
Abstract
Enhanced matrix metalloproteinases (MMPs) can cause vasogenic edema and hemorrhagic transformation after cerebral ischemia, and affect the extent of ischemic injury. We hypothesized that the endogenous MMP inhibitors, tissue inhibitor of MMPs (TIMPs), were essential to protect against blood—brain barrier (BBB) disruption after ischemia by regulating the activities of MMPs. We confirmed the transition of MMP-2 and MMP-9, and the TIMPs family after 30 mins of middle cerebral artery occlusion, and elucidated the function of TIMP-1 and TIMP-2 in focal ischemia, using TIMP-1−/−and TIMP-2−/− mice. TIMP-1 mRNA expression was gradually increased until 24 h after reperfusion. In TIMP-1−/− mice, MMP-9 protein expression and gelatinolytic activity were significantly more augmented after cerebral ischemia than those in WT mice, and were accompanied by exacerbated BBB disruption, neuronal apoptosis, and ischemic injury. In contrast, TIMP-2 gene deletion mice exhibited no significant difference in MMP expressions and the degree of ischemic injury despite an increased Evans blue leakage. These results suggest that TIMP-1 inhibits MMP-9 activity and can play a neuroprotective role in cerebral ischemia.
Keywords
Introduction
Cerebral ischemia and postischemic reperfusion cause edema formation and hemorrhagic conversion by the breakdown of the blood—brain barrier (BBB). Brain edema is a significant cause of early neurologic deterioration and mortality after ischemia (Schwab et al, 1998), and although several cascades have been proposed, matrix metalloproteinases (MMPs) are considered key molecules involved in the degradation of the basement membrane, leading to vasogenic edema (Simard et al, 2007).
Matrix metalloproteinases are zinc-dependent endopeptidases that are involved in a variety of cellular activities. The gene expressions of MMPs at the transcriptional level are controlled by growth factors, cytokines, and redox. Matrix metalloproteinases are secreted in the interstitial space or membrane-bound as zymogen and are cleaved to the active enzyme by other MMPs and plasmin through proteolytic processing. Active and latent MMPs are stringently regulated by endogenous tissue inhibitor of metalloproteinases (TIMPs), which, in general, broadly inhibit the activities of MMPs. A balanced interaction between MMPs and TIMPs is essential for the development and extracellular matrix homeostasis. Enhanced expression and activity of MMPs have been observed under numerous pathologic conditions, including not only cerebral ischemia, but also diabetes, atherosclerosis, cancer, and Alzheimer's disease. Thus, inhibition of MMPs is considered a potential therapeutic target (Cunningham et al, 2005; Gasche et al, 2006).
In vertebrates, the TIMPs family consists of four members, TIMP-1, −2, −3, and −4. Tissue inhibitor of metalloproteinase-1, −2, and −4 are secreted types, and only TIMP-3 is bound to the extracellular matrix to impinge on membrane-associated proteins (Lee et al, 2007). The N-terminal domain of the TIMPs broadly inhibits the activity of MMPs via high-affinity, noncovalent binding to the MMP catalytic domain, whereas the C-terminal domain of TIMPs forms a complex with pro-MMPs, and plays diverse roles. For example, the TIMP-1-proMMP-9 complex inhibits proMMP-9 activation. In contrast, the TIMP-2-proMMP-2 complex facilitates proMMP-2 activation (Brew et al, 2000; Gasche et al, 2006). Tissue inhibitor of metalloproteinases have also been shown to play roles in cell proliferation, differentiation, and angiogenic processes through mechanisms independent of MMP inhibition (Reed et al, 2003; Perez-Martinez and Jaworski, 2005).
In an acute cerebral ischemia, upregulated expression of MMP-2 and MMP-9 are associated with BBB disruption that may exacerbate neuronal damage (Rosenberg et al, 1998; Gasche et al, 1999; Copin et al, 2005; Yang et al, 2007) and hemorrhagic transformation following recombinant tissue plasminogen activator (Wang et al, 2003). Furthermore, hypothermia was shown to reduce ischemic injury by a decrease in MMP activities (Wagner et al, 2003). Leukocytes are considered as a key source of MMP-9 (Gidday et al, 2005). With regard to TIMPs, the expression of TIMP-1 in the normal central nervous system is very low, whereas TIMP-2, −3, and −4 are constitutively expressed. TIMPs expressions in the central nervous system can also be upregulated in response to MMPs under various pathologic conditions (Pagenstecher et al, 1998; Aoki et al, 2007). However, the function of TIMPs in acute cerebral ischemia, especially with respect to their influence on MMP levels and BBB disruption, is less well understood.
In hippocampal primary and organotypic cultures, TIMP-1 protected against glutamate-induced cytotoxicity by decreasing neuronal calcium influx (Tan et al, 2003). Adenovirus-mediated gene transfer of TIMP-1 and TIMP-2 were neuroprotective in global ischemia (Magnoni et al, 2007). Additionally, transplantation of autologous bone marrow cells overexpressing TIMP-1 and TIMP-2 reduced focal ischemic damage and promoted functional recovery with enhanced neurogenesis and angiogenesis (Baker et al, 2007). However, in those studies, the neuroprotective mechanisms of TIMP-1 and TIMP-2 were either independent of MMPs or unexplained.
Thus, in the present study, we evaluated the kinetics of MMPs and TIMPs after cerebral ischemia. Moreover, using TIMP-1−/− and TIMP-2−/− mice, we assessed the role of TIMP-1 and TIMP-2 on MMP proteolytic activities, BBB breakdown, neuronal apoptosis, and ischemic damage after ischemia/reperfusion injury.
Materials and methods
Mice
All experimental procedures were approved and carried out in accordance with the Institutional Animal Care and Use Committee of the Kyoto University. C57BL/6 male mice (WT), TIMP-1−/− mice (T1KO), and TIMP-2−/− mice (T2KO), weighing 20 to 30 g, were used. TIMP-1−/− and TIMP-2−/− mice, whose genetic background was C57BL/6, were generously gifted from Dr PD Soloway (Cornell University, NY, USA) (Lee et al, 2005; Wang et al, 2000).
Transient Focal Cerebral Ischemia
Cerebral ischemia was induced by using the standard intraluminal middle cerebral artery occlusion (MCAO) method as described previously (Longa et al, 1989). Briefly, anesthesia was induced in the mice by administration of 3% halothane in 30% oxygen and 70% nitrous oxide, and maintained using a mixture containing 1% halothane. Rectal temperature was maintained at 37.0°C ± 0.5°C with a thermostat-controlled heating pad. After 30 mins MCAO with a 8.0 nylon monofilament coated with silicon, the filament was withdrawn and the mouse placed in a thermally controlled incubator (32.0°C) for 2 h. Regional cerebral blood flow was measured with a laser-Doppler flowmetry (Omegawave, Tokyo, Japan) to confirm the induction of ischemia and reperfusion. The surgical procedure was considered successful as >80% reduction from baseline throughout ischemia and >60% recovery within 5 mins after reperfusion. In additional subgroups, the left femoral artery was cannulated for blood pressure monitoring (iWorx, Dover, NH, USA) and sampling. Blood samples were taken before ischemia, during (15 mins after occlusion), and after reperfusion. The samples were analyzed for blood gasses (PaO2 and PaCO2) and pH using a blood gas analyzer (FUSO, Osaka, Japan).
Cerebrovascular Angioarchitecture
The vascular casting method was performed as described previously (Maeda et al, 1998). Mice were anesthetized with halothane and maintained at 37°C using a heating pad. Afterward, papaverine hydrochloride (40 mg/kg, intravenously) was injected to induce maximal vasodilatation. A white Latex casting compound (Vultex, catalog number 563; Chicago Latex Production) was mixed with 50 μL/mL carbon black (Pelikan 4001, Brilliant—Schwarz Brilliant black) and was perfused transcardially. Next, mice were placed on ice for 20 mins. After 10% formalin fixation for overnight, the brains were removed and digitally imaged, using image analysis software (Image Pro Plus 6.0, Olympus). The posterior communicating artery plasticities were graded as follows: Grade 0, no connection between posterior and anterior circulation; Grade 1, capillary connection; Grade 2, small truncal connection; and Grade 3, truncal connection (Kawase et al, 1999). To evaluate the cortical surface area supplied by MCA, we delineated vascular boundaries between the anterior cerebral artery and MCA, counted the number of anastomoses, and measured the distance from midline at 2, 4, and 6 mm from the frontal pole (Asahi et al, 2001b).
Reverse Transcription-PCR
Under normal conditions and at 3 and 24 h after reperfusion, mice received an overdose of sodium pentobarbital and halothane. Total RNA was isolated from hemispheric tissue and converted into cDNA using a RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and Omniscript Reverse Transcriptase (Qiagen). Polymerase chain reaction was performed using QIAGEN Fast Cycling PCR Kit (Qiagen). Expression probes were used for target genes TIMP-1, TIMP-2, TIMP-3, TIMP-4, and GAPDH. The primer sets used were as follows: forward 5‘-GGCATCCTCTTGTTGCT ATCACT-3’, reverse 5‘-CTTATGACCAGGTCCGAGTTGC-3’ for TIMP-1; forward 5‘-GGCTGTGAGTGCAAGATCACTCG CT-3’, reverse 5‘-TCTTGATGCAGGCGAAGAACTTGGC-3’ for TIMP-2; forward 5‘-TGAGCTCGGACTGTAGCATCA-3’, reverse 5‘-AGGCTCCAACAGCTCAGGAG-3’ for TIMP-3; forward 5‘-TCACCACTTGCTATGCAGTGCCATGTA-3’, reverse 5‘-CTGCAGATGCCATCAACATGCTTCA-3’ for TIMP-4; and forward 5‘-TTGTCAAGCTCATTTCCTGGT ATG-3’, reverse 5‘-GGATAGGGCCTCTCTTGCTCA-3’ for GAPDH. The polymerase activation steps were 35 cycles of 96°C for 5 secs for denaturation, 54°C for 5 secs for annealing, and 68°C for 9 secs for the extension. All of the data were analyzed using GAPDH as a reference.
Western Blot Analysis
Under normal conditions and at 3 and 24 h after reperfusion, mice were deeply anesthetized as described above, then transcardially perfused with ice-cold phosphate-buffered saline. Hemispheric tissue was frozen immediately in liquid nitrogen and stored at −80°C. The tissue was homogenized on ice in lysis buffer containing protease inhibitors. After centrifugation, the supernatant was collected and the protein concentration of each sample was estimated by the Lowry method using a protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Aliquots of the lysate containing 20 μg protein underwent electrophoresis on 4% to 15% sodium dodecyl sulfate Polyacrylamide gel, and then transferred to nitrocellulose membranes (Bio-Rad Laboratories). After blocking for 1 h at room temperature with 5% ECL Blocking Agent (GE Healthcare, Buckinghamshire, UK), blots were incubated overnight at 4°C with the following primary antibodies: rabbit anti-MMP-2 (1:2,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-MMP-9 (1:5,000, Chemicon, Billerica, MA, USA), goat anti-collagen type IV (1:500, Southern Biotechnology Associates, Birmingham, AL, USA), rabbit anti-ZO-1 (1:125, Zymed Laboratories, South San Francisco, CA, USA), rabbit anti-Claudin5 (1:250, Zymed Laboratories), and rabbit anti-Cleaved Caspase-3 (1:1,000, Cell Signaling, Danvers, MA, USA). Peroxidase-conjugated anti-rabbit antibody was used as the secondary antibody and then antigens were detected using the standard chemical luminescence method (ECL; Amersham Pharmacia Biotech, Buckinghamshire, UK). ECV304 cell lysate (Santa Cruz Biotechnology) and murine MMP-9 (Chemicon) were used as positive controls for MMP-2 and MMP-9, respectively.
Gel Zymography
Similarly prepared protein samples as for western blot analysis were subjected to gelatin zymography with Gelatin Zymo-Electrophoresis Kit (Primary Cell, Hokkaido, Japan) according to the manufacturer's directions.
Blood—Brain Barrier Permeability
Blood—brain barrier disruption after ischemia was quantitatively evaluated using fluorescent detection of extravasated Evans blue dye (Asahi et al, 2001b). Briefly, 10 mins after reperfusion, mice received an intravenous injection of Evans blue dye (4 mL/kg of a 2% Evans blue solution in phosphate-buffered saline). At 24 h after reperfusion, ischemic hemispheres from transcardially perfused mice were homogenized in 1 mL of 50% trichloroacetic acid and centrifuged. The supernatant was diluted four-fold with ethanol. A fluorescent plate reader (620 nm excitation and 680 nm emission) was used to measure dye concentrations. The amount of extravasated Evans blue was expressed as nanograms per ischemic hemisphere.
In Situ Labeling of DNA Fragmentation
Mice were transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (PFA) at 24 h after reperfusion. Brains were postfixed for 12 h in 4% PFA, and then frozen in Tissue-Tek OCT Compound (Sakura Finetechnical, Tokyo, Japan) and cut at 40 μm thickness. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed using a commercially available detection kit (Chemicon). The sections were incubated in a TdT-labeling reaction mixture for 60 mins at 37°C and then with anti-digoxigenin conjugate. The number of TUNEL-positive cells was counted in four regions, the caudate putamen (medial and lateral) and the cerebral cortex (somatosensory and piriform cortex) in each of three sections at 0.8 mm intervals. Photomicrographs were taken with an Olympus confocal laser scanning microscope (FV-300, Olympus) and positive cell density was expressed as number per mm2.
Infarct Volume
Serial coronal brain sections separated by 400 μm intervals at various coronal levels (+ 1.2, +0.8, +0.4, +0, −0.4, −0.8, and −1.2 mm from the bregma) were obtained for immunohistochemistry analysis. The infarct areas were quantified from cresyl violet-stained sections using Image Pro Plus 6.0. The infarct volume was corrected for edema and expressed in mm3 units (McColl et al, 2007).
Neurologic Evaluation
Neurologic deficits were scored at 24 h after reperfusion on a 5-point scale: 0, no observable neurologic deficit; 1, failure to extend the right forepaw fully; 2, spontaneous turning to the right side; 3, circling to the right side; and 4, no spontaneous movement and/or inability to maintain upright posture.
Statistical Analysis
Quantitative data were expressed as mean ± s.d. Statistical comparisons were conducted using the Kruskal—Wallis test for intergroup comparisons. P-values of < 0.05 were considered statistically significant.
Results
Tissue Inhibitor of Metalloproteinases, and MMP-2 and MMP-9 Expression in Wild-Type Mice After Focal Ischemia
The transcriptional activities of TIMPs, MMP-2 and MMP-9 in ischemic hemispheres were analyzed in wild-type (WT) mice until 24 h after focal ischemia. Upregulated MMP-2 at 3 h (P < 0.03) and MMP-9 at 3 and 24 h (P < 0.03) after ischemia were confirmed. Additionally, the TIMP-1 mRNA level was quite low under normal conditions, and gradually increased up to 24 h after ischemia (P < 0.03) (Figures 1A and 1B). There was no significant difference in TIMP-2, −3, and −4 expressions during procedure (data not shown). From these results, we focused on TIMP-1 and TIMP-2, which are involved in the activation of MMP-2, and estimated the association with MMP-2 and MMP-9 using TIMP-1−/− and TIMP-2−/− mice.
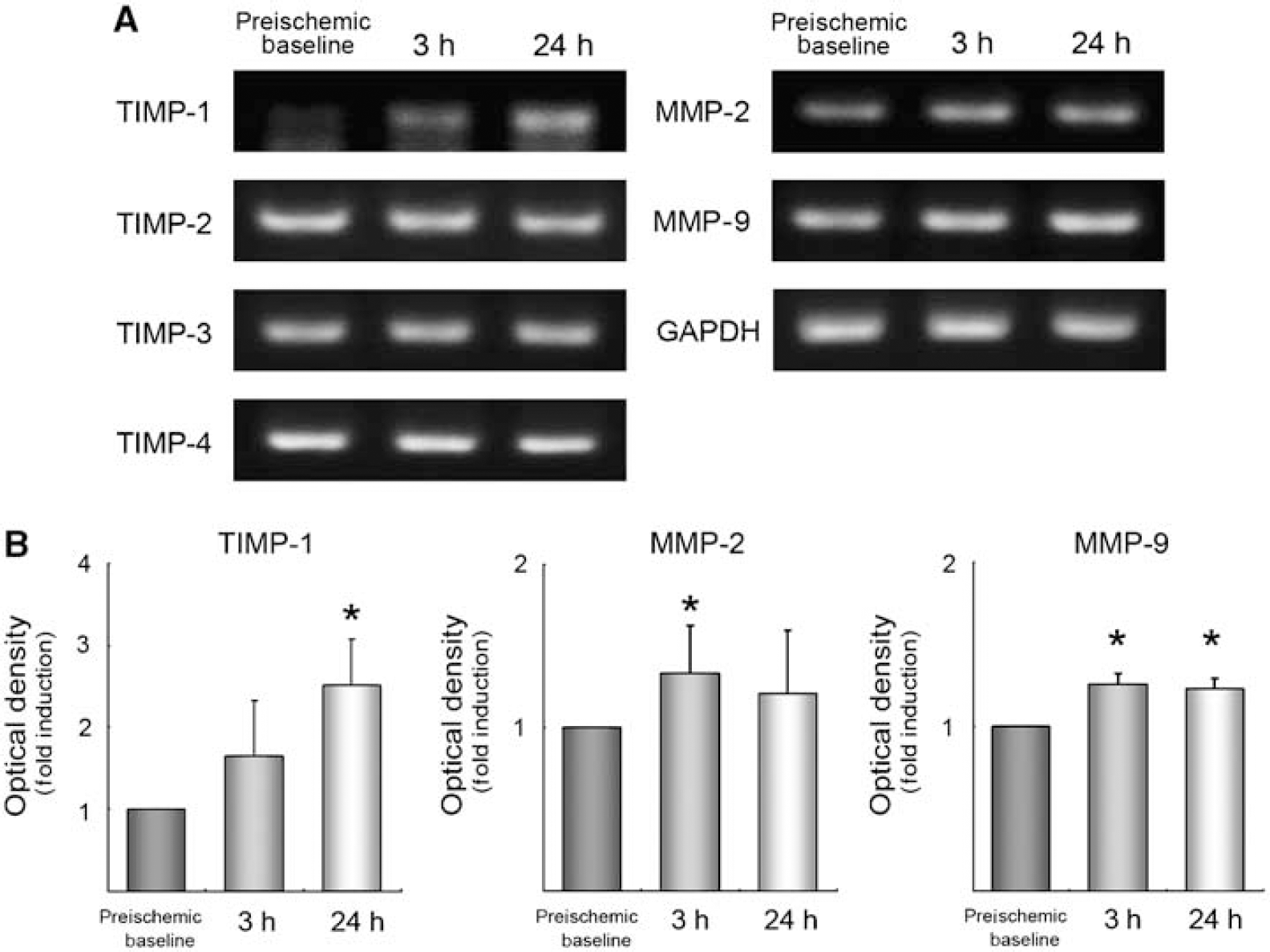
Induction of TIMPs/MMPs after cerebral ischemia. RT-PCR (
Physiologic Parameters and Angioarchitecture
There were no differences in physiologic parameters among WT, TIMP-1−/−, and TIMP-2−/− mice before, during, and after ischemia (Table 1A). The ratio of regional cerebral blood flow in procedure to baseline measured by laser-Doppler flowmetry did not differ significantly (Table 1B).
Physiologic parameters before, during, and after transient focal ischemia
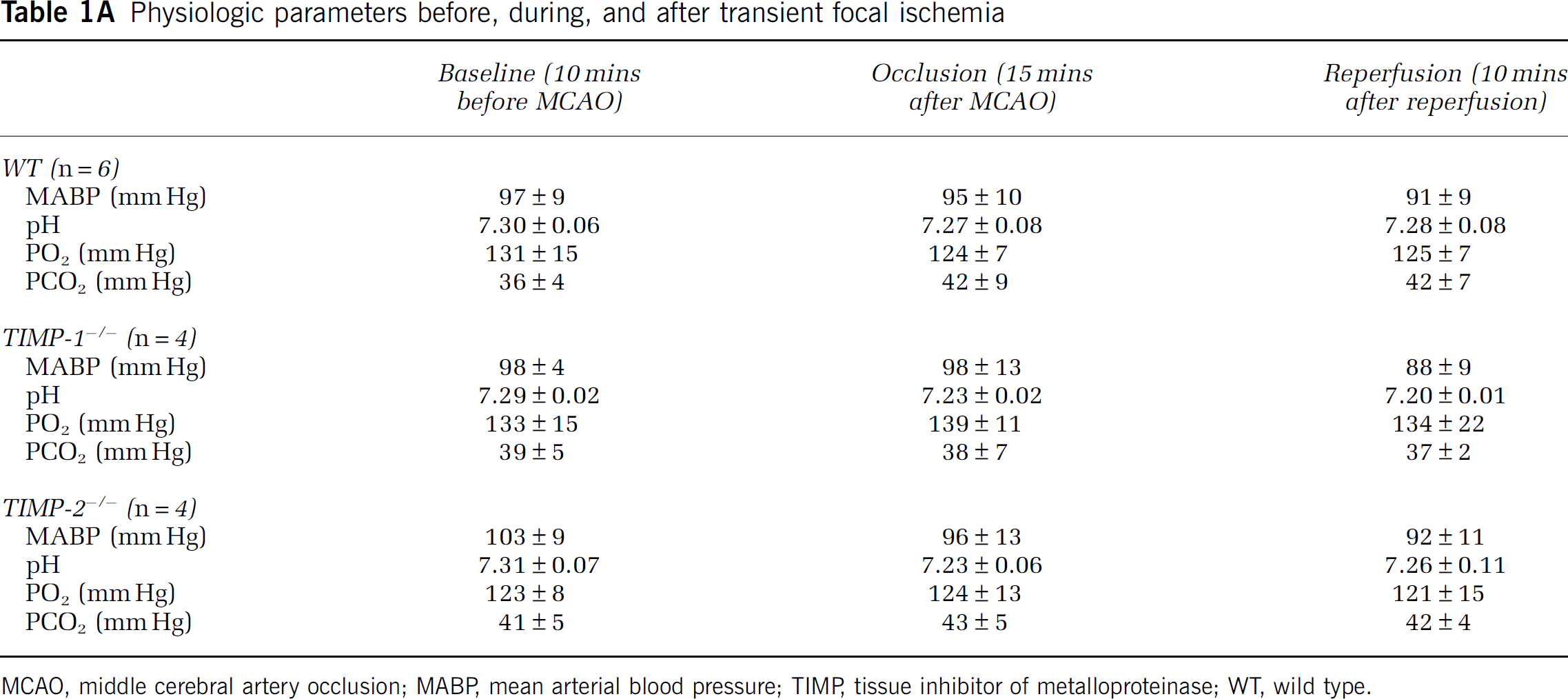
MCAO, middle cerebral artery occlusion; MABP, mean arterial blood pressure; TIMP, tissue inhibitor of metalloproteinase; WT, wild type.
Cortical blood flow by laser Doppler flowmetry during and after ischemia
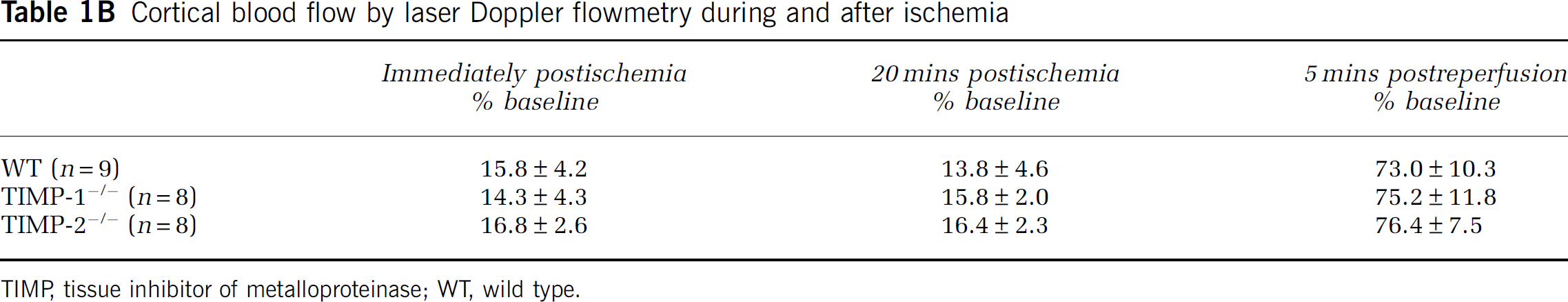
TIMP, tissue inhibitor of metalloproteinase; WT, wild type.
Differences in cerebrovascular angioarchitecture between the groups may affect the degree of ischemic damage when using the intraluminal MCAO method. However, it has been previously shown by light microscopy that there are no anatomic differences in the vascular anatomy of the circle of Willis among the WT, TIMP-1−/−, and TIMP-2−/− mice (Aoki et al, 2007). Furthermore, using vascular casting method, we evaluated the posterior communicating artery patencies. The grading of posterior communicating artery plasticity was not different among groups (Table 1C). We compared the vascular boundaries between anterior cerebral artery and MCA, and counted the number of anastomoses (Table 1C, Supplementary Figure 1). These data showed that gross vascular anatomy was not different among groups.
Angioarchitecture parematers (n=8 per group)
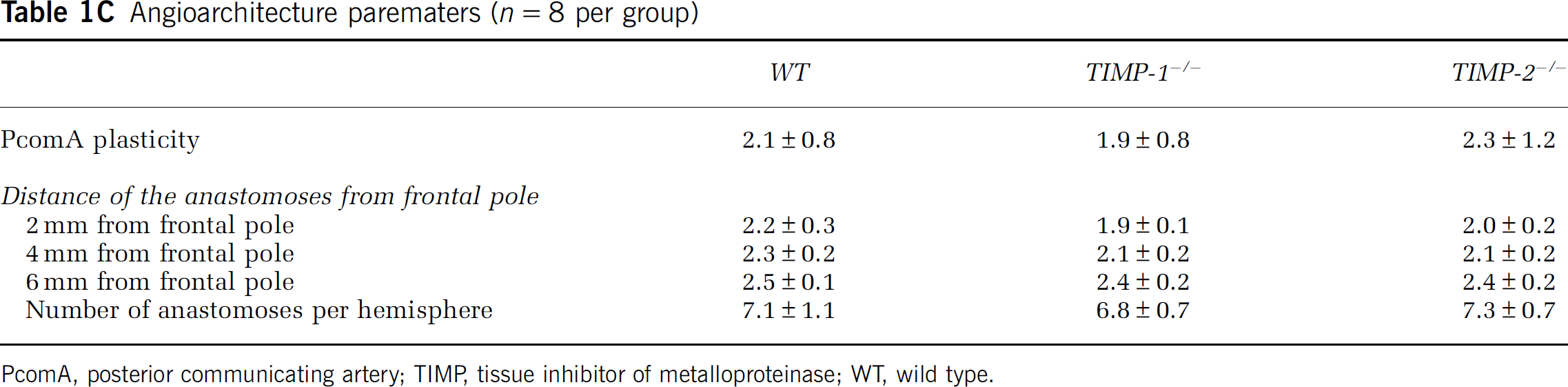
PcomA, posterior communicating artery; TIMP, tissue inhibitor of metalloproteinase; WT, wild type.
MMP-2 and MMP-9 Protein Expression and Activity in Wild-Type, TIMP-1−/−, and TIMP-2−/− Mice
In initial studies, we observed no obvious compensatory mRNA expression of other TIMPs in TIMP-1−/− and TIMP-2−/− gene deletion mice at any time during the experiments (data not shown).
To evaluate the effect of deleted TIMP-1 or TIMP-2 genes on MMPs after cerebral ischemia, using western blotting and zymography, we compared MMP-2 and MMP-9 under preischemic baseline and at 3 and 24 h after ischemia. Zymography was performed to validate gelatinolytic activities of MMP-2 and MMP-9. There were no significant differences in MMP-2 and MMP-9 protein expressions and gelatinolytic activities among different groups under preischemic condition (Figures 2A and 3A). Western blotting analysis showed that MMP-9 protein expression was more augmented in TIMP-1−/− mice than in WT mice at 24 h after ischemia (P < 0.02). There was no significant difference in the MMP-9 expression at 3 h after ischemia and in the MMP-2 expression between WT and knockout groups (Figure 2).
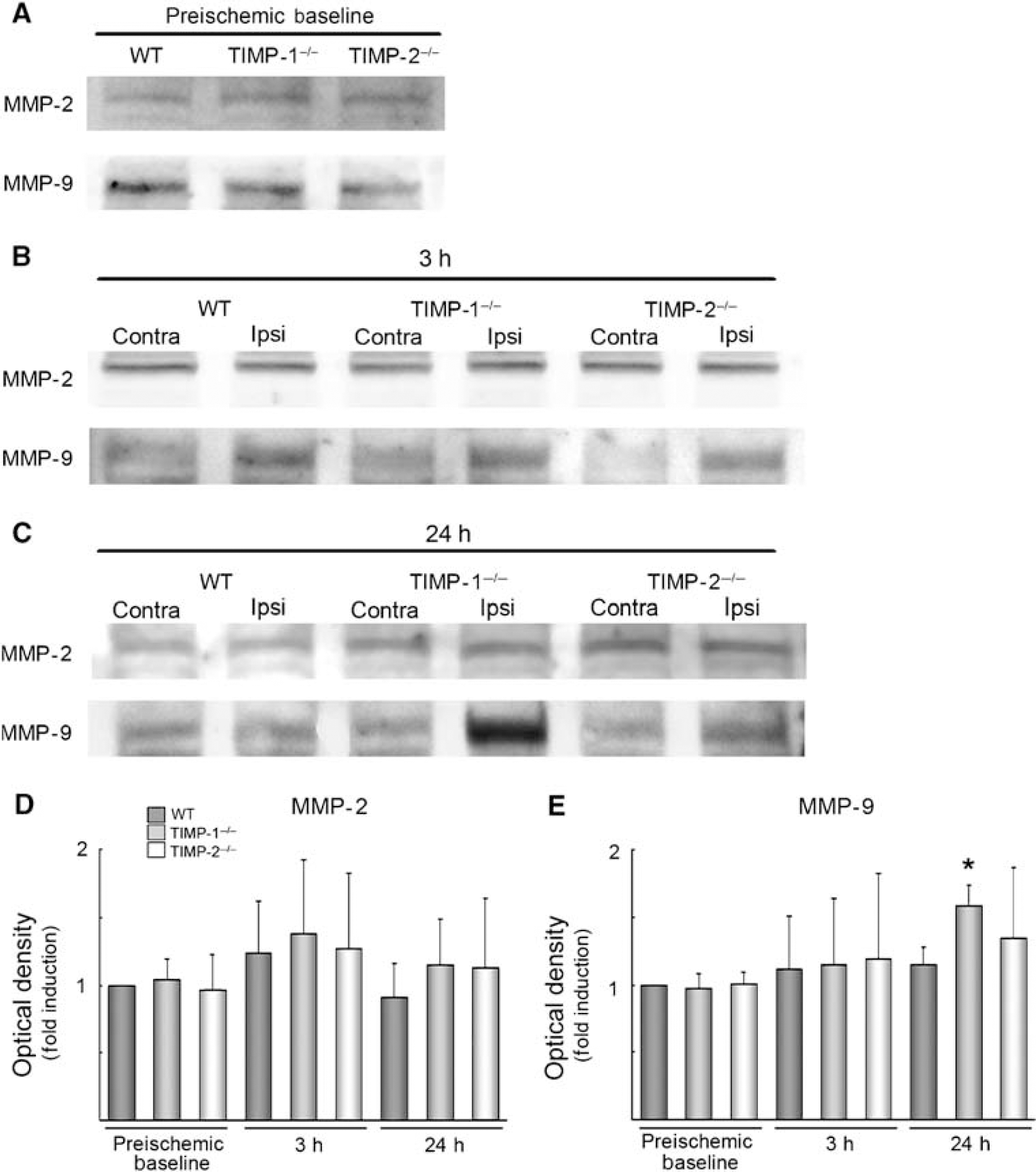
The protein expression of MMP-2 and MMP-9 after focal ischemia in WT, TIMP-1−/−, and TIMP-2−/− mice. Western blot under preischemic baseline (
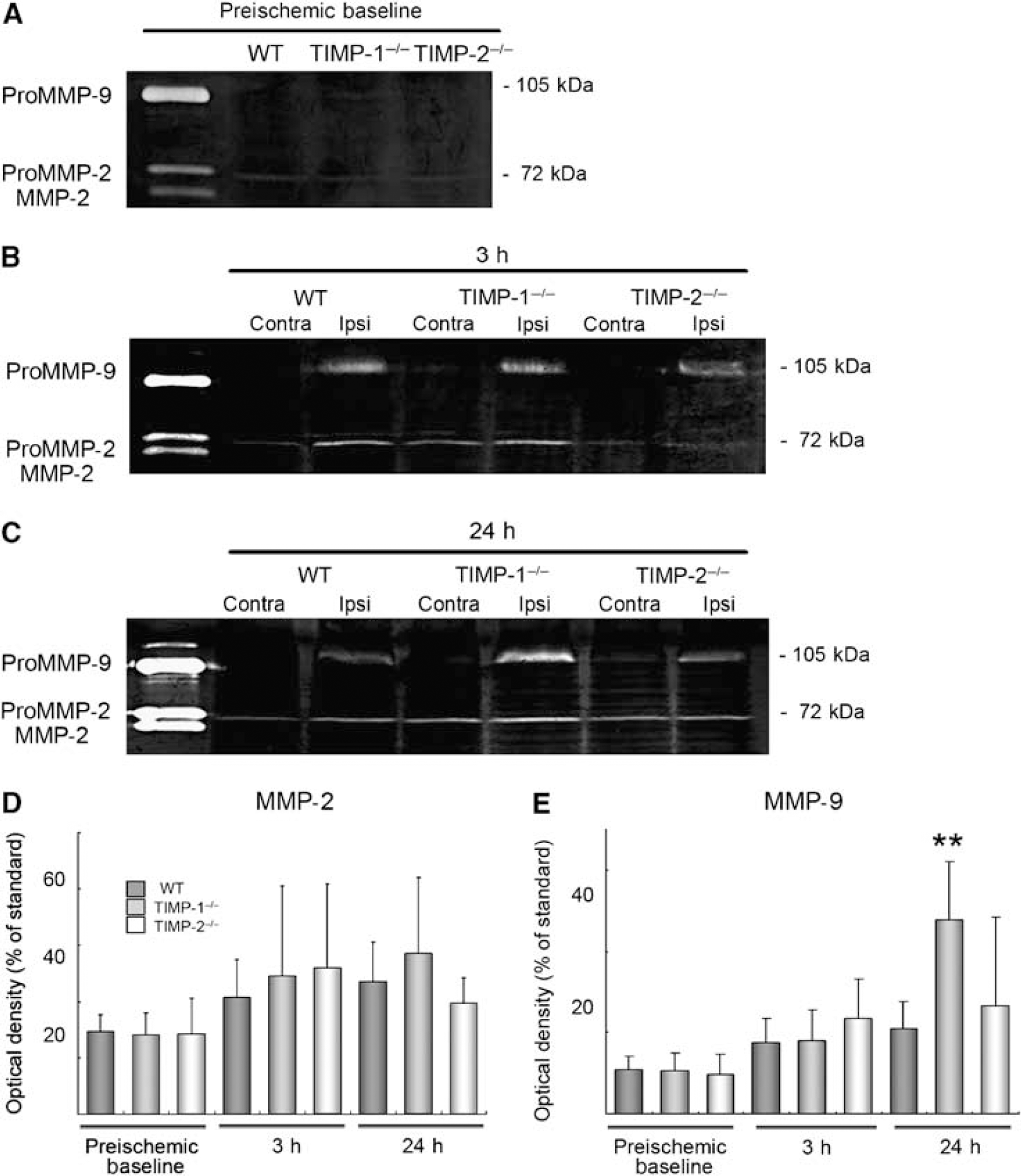
Gelatinolytic activities of MMP-2 and MMP-9 after focal ischemia in WT, TIMP-1−/−, and TIMP-2−/− mice. Gel zymographies under preischemic baseline (
As for results from western blots, zymographic analysis showed that only MMP-9 activity in TIMP-1−/− mice at 24 h after ischemia was more enhanced than in WT mice (P < 0.002) (Figure 3).
Blood—Brain Barrier Leakage and Degraded Tight Junction Protein
We quantified BBB disruption using Evans blue leakage per ischemic hemisphere. Evans blue leakage was used to estimate the extent of postischemic vasogenic edema, where increased permeability is correlated with ischemic injury (Gidday et al, 2005). First, the level of Evans blue leakage before ischemia was not different among groups (mean ± s.d.; WT: 0.26 ± 0.05, TIMP-1−/− mice: 0.29 ± 0.03, and TIMP-2−/− mice: 0.29 ± 0.04). At 24 h after focal ischemia, Evans blue extravasation was significantly increased in TIMP-1−/− (P < 0.002) and TIMP-2−/− mice (P < 0.001) when compared with WT mice. There was no significant difference between TIMP-1−/− and TIMP-2−/− mice (Figure 4A).
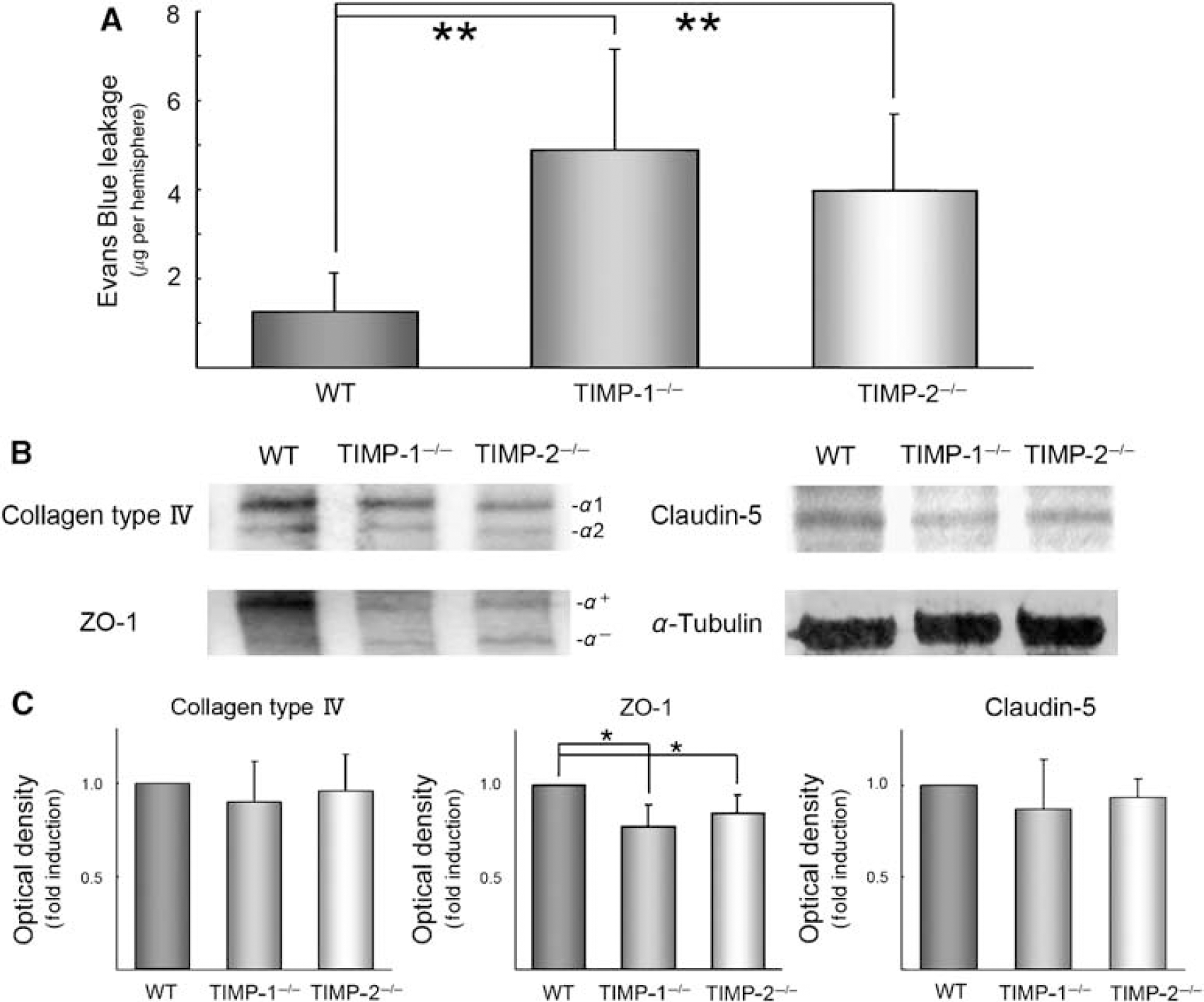
Blood—brain barrier dysruption evaluated by Evans blue leakage (
Western blot analysis of the ischemic hemisphere showed a significant decrescence of ZO-1 expression in TIMP-1−/− and TIMP-2−/− mice compared with WT mice (P < 0.02). However, we could not detect the diminished collagen type IV and Claudin-5 among intergroups (Figures 4B and 4C).
Neuronal Apoptosis and Cleaved Caspase-3
Matrix metalloproteinases and TIMP-3 have been associated with neuronal apoptosis in cerebral ischemia (Wallace et al, 2002; Copin et al, 2005; Wetzel et al, 2007). We evaluated DNA fragmentation by TUNEL staining and cleaved caspase-3 expression in the caudate putamen and the cortex at 24 h after ischemia. Caspase-3 is a major downstream enzyme of apoptosis and responsible for the proteolytic cleavage of key proteins such as the nuclear enzyme poly polymerase. TUNEL-positive cells with chromatin condensation and fragmented nuclei were considered as probable apoptotic cells. Quantitative analysis showed that TUNEL-positive cells before ischemia was not different in both putamen and cortex among groups (the putamen, the cortex (mean ± s.d.); WT: 0.22 ± 0.43, 0.87 ± 1.23; TIMP-1−/− mice: 0.43 ± 0.5, 0.87 ± 0.71; and TIMP-2−/− mice: 0.22 ± 0.43, 0.43 ± 0.5). At 24 h after ischemia, there was a significant increase of TUNEL-positive cells in the cerebral cortex in TIMP-1−/− (P < 0.007) and TIMP-2−/− mice (P < 0.004) compared with WT mice, but no change in the caudate putamen (Figures 5A-5C). In addition, densitometric analysis presented the expression of cleaved caspase-3 in the cerebral cortex in TIMP-1−/− mice was significantly more enlarged than in WT mice (P < 0.05), consistent with TUNEL-staining results. We could not certify the difference between TIMP-2−/− mice and WT mice (Figures 5D and 5E).
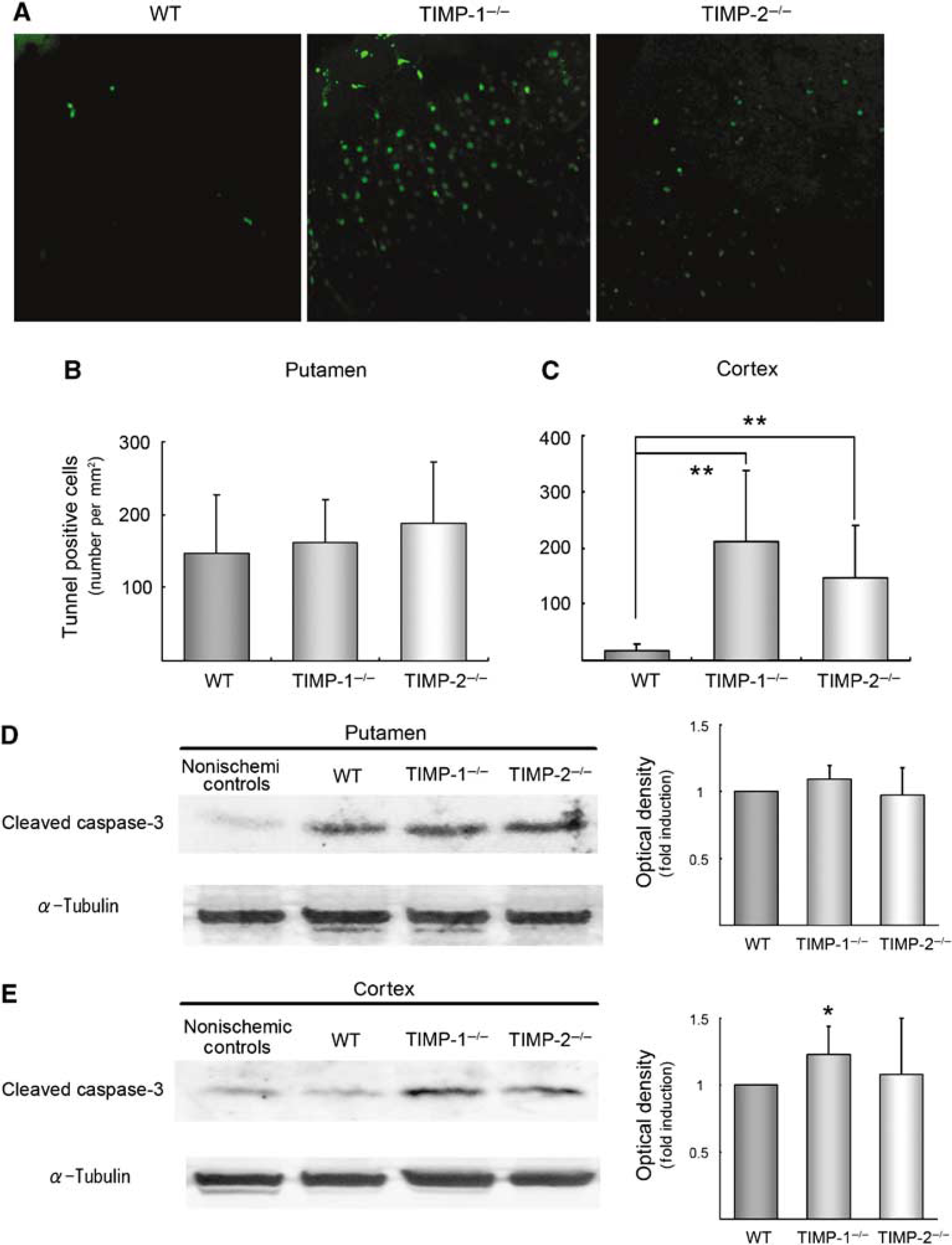
Neuronal apoptosis assessed by tunnel positive cells (
Ischemic Injury
Injury in the 30 mins MCAO cerebral ischemia model was predominantly confined to the ipsilateral putamen. Conversely, the infarct area was amplified to the cerebral cortex and more augmented in TIMP-1−/− mice than in WT mice at 24 h after ischemia (P < 0.04). TIMP-2 gene deletion mice also exhibited a slightly exacerbated infarct area, although this difference did not reach statistical significance (P < 0.11) (Figure 6). Neurologic deficits showed apparent difference among different mouse groups (data not shown).
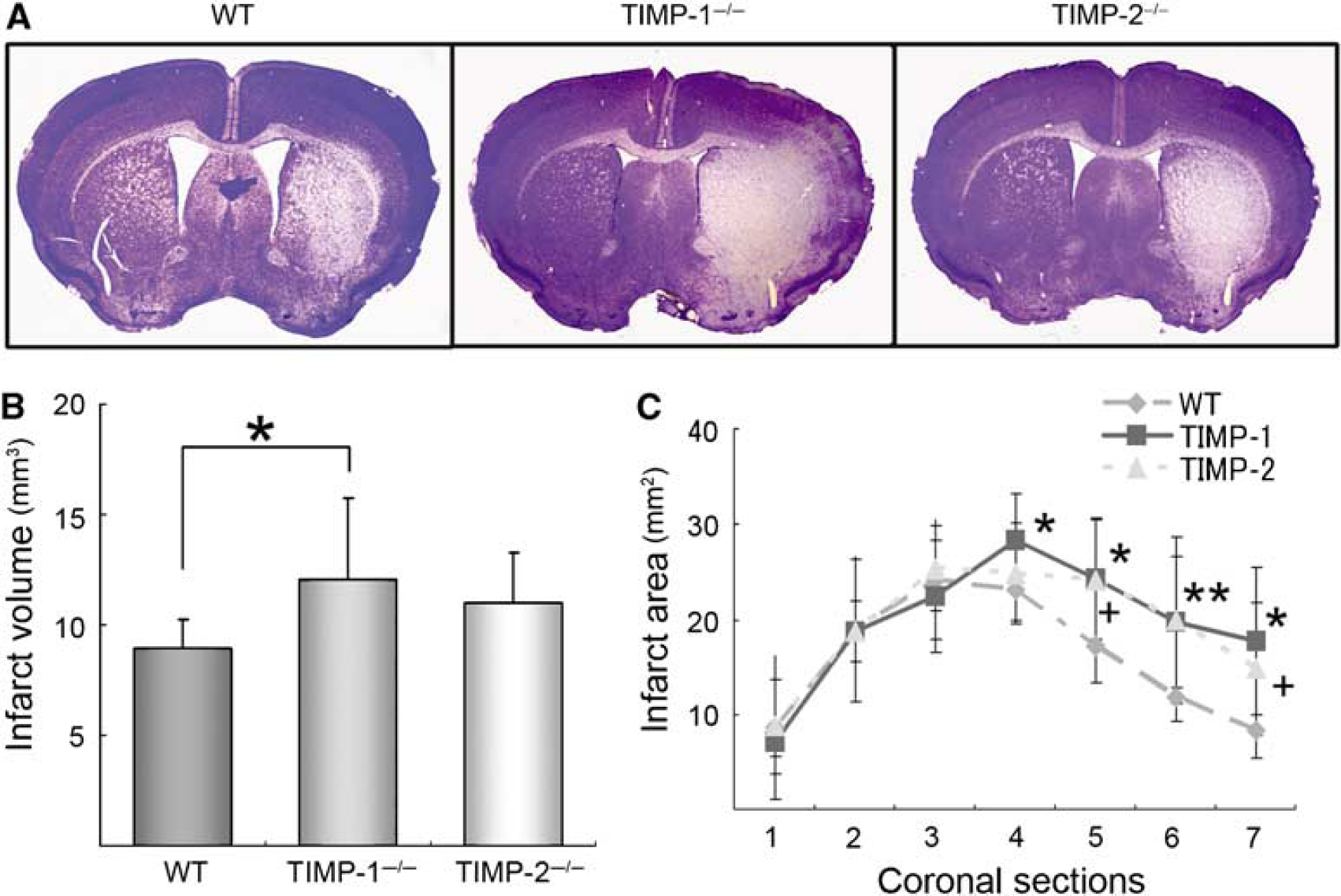
Ischemic damage at 24 h after ischemia. Nissl stainings in WT, TIMP-1−/−, and TIMP-2−/− mice are shown (
Discussion
In the present study, we affirmed upregulated MMP-2, MMP-9, and TIMP-1 expressions in WT mice after focal ischemia, and we showed that MMP-9 protein expression and gelatinolytic activity after cerebral ischemia were significantly more augmented in TIMP-1−/− mice than in WT mice, which was accompanied by exacerbated BBB disruption, neuronal apoptosis, and ischemic injury. In contrast, there was no significant difference in levels of MMPs and in ischemic injury in TIMP-2 gene deletion mice, despite an observed increased Evans blue leakage.
It has been shown that TIMPs play essential roles with MMPs on central nervous system plasticity (Cunningham et al, 2005; Gasche et al, 2006). The present study is the first to show the effects of TIMP-1 and TIMP-2 on BBB dysfunction and ischemic injury, using TIMP-1−/− and TIMP-2−/−mice. Following the ischemic reperfusion model, biphasic BBB openings were observed (Kuroiwa et al, 1985). The initial opening of the BBB within several hours was involved in upregulated MMP-2 activities, and inhibited by broad-spectrum MMP inhibitors (Yang et al, 2007). However, No differences between MMP-2 gene knockout mice and WT mice were observed, thereby its contribution to ischemic injury remains to be validated (Asahi et al, 2001a). Secreted latent MMP-2 is activated by TIMP-2 and MT-MMPs through a unique process (Zhao et al, 2004). Activated MMP-2 is then naturally inhibited by TIMPs. In the present study, we detected an upregulation of MMP-2 at 3 h after ischemia, without a change of TIMP-2 expression in WT mice. In addition, TIMP-2 gene deletion caused no significant difference in MMP-2 and MMP-9 expressions and activities compared with WT mice. Using the gene knockout approach, TIMP-2 was shown to be required for efficient proMMP-2 activation both in vivo and in vitro (Wang et al, 2000). However, in the in vitro BBB model, TIMP-2 was dramatically decreased during reperfusion, accompanied by increased MMP-2 activity. Furthermore, administration of the TIMP-2 protein abrogated MMP-2 activity (Krizanac-Bengez et al, 2006). Tissue inhibitor of metalloproteinase-2 in endothelial cells and in astrocyte cultures are refractory to the modulation of proinflammatory cytokines (Hanemaaijer et al, 1993). The alternation of TIMP-2 expression in an acute stage of ischemia has not been identified in vivo. Tissue inhibitor of metalloproteinase-2 is not supposed to play a central role in MMPs dynamics at least of early phase. Small ischemic damage in the 30 mins MCAO model or sensitivity may be a responsible factor. The results from the present study showed increased Evans Blue extravasation in TIMP-2−/− mice, but was not accompanied by ischemic damage. In the hemorrhagic model induced by type IV collagenase, TIMP-2 diminished BBB damage (Rosenberg et al, 1992). Tissue inhibitor of metalloproteinase-2 may influence BBB permeability independent of MMPs.
The second phase of BBB opening at 24 to 48 h is widely considered to be MMP-9 related. Enhanced MMP-9 activities were confirmed at 1 to 2 days after ischemia in rodents, similar to that in human stroke (Montaner et al, 2001; Rivera et al, 2002). The expression of TIMP-1, a natural inhibitor of MMP-9, was also identified at 12 h after focal ischemia (Rosenberg et al, 1998; Wang et al, 1998), although adverse results were reported (Romanic et al, 1998; Gasche et al, 1999). Synthetic MMP inhibitors, neutralization antibody to MMP-9, and MMP-9 gene deletion reduce ischemic injury through decreased BBB leakage (Rosenberg et al, 1998; Gasche et al, 1999; Asahi et al, 2000). Inducted TIMP-1 expression in a brain endothelial cell line could preserve the brain endothelial barrier function in vitro (Förster et al, 2007). In brain microvascular endothelial cells, the expression of TIMP-1 was dramatically increased by major proinflammatory cytokines, especially by the combination with interleukin-1β and tumor necrosis factor-α (Bugno et al, 1999). Tissue inhibitor of metalloproteinase-1 has also been considered as an immediate early gene.
We confirmed that TIMP-1 gene deletion caused more augmented MMP-9 expression compared with WT mice. Moreover, BBB permeability and neuronal apoptosis were increased in TIMP-1−/− mice, leading to a worsening of ischemic injury. As a result, TIMP-1 is conceived to suppress MMP-9 activity in acute cerebral ischemia and to diminish vasogenic edema in acute cerebral ischemia.
Western blotting showed that MMP-9 expression did not change in WT mice by 24 h after ischemia, whereas zymography indicated the marked change, even by 3 h. In zymograms, we detected strong bands of pro-MMP-9, which do not have gelatinolytic activities. Zymography does not measure enzyme activity, and is another way to quantify protein amount. Thus, this discrepancy between the result of western blot and that of zymography may be due to the sensitivity of these methods. It is necessary to analyze the reason for this difference in the results in further experiments.
Matrix metalloproteinase-2 and MMP-9 digest extracellular matrix of cerebral microvessel basal lamina such as collagen type IV. Zonae occluding-1 (ZO-1), one of the tight junction proteins in the cerebral endothelium, was progressively decreased after ischemic injury. MMP-9 gene deletion reduced degraded ZO-1 expression with attenuation of BBB leakage (Asahi et al, 2001b). Degraded tight junction proteins, claudin-5 and occludin in focal ischemia, were reversed by MMP inhibitors (Yang et al, 2007). In our ischemic model, only ZO-1, but not collagen type IV and Claudin-5 was decreased in TIMP-1−/− mice compared with WT mice. TIMP-1 gene deletion could accelerate the degradation of ZO-1 through increased MMP-9 activity in cerebral ischemia.
Matrix metalloproteinase-2 and MMP-9 facilitate neuronal apoptosis through cell—matrix interactions (Gu et al, 2002). MMP-9 gene deletion did not affect neuronal apoptosis, and in MMP-9-deficient mice with a broad MMP inhibitor, DNA fragmentation was reduced. Involvement of MMPs other than MMP-9 may be possible (Copin et al, 2005). Tissue inhibitor of metalloproteinase-3 also has been associated with neuronal apoptosis after ischemia (Wallace et al, 2002). Recently, TIMP-3 gene deletion was shown to prevent Fas-mediated cell death in focal ischemia (Wetzel et al, 2007).
The functions of TIMP-1 and TIMP-2 on neuronal apoptosis are less well understood. We observed an increased number of TUNEL-positive cells in TIMP-1−/− and TIMP-2−/− mice, and cleaved caspase-3 expression in the ischemic cortex in TIMP-1−/− mice, when compared with WT mice. These data suggest that both TIMP-1 and TIMP-2 may suppress neuronal apoptosis, which may be due to increased MMP-9 activity, an anti-apoptotic effect of TIMPs independent of MMPs. Not only our ischemic damage was predominantly located in the putamen, but also larger injury in TIMP-1 knockout mice was also located in the cortex. Increased cortical TUNEL-positive cells and cleaved caspase-3 expression in TIMP-1−/− mice could be due to aggravated ischemic injury. Further studies are necessary to evaluate the association between TIMPs and apoptosis after ischemia.
In conclusion, using knockout mice we showed the physiologic roles of TIMP-1 and TIMP-2 associated with MMPs in acute cerebral ischemia. TIMP-1 exhibited a neuroprotective function in BBB dysruption, neuronal apoptosis, and ischemic damage after ischemia, possibly through the inhibition of MMP-9 activity. Tissue inhibitor of metalloproteinase-2 gene deletion increased BBB leakage and apoptosis without any change in ischemic damage. These data suggest that both TIMP-1 and TIMP-2 contribute to BBB dysruption, and TIMP-1 may be potential therapeutic targets for ischemic damage after transient focal ischemia.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.