
Abstract
Signal transducers and activators of transcription (STAT) proteins are a family of transcription factors that play a crucial role in growth and differentiation in a variety of cell types. Among them, STAT1, which is expressed in the brain and directly activated by reactive oxygen species, participates in the regulation of cytokine-signaling and cellular responses, particularly to interferon-γ. Very little, however, is known about the importance of STAT1 during brain injury. The authors found that STAT1 was phosphorylated at tyrosine701 and serine727 and translocated into neuronal nuclei within hours after middle cerebral artery occlusion. At later time points, STAT1 immunoreactivity colocalized with TUNEL-positive neurons, thereby suggesting a role in cell death. In mice genetically deficient in STAT1 expression, the volume of ischemic brain injury was reduced, neurologic deficits were less severe, and TUNEL-positive neurons were also less numerous compared with wild-type mice. STAT1-knockout mice showed increased phosphorylated Akt and decreased pro-***caspase-3 cleavage. Major strain differences in phosphorylated STAT3 or cyclooxygenase-2 protein expression were not found after ischemia. These results indicate that STAT1 is activated and translocated within ischemic neurons and may contribute to brain injury by regulating transcription and phosphorylation of proteins related to apoptosis and cell death.
Signal transducers and activators of transcription (STAT) proteins are a family of transcription factors that are present in many cell types and function as a major signal transduction pathway in cytokine signaling, playing a crucial role in both growth and differentiation within a variety of cell types (Akira, 1999; Horvath, 2000). Following the binding of ligands to their receptors, Janus kinases (JAK) are activated and, in turn, phosphorylate STAT proteins. Phospho-STATs dimerize and translocate into the nucleus, binding to promoters of specific target genes (Horvath, 2000). To date, seven mammalian members of the STAT family have been identified. Among them, STAT1 is activated by a number of cytokines, growth factors, and hormones (Akira, 1999; Horvath, 2000), and has been shown to play a dedicated role in interferon (IFN)-γ–dependent biologic responses (Heitmeier et al., 1999; Kim and Maniatis, 1996; Lee et al., 1999, 2000). STAT1 reportedly induces p21WAF1, a member of the cyclin-dependent kinase inhibitors able to induce cell growth arrest (Chin et al., 1996). Accordingly, IFN-γ–induced cell-cycle arrest is disturbed in STAT1-knockout (STAT1−/−) mice (Chin et al., 1996).
Several reports have demonstrated that STAT1 regulates apoptosis in cardiomyocytes and fibroblasts, including caspase transcriptional activation (Chin et al., 1997; Kumar et al., 1997; Stephanou et al., 2000). In accordance with this finding, STAT1−/− fibroblasts are resistant to tumor necrosis factor-α–induced apoptosis (Kumar et al., 1997). There is little information, however, on the role of STATs in mediating proapoptotic and anti-apoptotic signals in the CNS. JAK1 is expressed in astrocytes and neurons of the rat brain, and STAT3 is activated in these cells 1 hour after ischemia, and in immune cells at 3 days from the insult (Justicia et al., 2000). The pathophysiologic relevance of these events, however, remains to be clarified.
During brain ischemia, apoptosis is an important cell death mechanism. Many studies have demonstrated that ischemia modulates the expression of several signaling molecules and transcription factors, including c-fos, c-jun, c-Jun NH2 terminal kinase (JNK), extracellular signal-regulated mitogen-activated protein kinase (ERK) and nuclear factor-κ. Such changes may play a major role in the recovery process or in tissue injury during ischemia by regulating genes involved in apoptotic cell death (Sharp et al., 2000). Reactive oxygen species (ROS), which are typically generated during ischemic brain injury, reportedly activate STATs (Chan, 2001; De-Fraja et al., 1998; Madamanchi et al., 2001), strengthening the link between the brain ischemic insult and STAT activation. Nevertheless, it remains unclear which molecules are directly responsible for neuronal injury and how these molecules activate downstream targets.
Here, we report that cerebral ischemia promotes STAT1 phosphorylation and STAT1 nuclear translocation, and that STAT1−/− mice are more resistant to ischemic brain injury. In addition, we also report that this resistance is associated with two antiapoptotic processes, namely increased Akt phosphorylation and suppressed caspase-3 cleavage. This study suggests that transcriptional events regulated by STAT1 are important in ischemia-induced neuronal death.
MATERIALS AND METHODS
Animals
STAT1−/− mice were generated as described elsewhere (Durbin et al., 1996; Meraz et al., 1996). Homozygous STAT−/− 129S6/SvEv male mice (Taconic Farms, German-town, NY, U.S.A.) were used for experiments. Age- and sex-matched 129S6/SvEv mice (Taconic Farms) were used as controls.
Induction of focal ischemia
Mice (20 to 25 g) were anesthetized with 2% isoflurane and maintained on 1.5% isoflurane in 70% nitrous oxide and 30% oxygen. Regional cerebral blood flow (rCBF) was measured by laser-Doppler (PF2B; Perimed, Stockholm, Sweden) using a flexible probe as described (Hara et al., 1997; Huang et al., 1994). In randomly selected animals, the left femoral artery was cannulated with a PE-10 polyethylene tube for arterial blood pressure measurement and blood gas determination. Arterial blood samples (50 μL) were analyzed for pH, arterial oxygen pressure (PaO2), and partial pressure of carbon dioxide (PaCO2) using a Ciba-Corning 248 pH/blood gas analyzer (Ciba-Corning Diagnostics Corp., Medfield, MA, U.S.A.). Rectal temperature was maintained between 36.5°C and 37.5°C with a homeothermic blanket (Frederick Haer and Co., Brunswick, ME, U.S.A.). Focal cerebral ischemia was induced by occluding the middle cerebral artery using the intraluminal filament technique as previously reported (Hara et al., 1997). After surgery, mice were kept at 37°C for 30 minutes or 2 hours, after which time the filament was withdrawn to allow reperfusion.
Neurologic deficits
Neurologic deficit was scored according to the criteria of Hara et al. (1997), where a score of 0 indicated no deficits (normal), 1 indicated a failure to extend forepaw (mild); 2 indicated contralateral circling (moderate), and 3 indicated loss of walking or righting reflex (severe). The scorer was naive to the treatment group, and assessments were made 2 and 24 hours after the ischemic insult.
Determination of infarct size
Animals were killed 22 or 24 hours after reperfusion and the brains were snap-frozen in N2 vapor for cryostat sectioning. Infarction areas were quantitated with MCID M4 image analysis software (Imaging Research Inc., St. Catharine's, Ontario, Canada) on hematoxylin–eosin stained sections. In order to account for and eliminate swelling/edema, infarction volume was calculated using an indirect measurement (Hara et al. 1997) by summing the volumes of each section using the following formula: contralateral hemisphere (mm3) – undamaged ipsilateral hemisphere (mm3).
Western blotting
Ischemic and contralateral healthy tissues were collected and stored at −80°C. Proteins were isolated according to standard techniques, separated by a 4% to 20% SDS/PAGE gel, and transferred onto a nitrocellulose membrane. Subsequently, blots were incubated with rabbit polyclonal antibodies raised against the following proteins: STAT1, pTyr701-STAT1, pSer727-STAT1 (BioSource International, Camarillo, CA, U.S.A.), pTyr705-STAT3, pSer473-Akt, caspase-3, cleaved (Asp175) caspase-3, pThr183/Tyr185-c-Jun NH2 terminal kinase (JNK), pThr180/Tyr182-p38 MAPK, pThr202/Tyr204-ERK (Cell Signaling Technologies, Beverly, MA, U.S.A.), caspase-1 (M-19; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), and cyclooxygenase-2 (COX-2; Cayman Chemical, Ann Arbor, MI, U.S.A.). A donkey peroxidase-conjugated antirabbit antibody was used and binding was revealed by chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.). Densitometric data, obtained using the NIH image analysis system (National Institutes of Health, Bethesda, MD, U.S.A.), were normalized to the contralateral side and expressed as the mean ± SD.
Immunohistochemistry
Twelve-micrometer sections were mounted onto precleaned glass slides and kept at −80°C until use. Sections were post-fixed in 4% paraformaldehyde for 10 minutes. After several washes in 0.1 mol/L phosphate-buffered saline (PBS), sections were incubated with 10% normal goat serum containing 0.3% Triton X-100 for 1 hour at room temperature. Immunohisto-chemical staining for STAT1 and phosphorylated-STAT1 were performed using corresponding antibodies (BioScource International) diluted 1:100 in 2% normal goat serum, 0.3% Triton X-100, and 0.1% NaN3 in PBS, and then incubated at 4°C for 16 hours. After three rinses in PBS, sections were incubated for 1 hour at 25°C with biotinylated goat antirabbit IgG (1:200; Vector Laboratories, Burlingame, CA, U.S.A.). After washing, sections were incubated with streptavidin-conjugated Cy2 (1:500; Jackson ImmunoResearch, West Grove, PA, U.S.A.) in PBS for 30 minutes at room temperature. Sections were later incubated with a Cy3-labeled (Jackson ImmunoResearch) NeuN monoclonal antibody (1:300; Chemicon, Temecula, CA, U.S.A.) at 4°C overnight. Fluorescence was visualized using a krypton-argon laser-scanning confocal microscope (Bio-Rad MRC 1024; Bio-Rad, Hercules, CA, U.S.A.).
TUNEL staining
Terminal deoxynucleotidyl transferase (TdT)-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick-end labeling (TUNEL) staining was performed by using a kit from Boehringer Mannheim (Indianapolis, IN, U.S.A.), according to the manufacturer's instructions. Briefly, sections were incubated with TdT buffer (30 mmol/L Tris, pH 7.2; 140 mmol/L sodium cacodylate; and 1 mmol/L cobalt chloride) containing TdT enzyme (0.5 U/mL) and biotin-16-dUTP (0.04 mmol/L) for 1 hour at 37°C. The reaction was terminated by incubating with 300 mmol/L NaCl and 30-mmol/L sodium citrate for 15 minutes. After washing with 50 mmol/L Tris-HCl (pH 7.7), sections were incubated with streptavidin-conjugated Cy2 or Cy3 in PBS for 30 minutes. The total number of TUNEL-positive cells was counted on three tissue sections for each brain.
RESULTS
Activation of STAT1 after ischemia
Because STAT1 is constitutively expressed in protein extracts of brain homogenates (Fig. 1A) (De-Fraja et al., 1998; Neumann et al., 1997) and because STAT1 phosphorylation triggers its activation and nuclear translocation, we investigated whether 2-hour ischemia triggers STAT1 phosphorylation and nuclear localization by probing with site-specific phospho-STAT1 antibodies. As shown in Figs. 1A and 1B, STAT1 tyrosine701 phosphorylation significantly increased 30 minutes and 2 hours after reperfusion within ischemic tissue. The increase in ischemia-induced STAT1 phosphorylation at serine727 was evident 15 minutes after reperfusion and was sustained for at least 2 hours (Figs. 1A and 1C). Consistent with an increase in phosphorylation, STAT1 was activated and translocated into neuronal nuclei 2 hours after reperfusion (Fig. 2A). Nuclear translocation of STAT1 was found as early as 30 minutes after ischemia and persisted until the time of death at 24 hours (data not shown). STAT1 immunoreactivity colocalized within TUNEL-positive nuclei 22 hours after reperfusion (Fig. 2B), thereby suggesting an association between STAT1 activation and cell death. Moreover, TUNEL-positive cells were rarely positive for glial fibrillary acidic protein (data not shown), suggesting that STAT1 was phosphorylated selectively within neurons at these time points.
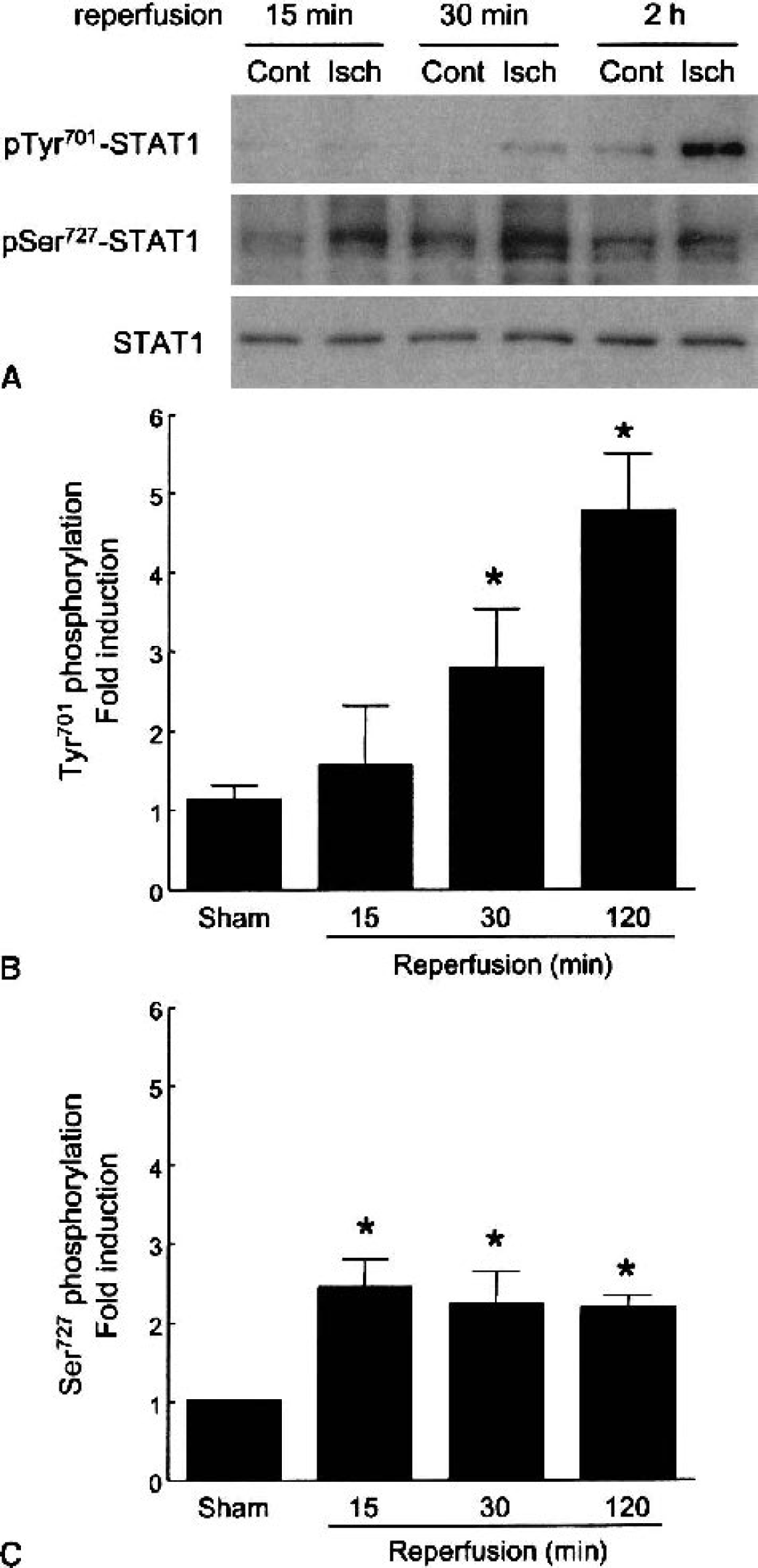
Activation of STAT1 after cerebral ischemia. Western blot analysis
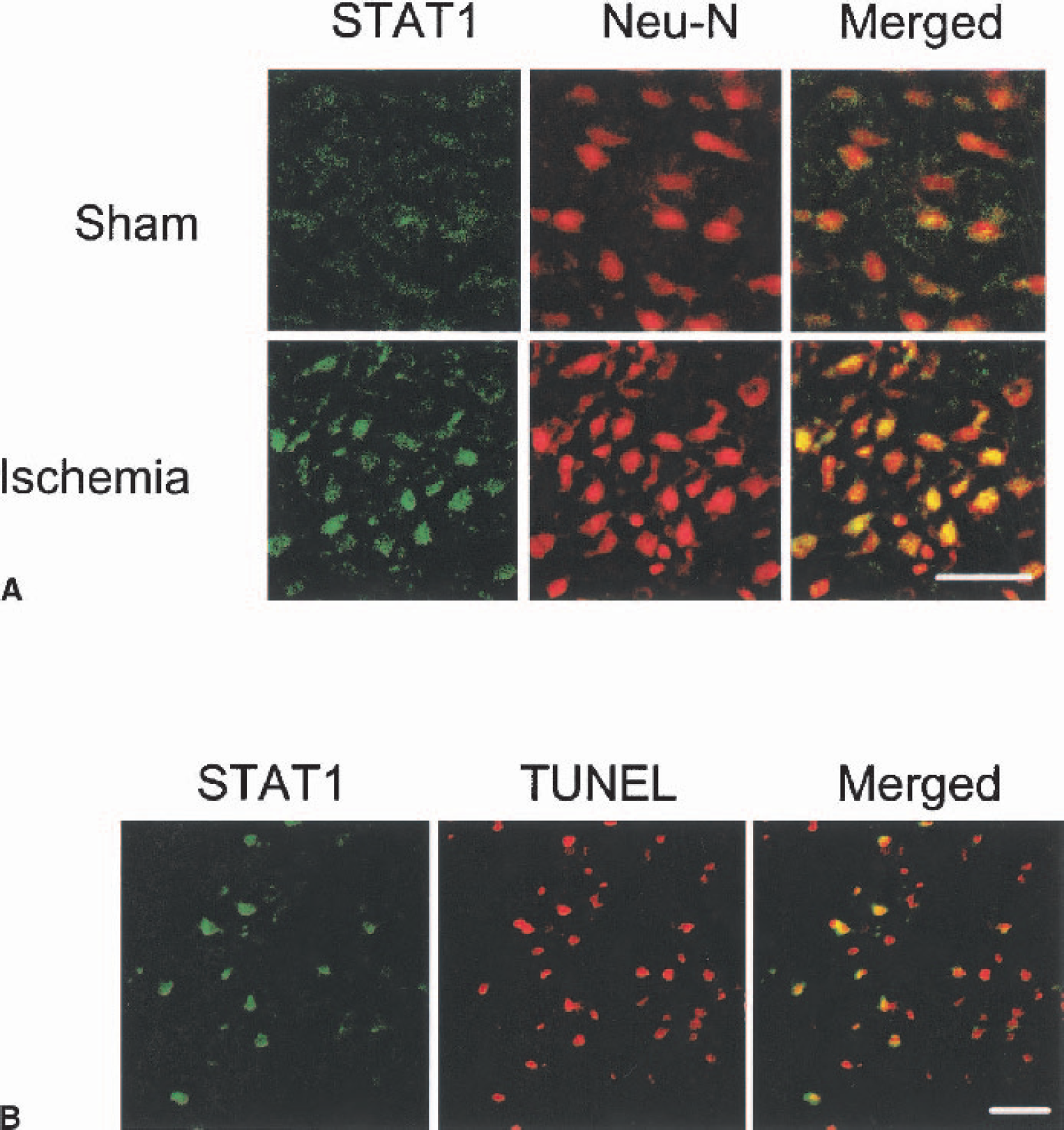
Immunohistochemical study of STAT1 activation after 2-hour middle cerebral artery occlusion and different times of reperfusion.
Phenotype of STAT1−/− mice
STAT1−/− mice develop normally and show no macroscopic or microscopic anatomic differences in the brain compared with wild-type (WT) mice (Durbin et al., 1996; Meraz et al., 1996). STAT1−/− mice do not respond to IFNs and are highly sensitive to infections (Meraz et al, 1996). In the present study, there also were no differences in brain vascular anatomy between strains, as detected by transcardiac carbon black infusion (10% in PBS; data not shown). Physiologic parameters such as rectal temperature, arterial blood pressure and pH, PaO2/PaCO2 did not differ between strains before, during, and 2 hours after ischemia (n = 4; Table 1). Although activation of STAT3, ERK, and JNK phosphorylation signaling pathways did not differ between strains, we cannot exclude the possibility that other STAT family members or different signaling pathways could perturb signaling in these mutants.
Physiologic parameters before, during, and 2 hours after middle cerebral artery occlusion in wild-type and STAT1−/− mice
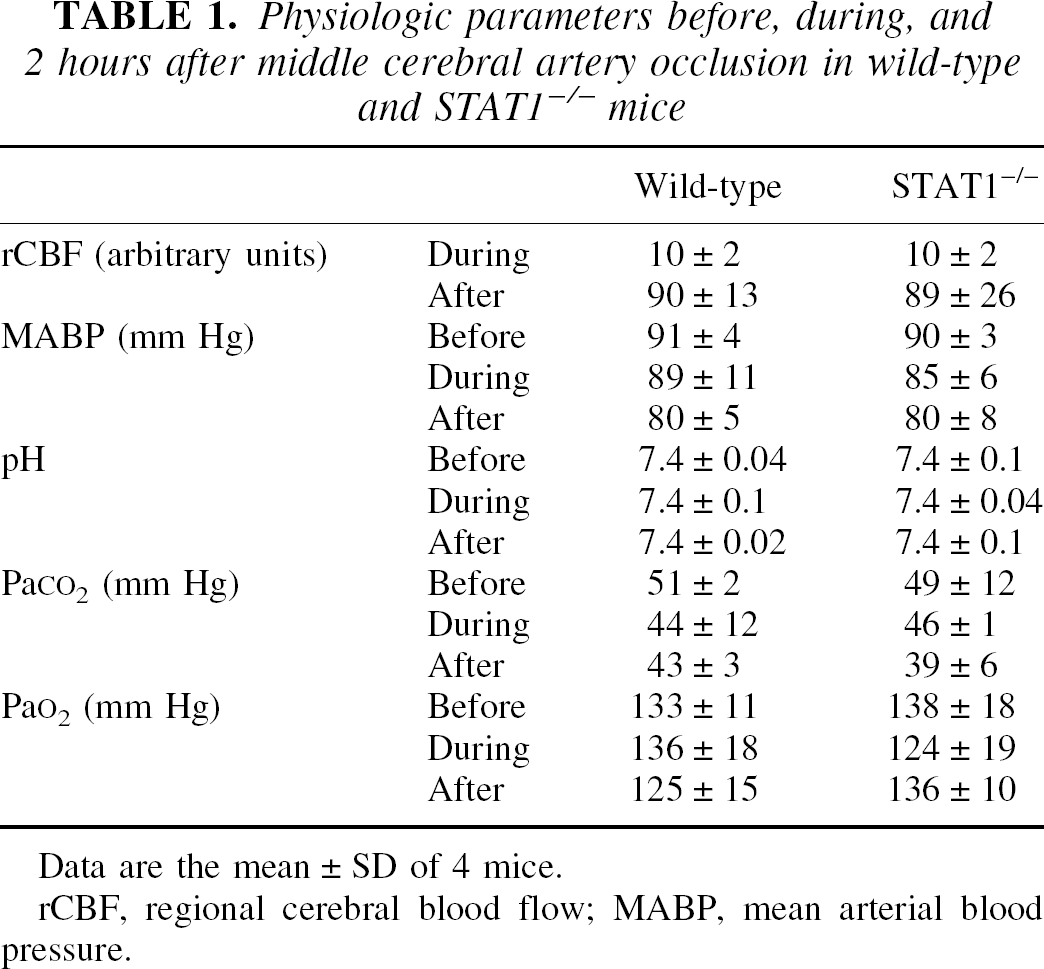
Data are the mean ± SD of 4 mice.
rCBF, regional cerebral blood flow; MABP, mean arterial blood pressure.
Ischemic brain injury in STAT1−/− mice
To address the role of STAT1 in ischemic brain injury, we measured lesion volume, neurologic score, as well as number of TUNEL-positive neurons at 22-hour reperfusion after 2-hour ischemia in STAT1−/− and WT mice. Both lesion size and neurologic deficits were significantly reduced in STAT1−/− mice (Figs. 3A through 3C). It is well established that reversible 2-hour ischemia causes necrosis and apoptosis in both striatum and cortex in the mouse. To investigate the role of STAT1 during a milder ischemic insult, we adopted the 30-minute ischemia/24-hour reperfusion model (known to induce cell death limited to the lateral striatum mainly through the induction of apoptosis). As shown in Fig. 3D, infarct volume was also reduced in STAT1−/− mice after milder ischemic injury (30-minute ischemia/22-hour reperfusion). Furthermore, the number of TUNEL-positive neurons within ischemic tissue of STAT1−/− mice was lower than that of WT mice (Fig. 4), confirming the involvement of STAT1 in neuronal apoptosis, as initially suggested by its activation in TUNEL-positive neurons (Fig. 2).
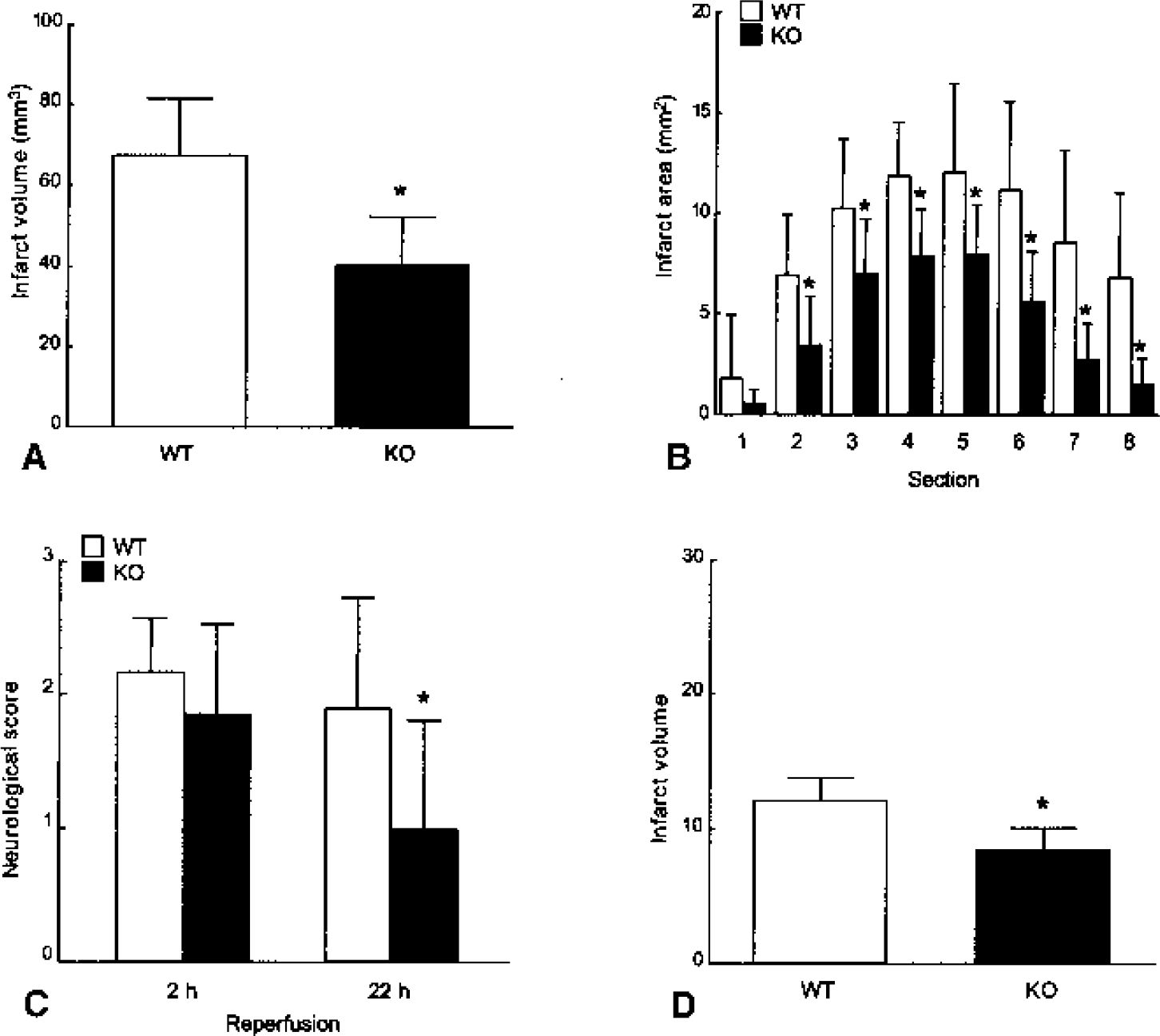
The effect of STAT1 gene deletion on ischemic brain injury. Twenty-two hours after 2-hour middle cerebral artery occlusion, both infarct volume
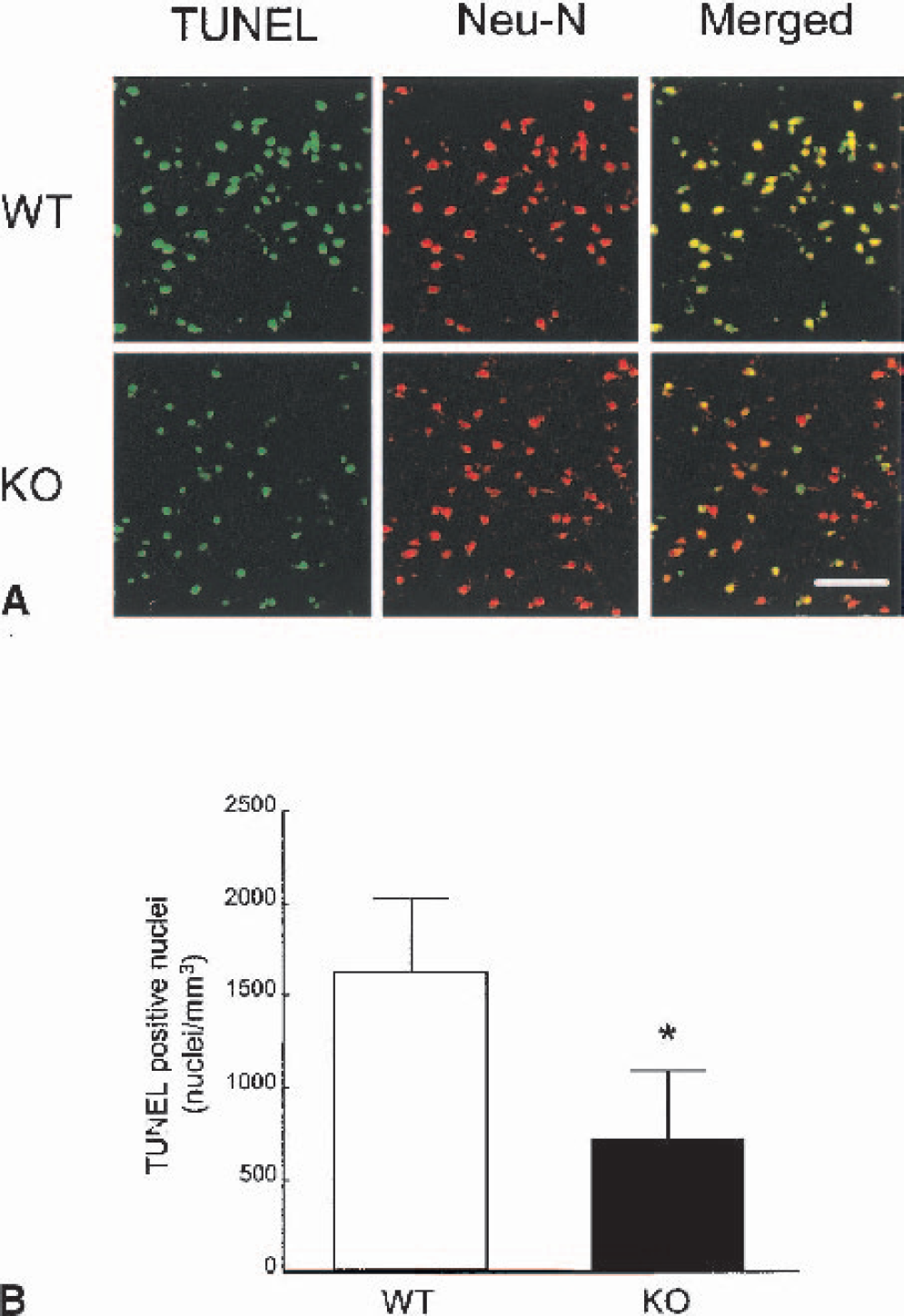
TUNEL staining in the cerebral cortex after 2-hour middle cerebral artery occlusion and 22-hour reperfusion.
Cell death-related kinase activation and cyclooxygenase-2 expression in STAT1−/− and wild-type mice
To understand the possible mechanisms conferring resistance to ischemic brain injury in STAT1−/− mice, we investigated whether activation/expression of several cell death-related proteins became altered after ischemiareperfusion. As revealed by immunohistochemistry, Akt phosphorylation at serine473 was more prominent in neurons of STAT1−/− mice compared with WT mice after 2-hour ischemia/2-hour reperfusion (Fig. 5). Western blotting confirmed that after 2-hour ischemia/2-hour reperfusion, Akt phosphorylation at serine473 was significantly enhanced in the ischemic brain of STAT1−/− mice (Figs. 6A and 6C). Remarkably, there were no differences in ischemia-induced STAT3, ERK, JNK, or p38 MAPK phosphorylation between strains (Fig. 6A and data not shown). Hence, a specific role for STAT1 as a regulator of Akt activation is suggested, although evaluation at a single time point may limit the impact of this finding. Interestingly, we found a significant decrease in caspase-3 activation in STAT1−/− mice after 2-hour ischemia/22-hour reperfusion, despite equivalent expression of procaspase-1 and 3 in mutant and WT mice (Figs. 6B and 6C). Considering the relevance of JAK/STAT signaling during immune cell activation and the role of inflammation in the pathogenesis of ischemic brain injury, we investigated whether STAT1 deletion affected induction of inducible nitric oxide synthase (iNOS) and COX-2 in the ischemic brain. After 2-hour ischemia/22-hour reperfusion, iNOS expression was undetectable, whereas no significant difference in COX-2 expression was found between STAT1−/− and WT mice (Fig. 5D).
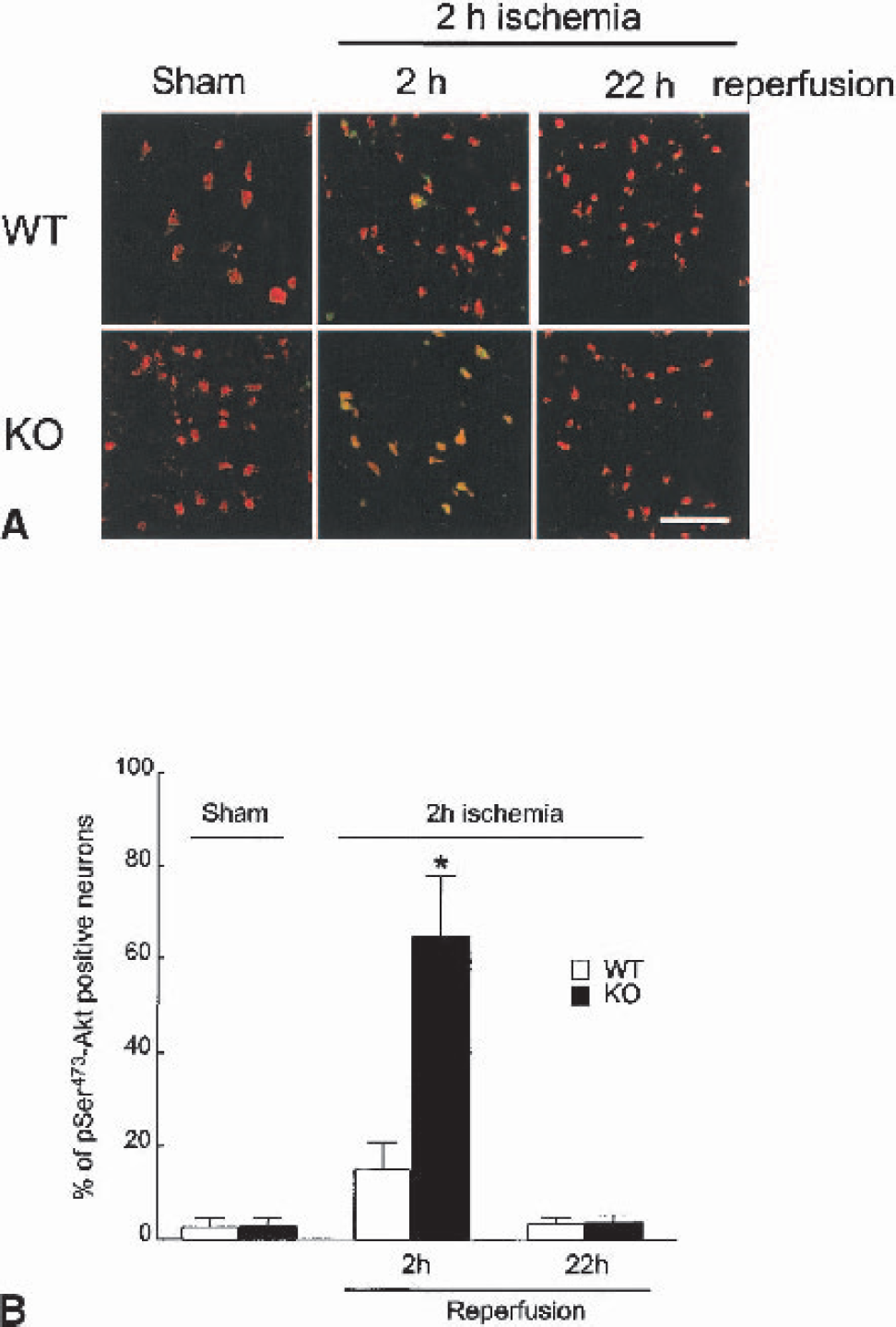
Akt phosphorylation in neurons after 2-hour middle cerebral artery occlusion and different times of reperfusion in STAT1−/− (knockout [KO]) and wild-type (WT) mice.
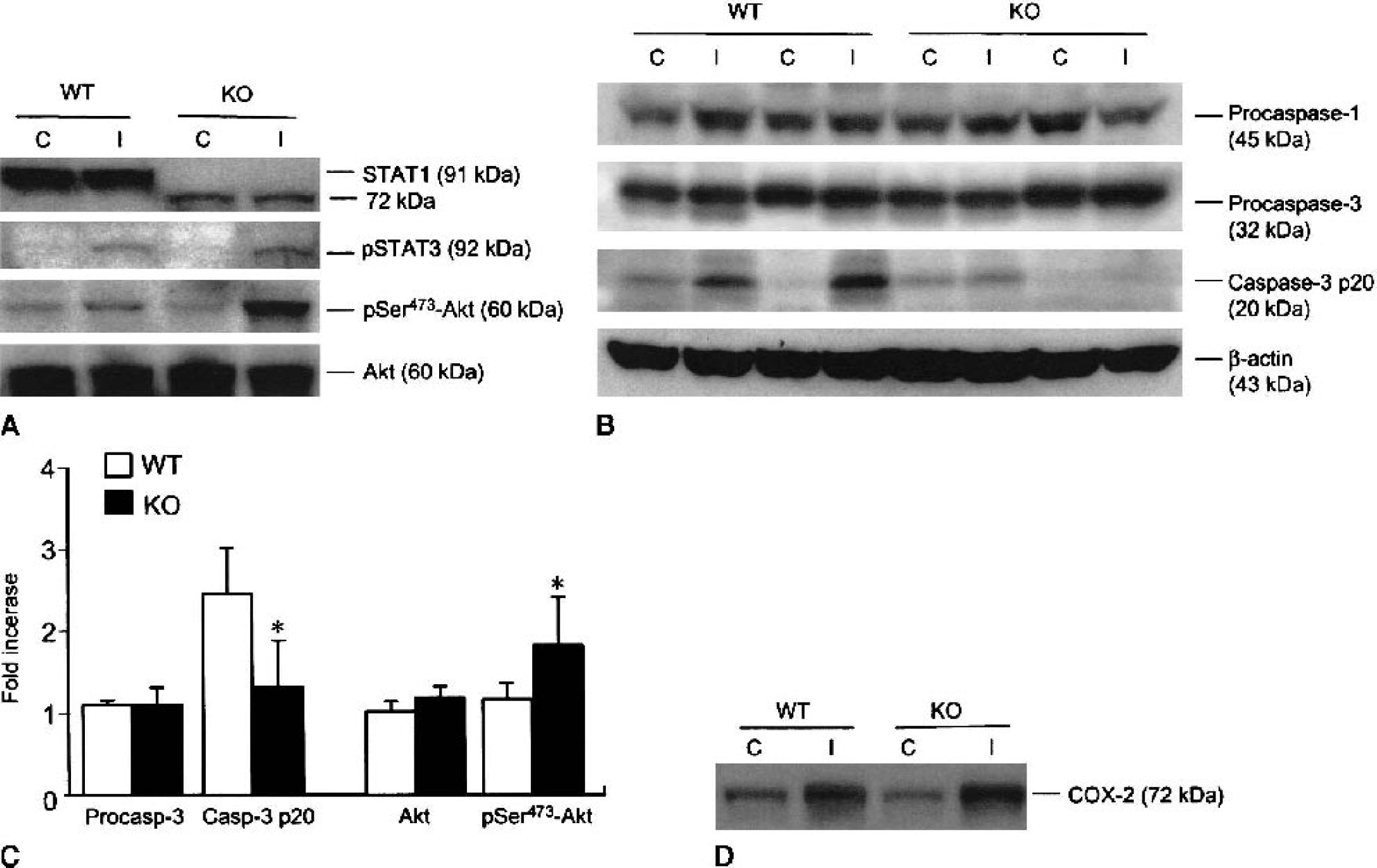
Akt phosphorylation levels in ischemic (I) and contralateral (C) cortex of STAT1−/− (knockout [KO]) and wild-type (WT) mice after 2-hour middle cerebral artery occlusion and 2-hour reperfusion. STAT3 phosphorylation is unaffected in STAT1−/− mice during ischemia/reperfusion. Akt is shown to confirm equal protein loading. As previously described (Meraz et al., 1996), STAT1−/− mice express an inactive STAT1 truncated protein of 72 kd.
DISCUSSION
This is the first report implicating a role for STAT1 in the evolution of ischemic brain injury. We found that STAT1, expressed in normal brain, was phosphorylated and translocated into neuronal nuclei during reperfusion and later colocalized in TUNEL-positive neurons. Deletion of the STAT1 gene rendered brain tissue resistant to ischemic injury, most likely by modulating mechanisms important to cleavage of executioner caspases, such as caspase-3. Hence, neuronal apoptosis may be an important target for STAT1 in ischemic injury and for mechanisms of protection in STAT1−/− mice, at least until 24 hours.
Growth factors (e.g., fibroblast growth factor, epidermal growth factor), IFN-γ, and ROS are classical STAT1 activators (Akira, 1999). Interestingly, IFN-γ–expressing cells are detected in ischemic tissue 12 to 24 hours after middle cerebral artery occlusion and increase with time along with the number of infiltrating mononuclear cells (Li et al., 2001). The ROS are prototypical mediators of ischemic neuronal death (Kovarik et al., 1999; Madamanchi et al., 2001; Maziere et al., 1999; Pan et al., 1999), and oxidative stress activates the STAT1 signaling pathway in vascular smooth muscle cells and fibroblasts (Madamanchi et al, 2001) with a time course reminiscent of ischemia-induced STAT1 phosphorylation reported here. Consistent with an active role of STAT1 in cell death, STAT1−/− fibroblasts are resistant to apoptosis induced by tumor necrosis factor-α (Kumar et al., 1997) or IFN-γ (Chin et al., 1997). Moreover, ischemia-induced Fas-mediated apoptosis in cardiomyocytes requires STAT1 activation (Stephanou et al, 2001) and is suppressed by STAT1 antisense (Stephanou et al., 2000).
We report here that STAT1 was phosphorylated at serine727 and tyrosine701, thereby suggesting a possible role during brain ischemia. Previous reports suggested that serine727 phosphorylation is mainly regulated by p38 MAPK or oxidative stress and that its phosphorylation promotes transcriptional activity, whereas tyrosine701 is selectively phosphorylated by JAKs and its phosphorylation induces dimerization and nuclear translocation of STAT1 (Decker and Kovarik, 2000; Horvath, 2000; Kovarik et al., 1999). The significance of different phosphorylation sites on STAT1 functioning, however, has not yet been clarified.
Reduced expression of procaspases 1, 2, and 3 in STAT1-deficient cells has been reported (Chin et al., 1997; Kumar et al., 1997; Stephanou et al., 2000). We did not find any difference, however, in the basal expression of procaspases 1 and 3 in whole brain homogenates; importantly, ischemia-induced activation of caspase-3 was reduced in the brain of STAT1 −/− mice (Fig. 6B). Decreased processing of caspase-3 may confer resistance to ischemia in the mutant strain. Caspase-3 is a major executioner caspase that cleaves important substrates including ICAD (an inhibitor of caspase-activated DNase), which in turn activates the apoptotic DNA ladderforming activity of CAD (caspase-activated DNase) (Nagata, 1997). During cerebral ischemia, caspase-3 is cleaved and promotes cell death (Namura et al., 1998). STAT1 may affect the cleavage of caspase-3 through pathways that lie upstream, such as processing of caspases 8 and 9. In apoptosis, the release of cytochrome c from mitochondria induces the formation of the cytochrome c/Apaf-1/caspase-9 complex (Yuan and Yankner, 2000). The interaction of cytochrome c, Apaf-1, and caspase-9 along with d-ATP triggers caspase-9 activation, which in turn activates procaspase-3 through direct proteolytic processing. During brain ischemia, caspases 1, 3, 8, 9, and 11 are reportedly cleaved (Hara et al., 1997; Kang et al., 2000; Namura et al., 1998; Noshita et al., 2001a).
Increased activity of the antiapoptotic kinase Akt may provide a second explanation for ischemic resistance in STAT1−/− mice, and may also be related to reduced caspase-3 processing. Indeed, Akt, also known as protein kinase B, is involved in cell survival as a downstream kinase in the phosphoinositide 3-kinase (PI3-K)-mediated signaling cascades upstream of caspase-3 (Datta and Greenberg, 1999). Phosphorylated Akt inactivates death-promoting molecules such as Bad, fork-head transcription factors, and caspase-9 by direct phosphorylation, thereby playing a key role in the inhibition of apoptosis (Datta and Greenberg, 1999). In addition, Akt induces the transcriptional activity of nuclear factor-κ (Yang et al., 2001), a well-known antiapoptotic transcription factor. Recently, ischemia-induced Akt phosphorylation was detected in hippocampal CA1 neurons (Ouyang et al., 1999) and cerebral cortex (Noshita et al., 2001b), possibly delaying the onset of cell death. Accordingly, a study by Yano et al. (2001) shows that the PI3-K inhibitor LY-294002 blocks both Akt phosphorylation and neuroprotection after ischemic preconditioning, indicating that Akt is important in the protection of brain after cerebral ischemia. We report here that Akt phosphorylation was more pronounced in neurons of STAT1−/− mice than in WT mice after reperfusion (Figs. 5 and 6). These novel findings suggest that STAT1 lies upstream and inhibits the PI3-K/Akt pathway during brain ischemia, thereby promoting insertion of Bad into the mitochondrial membrane, caspase-9 release from the organelle, and ensuing caspase-3 activation. These findings are especially interesting given the observation that JAKs are not only required for optimal activation of STAT signaling, but also for enhancing the PI3-K/Akt pathway after activation of cytokine/IFN receptors (Rane and Reddy, 2000). Furthermore, considering that the kinase responsible for serine473-Akt phosphorylation is as yet unidentified (Brazil and Hemmings, 2001), our findings provide the first evidence that the JAK/STAT pathway may directly regulate this elusive kinase. Although in our ischemic model STAT1 was selectively activated in neurons, we have not ruled out the possibility that STAT1 is activated in nonneuronal cells such as neutrophils, macrophages, or reactive astrocytes at later time points after the ischemic insult, as reported for STAT3 (Justicia et al., 2000).
In conclusion, we have shown that STAT1 is phosphorylated and translocated after cerebral ischemia and that its genetic deletion confers resistance to ischemic brain injury. An increase in Akt-mediated survival signaling reducing caspase-3 activation may underlie brain protection in these mutants.