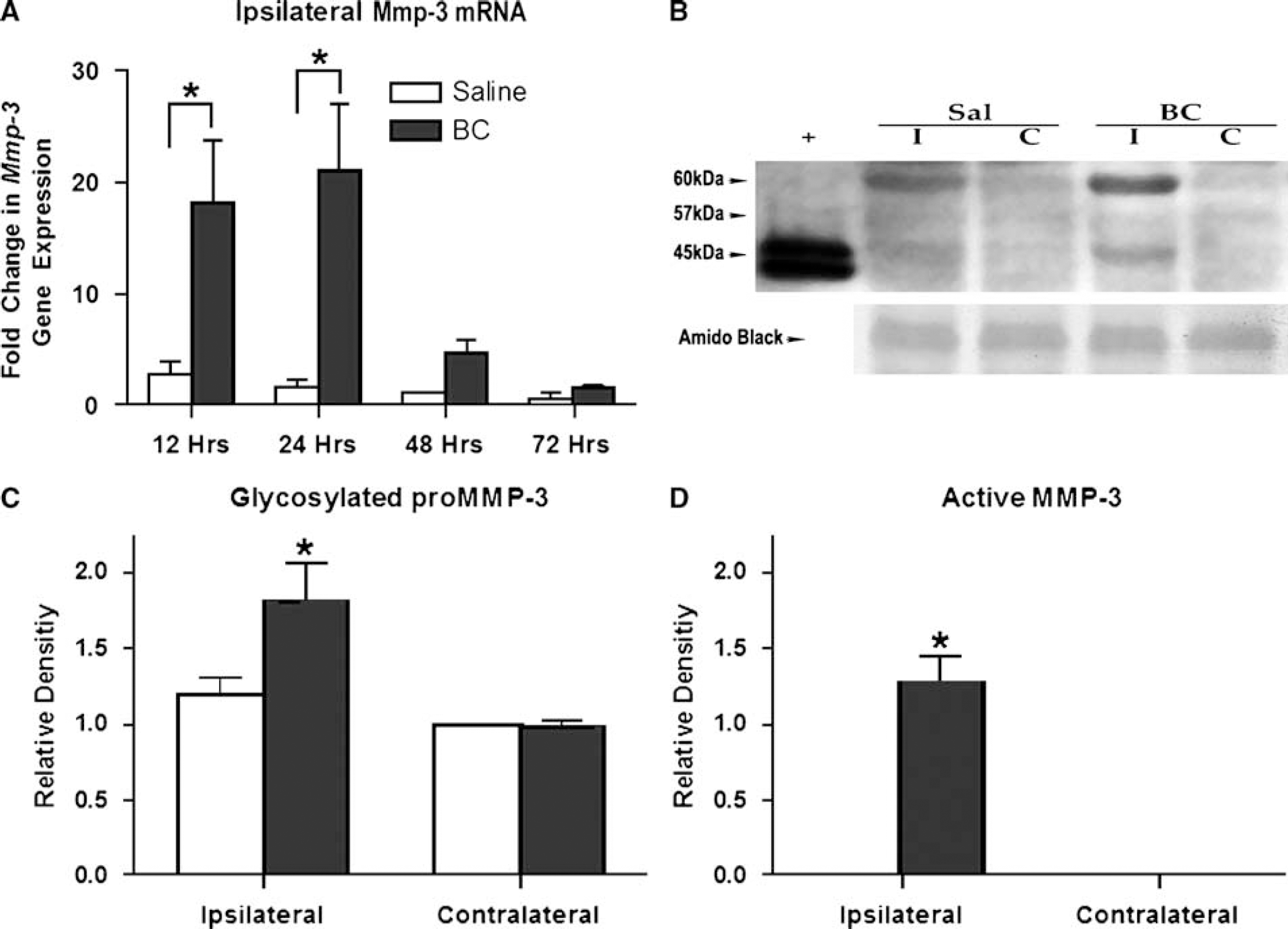
Real-time PCR time course for Mmp-3 mRNA and densitometric analysis of Western immunoblotting after 72 h of either saline or BC injection. (
TUNEL-Labeled Cells and Caspase Induction and Activation
We observed that TUNEL-labeled cells began to appear in the lateral regions of the hemorrhage at 24 h with a dramatic increase by 72 h within and surrounding the hemorrhage, and that time was selected for measurements of caspases and stereological analysis.
The presence of TUNEL-labeled cells suggested caspase-mediated cell death; therefore, we measured caspase-3, -8, and -9, which are increased in apoptotic cell death. Western blots showed the presence of caspase-3 (Figure 4A). By 2 h after ICH, we observed an increase in pro-caspase-3 levels in the BC-injected side compared to the saline-injected side, whereas no changes were found in either the BC contralateral or the saline-injected hemisphere (Figure 4B). For the BC-injected striata, there was a decrease in caspase-3 levels by 24 h, which increased again at 72 h. Active forms of caspase-3 were not observed.
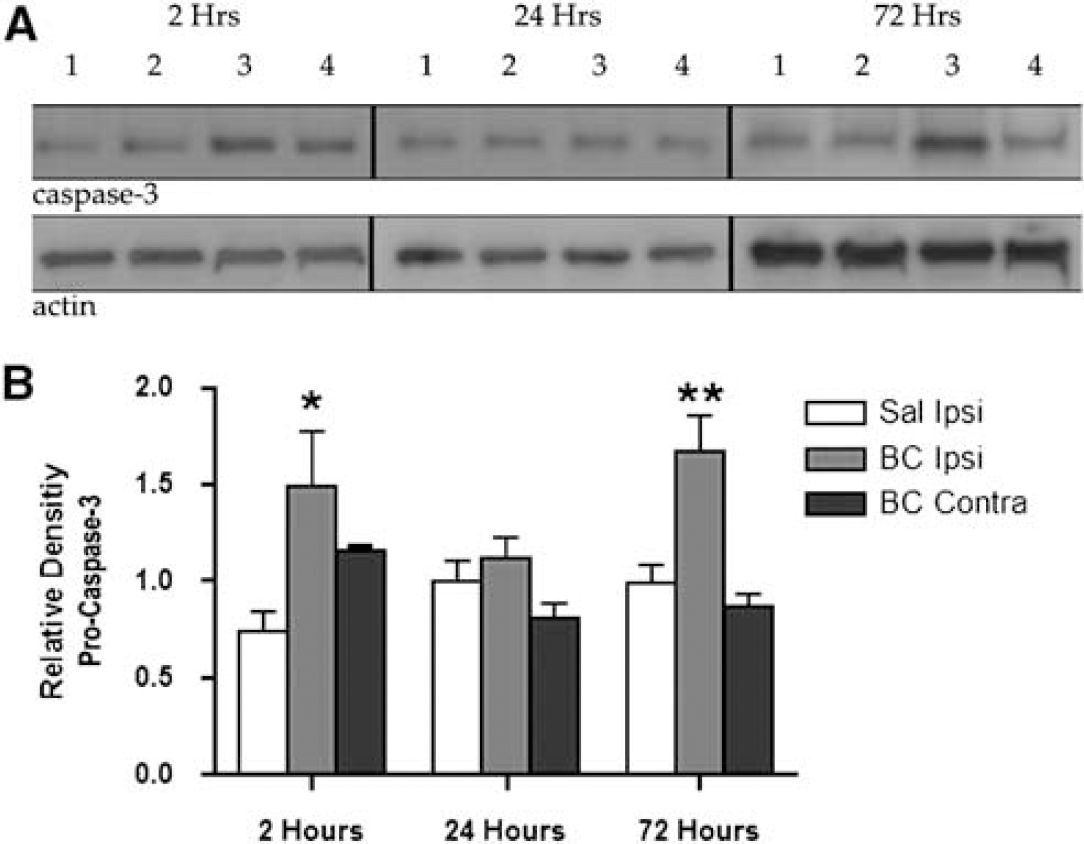
Caspase-3 analysis after saline or BC injection. (
Caspase-3 is an end product of both the extrinsic and intrinsic caspase cascades. However, caspase-8 is an initiator caspase involved in the extrinsic death receptor-mediated pathway. We found that pro-caspase-8 (57 kDa) was expressed at all time points in injected and uninjected regions measured (Figure 5A) and that the BC-injected hemisphere showed statistically elevated levels of pro-caspase-8 at the 2 h time point (Figure 5B). More importantly, we found that cleaved, active caspase-8 (18 kDa) was present in the BC-injected tissue and was statistically significant at 2 and 24 h (Figure 5C). Caspase-9 was not induced at 2 and 24 h; however, by 72 h, both the saline-injected ipsilateral and BC-injected contralateral hemispheres were significantly elevated compared with the BC-injected striata. The active species, present under all conditions, was not significantly altered after saline or BC injection (data not shown).
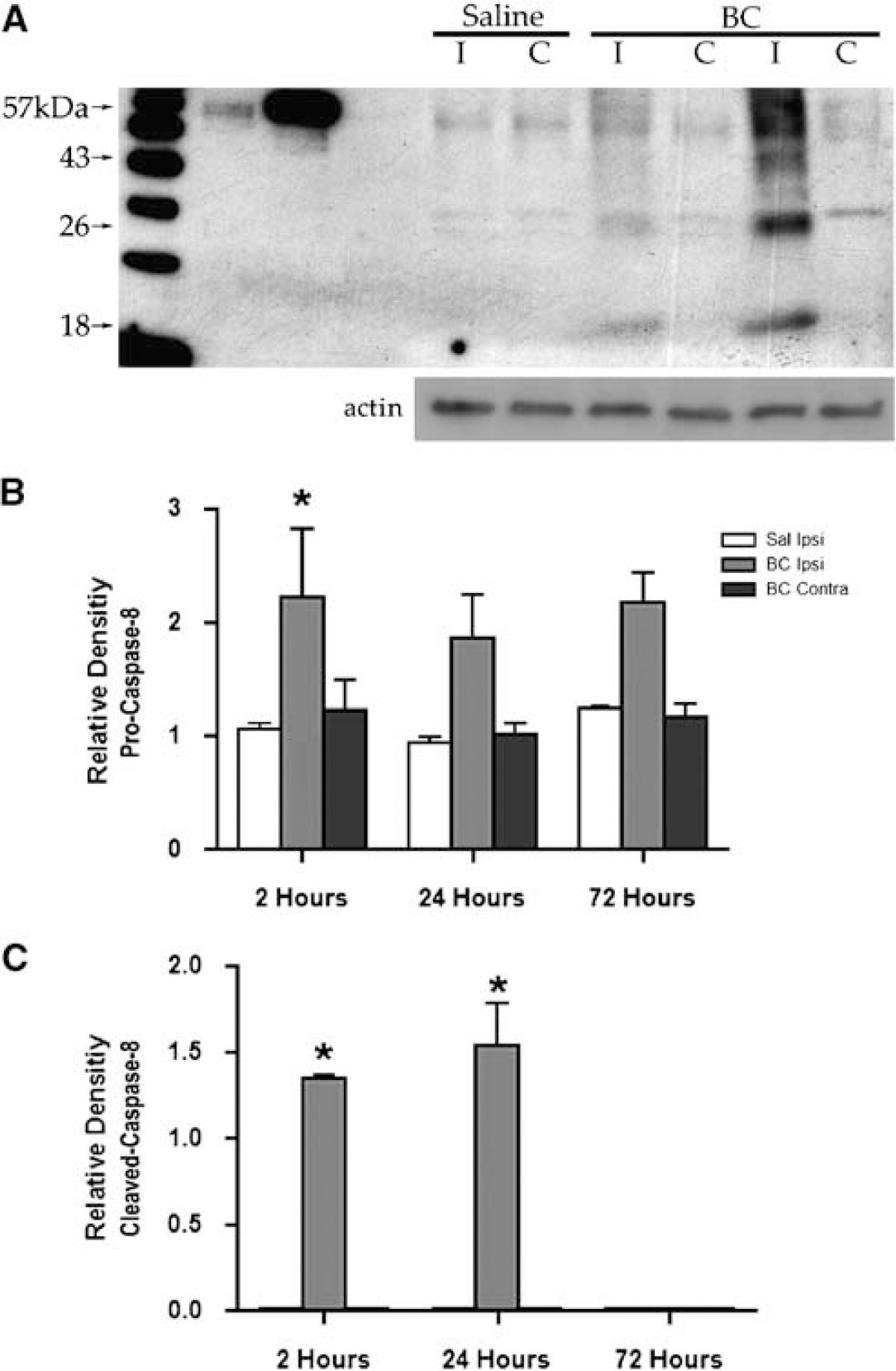
Western immunoblot analyses for pro (57 kDa) and active caspase-8 (18 kDa). (
Tissue Inhibitor of Metalloproteinase-3 Does Not Facilitate Cell Death
Two approaches were used to investigate the role of TIMP-3 in cell death: we measured the expression of Timp-3 mRNA with real-time PCR after CIH and the contribution of TIMP-3 to cell death and hemorrhage volume using Timp-3 knockout mice. Timp-3 null mice did not show a change in either hemorrhage volume or the number of TUNEL-labeled cells measured (data not shown). Furthermore, gelatinase production and activation were similar in the Timp-3-deficient mice compared to wild-type mice (data not shown). A comparison of Timp-3 mRNA levels after either saline or BC intracerebral injection over the time course of 12 to 72 h was made in the ipsilateral injected and contralateral noninjected striata. No increase in mRNA production was observed over the time course measured (data not shown).
BB-94 Increases Cell Death and Does Not Reduce Edema
As TIMP-3 did not appear to play a role in CIH-induced cell death, we investigated the role of MMPs by the use of a broad-spectrum MMP inhibitor, BB-94, after 72 h of CIH. Without treatment the CIH volume was approximately 11 mm3 (Figure 6A), and the number of TUNEL-labeled cells was stereologically estimated to be 100,000 cells (Figure 6B). We found that BB-94 significantly increased the size of the hemorrhage (Figure 6A) and significantly increased the number of TUNEL-labeled cells (Figure 6B).
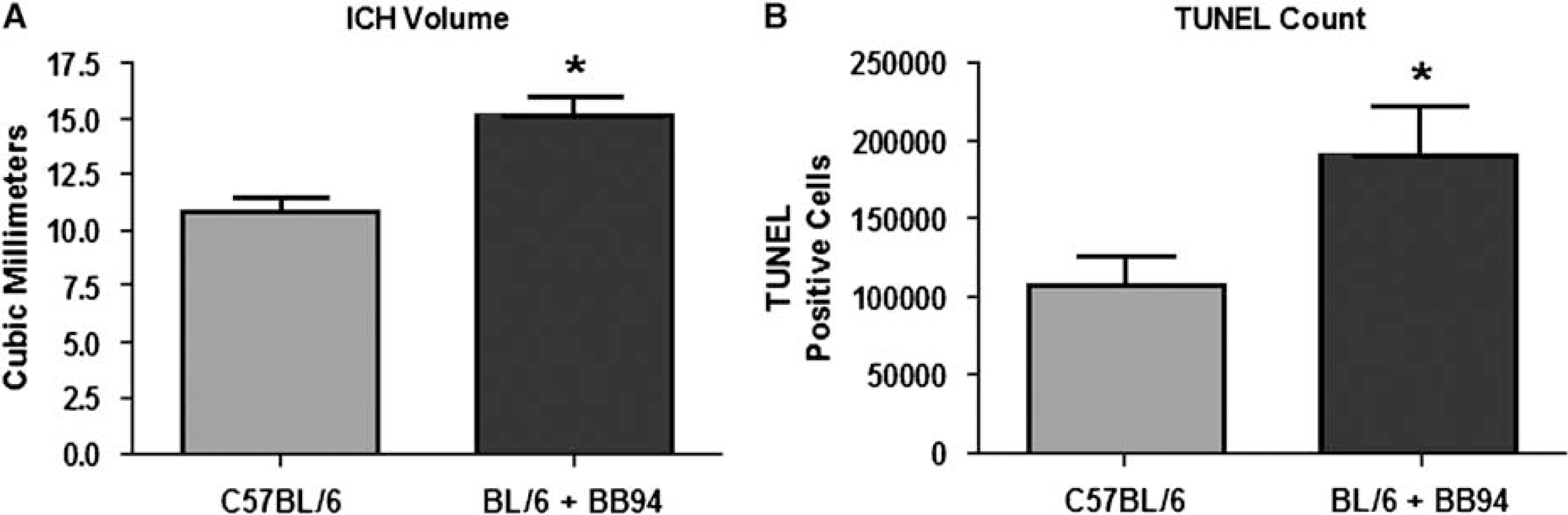
Matrix metalloproteinase inhibition increases cell death and hemorrhage volume after 72 h of CIH. (
Immunohistochemical staining to colocalize TUNEL-labeled cells with cell type revealed dual labeling for neurons, astrocytes, and neutrophils after 72 h of CIH in BB-94-treated mice (Figures 7B to 7D, respectively). Endothelial cells and microglia/macrophages failed to show colocalization with TUNEL (data not shown).
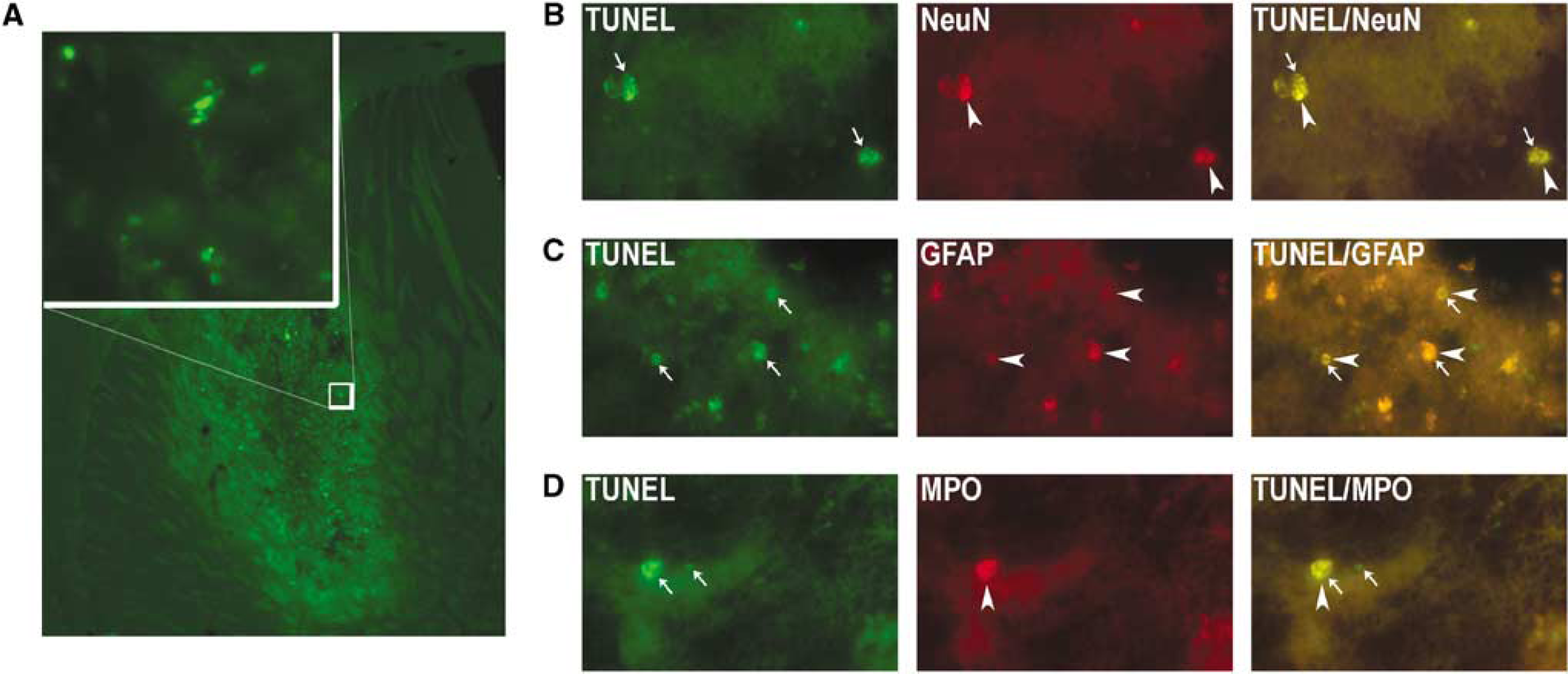
TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeled)-labeled cells after 72 h of CIH and BB-94 treatment. (
Brain edema was measured after 24 h of CIH in animals with and without BB-94 treatment. Five of the animals were treated with the broad-spectrum hydroxamate MMP inhibitor BB-94 (30 mg/kg, intraperitoneally) 10 mins before BC was injected intracerebrally and compared with seven untreated CIH mice. No significant reduction in brain water content was seen with treatment (data not shown).
Discussion
Intracerebral hemorrhage leads to a progressive growth of the mass lesion that is associated with an increase in MMPs. We found a significant increase in mRNA and protein for MMP-3. Although the mRNA for MMP-2 and -9 failed to show a statistical increase after BC injection, elevation of pro-MMP-9 suggested an influx of MMP-9-laden neutrophils. Caspase-mediated cell death occurred with an increase in latent caspase-3 and latent and active caspase-8. The presence of cleaved caspase-8 suggested involvement of the extrinsic cell death pathway and the cell surface death receptors of the tumor necrosis factor (TNF)-α superfamily. Tissue inhibitor of metalloproteinase-3, which enhances cell death in ischemic injury, failed to affect cell death in CIH. However, a broad-spectrum MMP inhibitor caused an increase in TUNEL-labeled cells. Our results suggest that inhibition of the sheddase MMP-3 by the synthetic inhibitor BB-94 in the absence of TIMP-3 induction could have contributed to cell death.
Cell death has been associated with the extrinsic death receptor-mediated pathway, which includes the death receptors of the TNF superfamily, namely Fas (CD95) with its cognate ligand, FasL, and the p55 (TNFR1) TNF receptor and its ligand, TNF-α. Shedding of death receptors by MMPs attenuates the death signal; MMP inhibitors stabilize the receptors through sheddase inhibition, facilitating cell death (Powell et al, 1999; Mitsiades et al, 2001; Cunningham et al, 2005). Tissue inhibitor of metalloproteinase-3 promotes cell death by inhibiting MMP-3, which blocks the shedding of Fas/FasL, and by inhibiting TNF-α-converting enzyme (TACE), which activates TNF-α and causes the shedding of TNFR1 (Black et al, 1997; Amour et al, 1998; Cunningham et al, 2005). Tumor necrosis factor-α signaling through its receptors may either facilitate cell death or promote cell survival (Hallenbeck, 2002). As MMP-3 was increased and TIMP-3 was unaffected in CIH, it is possible that the MMP inhibitor, BB-94, promoted cell death by inhibiting the protective sheddase function of MMP-3.
Matrix metalloproteinases are induced in the brain in response to hypoxic/ischemic and inflammatory lesions. We found elevated levels of pro-MMP-9 activity by zymography despite the absence of a statistically significant mRNA induction between BC and saline injection; the lack of mRNA transcripts with the coincident elevated protein production suggests an exogenous source, such as neutrophils, which contain prepackaged MMP-9 protein (Opdenakker et al, 2001a). Matrix metalloproteinase-3, however, is an endogenously produced MMP, which has been shown to be important in lipopolysaccharide-induced inflammatory opening of the blood—brain barrier and in protecting neurons from apoptosis (Wetzel et al, 2003; Gurney et al, 2006). Additionally, MMP-3 and -7 cleave membrane-bound FasL to possibly attenuate the death signal (Cunningham et al, 2005). Direct action of the MMPs has been implicated in cell death in ischemia, but this seems unlikely in CIH because the MMP inhibitor, BB-94, promoted cell death.
We have shown in ischemic stroke that TIMP-3 is associated with TUNEL-labeled neurons, and that mRNA and protein for Timp-3 are expressed within hours after an ischemic insult (Wallace et al, 2002). The presence of TIMP-3 in the adult brain under normal conditions is very low; however, after cerebral ischemia, it becomes elevated in the cortical neurons that undergo death in the 90 mins focal ischemia model in the rat (Rosenberg et al, 2001; Wallace et al, 2002; Cunningham et al, 2005). However, in cultured astrocytes and neurons, TIMP-3 is constitutively expressed (Wetzel et al, 2003; Liu et al, 2007). Unlike the induction seen for Timp-3 mRNA after 90 mins of middle cerebral artery occlusion in rat, (Wallace et al, 2002) our results show that Timp-3 mRNA levels are unaltered after saline or BC injection from 12 to 72 h. The Timp-3 knockout animal shows similar MMP activity compared to the wild-type mice owing to the lack of any Timp-3 induction after CIH.
In vitro studies, using the chemotherapeutic agent Dox to induce Fas-mediated apoptosis, showed that TIMP-3 facilitates cell death through the extrinsic pathway by inhibiting the cleavage of Fas/FasL from the cell surface by MMP-3 (Wetzel et al, 2003). Although evidence from ischemic infarctions suggested a role of TIMP-3 in apoptosis, when we induced a CIH in the Timp-3 knockout mouse, there was no effect on the numbers of apoptotic cells or hemorrhage volume measured compared to wild-type mice. The absence of Timp-3 mRNA induction in CIH suggests that a hypoxic stimulus is necessary for the induction of Timp-3 mRNA.
In an earlier study, we found a beneficial effect of the MMP inhibitor BB-1101 in the CIH model in the rat, and more recently another group showed that the broad-spectrum MMP inhibitor GM6001 reduced hemorrhage size and improved recovery in a mouse model of CIH (Rosenberg and Navratil, 1997; Wang and Tsirka, 2005). BB-1101 was effective in reducing posterior edema in the rat CIH model at 24 h, which was coincident with elevated MMP-9 levels (Rosenberg and Navratil, 1997). Additionally, GM6001 reduced brain edema in a murine CIH model in the lesioned hemisphere by 3 days, which paralleled increased gelatinase induction and activation, thereby suggesting a role for MMPs in edema after hemorrhage (Wang and Tsirka, 2005). We were unable to show a reduction in either hemorrhage size or apoptotic cell death with another broad-spectrum MMP inhibitor, BB-94.
There are other possible explanations for the differences in the response to MMP inhibitors in the earlier studies and in our study. BB-94, BB-1101, and GM6001 are able to broadly inhibit the MMPs; however, they have variable inhibitory effects on TACE (Barlaam et al, 1999; Skiles et al, 2004; Rosenberg et al, 2007). Additionally, in a study of lipopolysaccharide-induced inflammation in the mouse, we found differences in response to blood—brain barrier opening between BB-1101 and BB-94 (Rosenberg et al, 2007). Depending on the assay conditions, BB-94 is less potent than BB-1101 as an inhibitor of TACE. Although GM6001 is reported to inhibit TACE, consistent inhibitory constants are difficult to obtain from the literature and differences in bioavailability and pharmacokinetics of the inhibitors in the systems tested contribute to the variances. We did not measure TNF-α levels in our study; however, others have shown that it is elevated in the CIH model (Mayne et al, 2001). It is possible that the beneficial effect of BB-1101 and GM6001 was due to their ability to block the action of TACE, which would reduce the cell death induced by TNF-α (Hallenbeck, 2002). It remains possible that negligible TACE inhibition by BB-94 could leave the cell vulnerable to the adverse effects of TNF-α, resulting in increased edema and cell death through TNFR1.
In a study of CIH in Mmp-9 null mice, there was an increase in hemorrhage volume and edema in the knockout mice compared to wild-type mice, which correlated with diminished neurologic function and increased mortality (Tang et al, 2004). The finding that Mmp-9 null mice develop larger hemorrhage volumes compared to wild-type mice parallels our data, which show increased cell death and hemorrhage size after MMP inhibition with the administration of BB-94. This finding supports the notion that long-term MMP inhibition may be deleterious. More recently, it was found that autologous blood injection caused less edema in Mmp-9 null mice compared to wild-type mice; the different methods of hemorrhage induction may account for the opposite findings (Tejima et al, 2007; Xue et al, 2006).
We were unable to prove the hypothesis that TIMP-3 affects cell death in CIH, as it does in hypoxic/ischemic insults. However, we observed a significant increase in both mRNA and protein for MMP-3 along with an active species. Tumor necrosis factor-α, Fas, and FasL have previously been shown to be elevated after ICH (Lee et al, 2006). Matrix metalloproteinase-3 may shed Fas/FasL and attenuate the death signal. However, cleavage of membrane-bound TNF-α by TACE to form the active soluble form could have occurred as TIMP-3 was not elevated (Hua et al, 2006). The TNF-α-mediated cell death pathway may be responsible for the approximately 100, 000 TUNEL-labeled cells present by 72 h. However, when the mice are treated with the MMP inhibitor BB-94 the number of dying cells is doubled to approximately 200,000 cells with a concomitant increase in hemorrhage volume. Treatment with BB-94 may inhibit cleavage of Fas/FasL by MMP-3, thus facilitating the TNF and Fas death pathways to operate simultaneously and increase cell death.
Footnotes
Acknowledgements
We thank Ed Estrada for surgical support and edema data, Jeffery Thompson for performing the genotyping, and Justin Tibbitts for his assistance with the RT-PCR data.