
Abstract
The involvement of caspase-3 in cell death after hypoxia–ischemia (HI) was studied during brain maturation. Unilateral HI was produced in rats at postnatal day 7 (P7), 15 (P15), 26 (P26), and 60 (P60) by a combination of left carotid artery ligation and systemic hypoxia (8% O2). Activation of caspase-3 and cell death was examined in situ by high-resolution confocal microscopy with anti-active caspase-3 antibody and propidium iodide and by biochemical analysis. The active caspase-3 positive neurons were composed of more than 90% HI damaged striatal and neocortical neurons in P7 pups, but that number was reduced to approximately 65% in striatum and 34% in the neocortex of P15 pups, and approximately 26% in striatum and 2% in neocortex of P26 rats. In P60 rats, less than 4% of the damaged neurons in striatum and less than 1% in neocortex were positive for active caspase-3. Western blot analysis demonstrated that the level of inactive caspase-3 in normal forebrain tissue gradually declined from a high level in young pups to very low levels in adult rats. Concomitantly, HI-induced active caspase-3 was reduced from a relatively high level in P7, to moderate levels in P15 and P26, to a barely detectable level in P60 rats. The authors conclude that the involvement of caspase-3 in the pathogenesis of cell death after HI declines during neuronal maturation. The authors hypothesize that caspase-3 may play a major role in cell death in immature neurons but a minor role in cell death in mature neurons after brain injury.
Keywords
Neuronal apoptosis has been repeatedly and consistently observed in neonatal hypoxia–ischemia (HI) models in numerous studies (Mehmet et al., 1998; Pulera et al., 1998; Cheng et al., 1998; Renolleau et al., 1998). Inhibition of caspases protects neurons against HI-induced injury (Cheng et al., 1998). However, there is a controversy as to whether neuronal death induced by ischemia is apoptosis or necrosis in adult animals. It seems important to distinguish apoptosis from necrosis in ischemic conditions as therapeutic intervention may aim at key molecular events involved in the well-established apoptotic pathways. Classic apoptotic DNA fragmentation (MacManus et al., 1999) and morphology of ischemic dead neurons in adult brains have not been consistently observed in previous studies (Deshpande et al., 1992; van Lookeren Campagne and Gill, 1996; Colbourne et al., 1999). However, recent biochemical evidence from several laboratories suggests that some apoptotic components may be involved in ischemic neuronal death (MacManus and Linnik, 1997; Chen et al., 1998; Namura et al., 1998; Velier et al., 1999). The discrepancy between the morphologic and biochemical results suggests that both apoptosis and necrosis may take place in the same, or in different neurons, or both, dying from ischemia in adult animals. To understand the pathogenesis of neuronal death after injury during brain maturation, the authors investigated caspase-3 activation during neuronal death in rats of different ages. The authors found that involvement of caspase-3 in cell death after HI gradually declined during brain development and maturation. The decline may be because the genetic components for apoptosis, such as caspase-3 protein, are less expressed in postmitotic adult neurons.
MATERIALS AND METHODS
Hypoxia–ischemia model
Pregnant Wistar rats were purchased from Charles River Laboratories (Wilmington, MA, U.S.A.) and housed in individual cages. The newborn rats were housed with their dams until weaning at postnatal day 21. All procedures for the animal study were approved by The Animal Care Committee at The Queen's Medical Center. Brain HI was produced by a combination of ligation of the left common carotid artery and systemic hypoxia (8% O2) according to the method of Rice et al. (1981). Briefly, rats at postnatal day 7 (P7), 15 (P15), 26 (P26), and 60 (P60) were used. Hypoxia–ischemia was induced for 60 minutes in P7 and P15 pups, and for 30 minutes in P26 and P60 rats. In addition, the authors used 20 minutes and 60 minutes of HI in P60 rats to study the severity of ischemia on activation of caspase-3. The hypoxic duration in different groups of rats was chosen based on the authors' preliminary experiments and the previous studies, which showed that these periods produced a similar severity of brain damage in the different age groups (Blumenfeld et al., 1992; Towfighi et al., 1997; Towfighi and Mauger, 1998). The contralateral sides of the brains after HI were used as controls. In some cases, rats subjected to carotid ligation without subsequent hypoxia were also used as controls. There was no difference between the two controls and untreated rats in the authors' biochemical and microscopic studies. Brains were collected at 6, 24, and 48 hours after HI. Tissue samples for biochemical analyses were obtained by freezing the brain in situ with liquid nitrogen. The brains were perfused with ice-cold 4% phosphate-buffered paraformaldehyde for confocal microscopy.
Electron microscopy
Tissue sections from P7, P15, P26, and P60 rats subjected to HI were stained by the conventional osmium-uranium-lead method, and thin sections were cut and examined with an electron microscope as described previously (Hu et al., 1998).
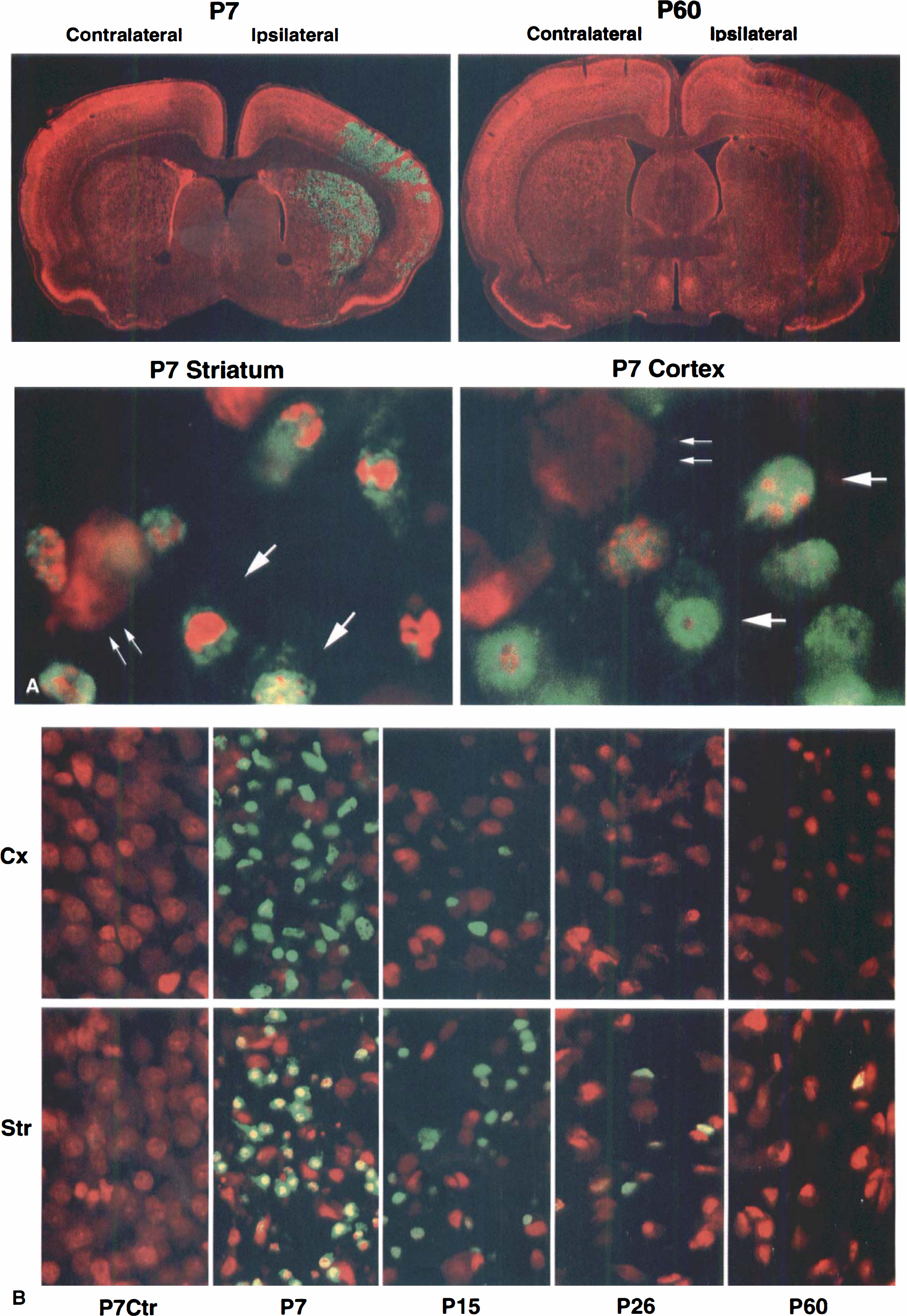
Laser-scanning confocal microscopy and histology
Double-labeled fluorescence confocal microscopy was performed on 50 μm vibratome brain sections as described previously (Hu et al., 1999). Brain sections were taken from sham-control rats and rats subjected to HI followed by 48 hours of recovery for all age groups, and in separate experiments, by 6, 24, and 48 hours of recovery for P15 and P60 rats. A specific antibody against active caspase-3 was obtained from New England Biolab (NEB, Beverly, MA, U.S.A.). A polyclonal antibody to 32-33 kDa caspase-3 was obtained from Upstate Biotechnology (UBI, Lake Placid, NY, U.S.A.). A monoclonal antibody against neuronal nuclear protein NeuN was purchased from Chemicon (Temecula, CA, U.S.A.). Brain sections were labeled with the antibodies and analyzed on a BioRad MRC 1024 laser-scanning confocal microscope (Hercules, CA, U.S.A.). The vibratome sections were also stained with acid fuchsin and thionin. The active caspase-3 positive neurons were dead as they were shrunken and acidophilic. Cell counting was performed to determine the absolute and percentage numbers of active caspase-3 positive and negative neurons among total damaged cells in each brain region at 48 hours after HI. The brain sections at the level of striatum (approximately Bregma −0.35 mm;Fig. 1A) were double-stained with antiactive caspase-3 and propidium iodide and examined by confocal microscopy using the 60× objective. The counting was conducted in the core of the HI-damaged neocortical and striatal regions where more than 95% neurons were damaged, as determined with acid fuchsin and thionin staining (Fig. 1). Five confocal microscopic fields with the 60x objective were taken in each brain region with the BioRad 1024 confocal microscope, and the neurons in each field (175 μm × 175 μm) were counted. Data were obtained from four animals in each group (n = 4) and expressed as mean ± SD. Two-way analysis of variance was used to assess the effects of age and recovery time on active caspase-3 induction and neuronal death.
(
Biochemical analyses
Brain homogenates and subcellular fractions were prepared from sham-operated, ipsilateral (ischemic), and contralateral (nonischemic) brain tissues after HI according to the method of Hu and Weiloch (1994). Each experimental group consisted of 3 individual samples derived from 3 rats. Forebrain tissue blocks between Bregma 1.70 mm and Bregma −4.52 were collected and divided into ipsilateral and contralateral sides to prepare the samples. The homogenates were analyzed by Western blotting for caspase-3 and active caspase-3. The protein concentration was determined by the method of Lowry.
RESULTS
The histopathology of the brain sections stained with acid fuchsin and thionin were examined by light microscopy. As in mature rats, a period of HI caused delayed neuronal death occurring in the ipsilateral side of the dorsal striatum and dorsal-lateral neocortex at 24 to 48 hours after HI in immature rats (Towfighi and Mauger, 1998; data not shown but see Fig. 1).
To study involvement of caspase-3 activation in the pathogenesis of neuronal death after HI among different age groups, the brain sections after HI were double-labeled with a specific antibody against active caspase-3 (green) and propidium iodide (red), and were then examined with confocal microscopy (Fig. 1). The specificity of active caspase-3 immunoreactivity was proved by adding the antigen peptide and by omitting the primary antibody during the incubation; both treatments eliminated the active caspase-3 labeling from the brain sections (data not shown). The active caspase-3 positive regions after HI were colocalized with the condensed and fragmented ischemic neurons in the brain sections double-stained with the active caspase-3 (green) and either fuchsin (data not shown) or propidium iodide (red;Fig. 1). Active caspase-3 positive neurons were condensed and fragmented as illustrated by high-resolution confocal microscopy (Fig. 1A, lower panel).
The distribution of active caspase-3 in the neocortex and striatum of different age groups after HI is shown in Fig. 1B. Active caspase-3 induction (green) in the ipsilateral regions was found in most HI-damaged neurons in P7 pups but was markedly decreased in P15 pups. By P26, some of the active caspase-3 positive neurons were still present on the ipsilateral striatum, but they were only occasionally found in the neocortex. In P60 rats, most of ischemic dying neurons were negative for active caspase-3 (Fig. 1B).
To follow up the progressive decrease in active caspase-3 positive neurons with increasing animal age after HI, the authors conducted a quantitative study. The number of normal neurons in the corresponding contralateral regions declined with increasing animal age (Table 1), which was consistent with the neuronal elimination during brain maturation (Bayer and Altman, 1995). However, the percentage of active caspase-3 positive neurons among the total HI-damaged neurons declined with increasing animal age at 48 hours after HI (Fig. 2, upper panel). The 48-hour time period was chosen because neuronal death reached a plateau at 48 hours after HI (see below) and active caspase-3 in these dead neurons were strongly labeled with the antibody (Fig. 1). In the striatum and cortex, active caspase-3 positive neurons were reduced from 92.63 ± 3.92% and 91.97 ± 2.42% in P7 rat pups, to 65.14 ± 10.92% and 37.99 ± 8.87% in P15 rat pups, to 26.89 ± 11.35% and 2.04 ± 4.30% in P26 rats, to 4.01 ± 2.98% and 0.79 ± 3.23% in P60 rats, respectively (Fig. 2, upper panel).
Average number of neurons in the contralateral brain regions of different age rats after hypoxia–ischemia
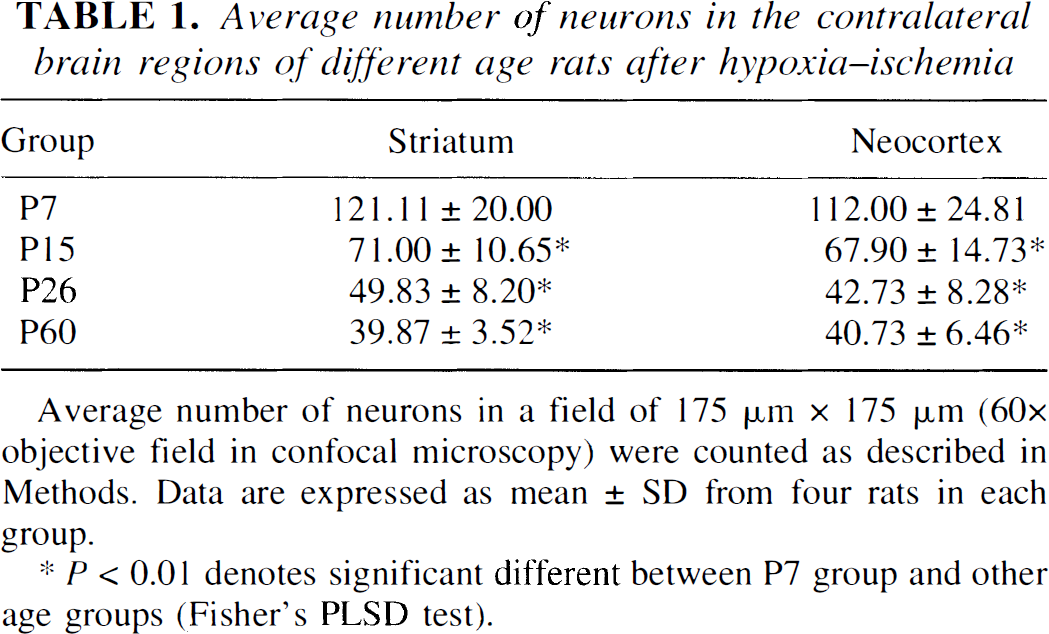
Average number of neurons in a field of 175 μm × 175 μm (60×objective field in confocal microscopy) were counted as described in Methods. Data are expressed as mean ± SD from four rats in each group.
P < 0.01 denotes significant different between P7 group and other age groups (Fisher' PLSD test).
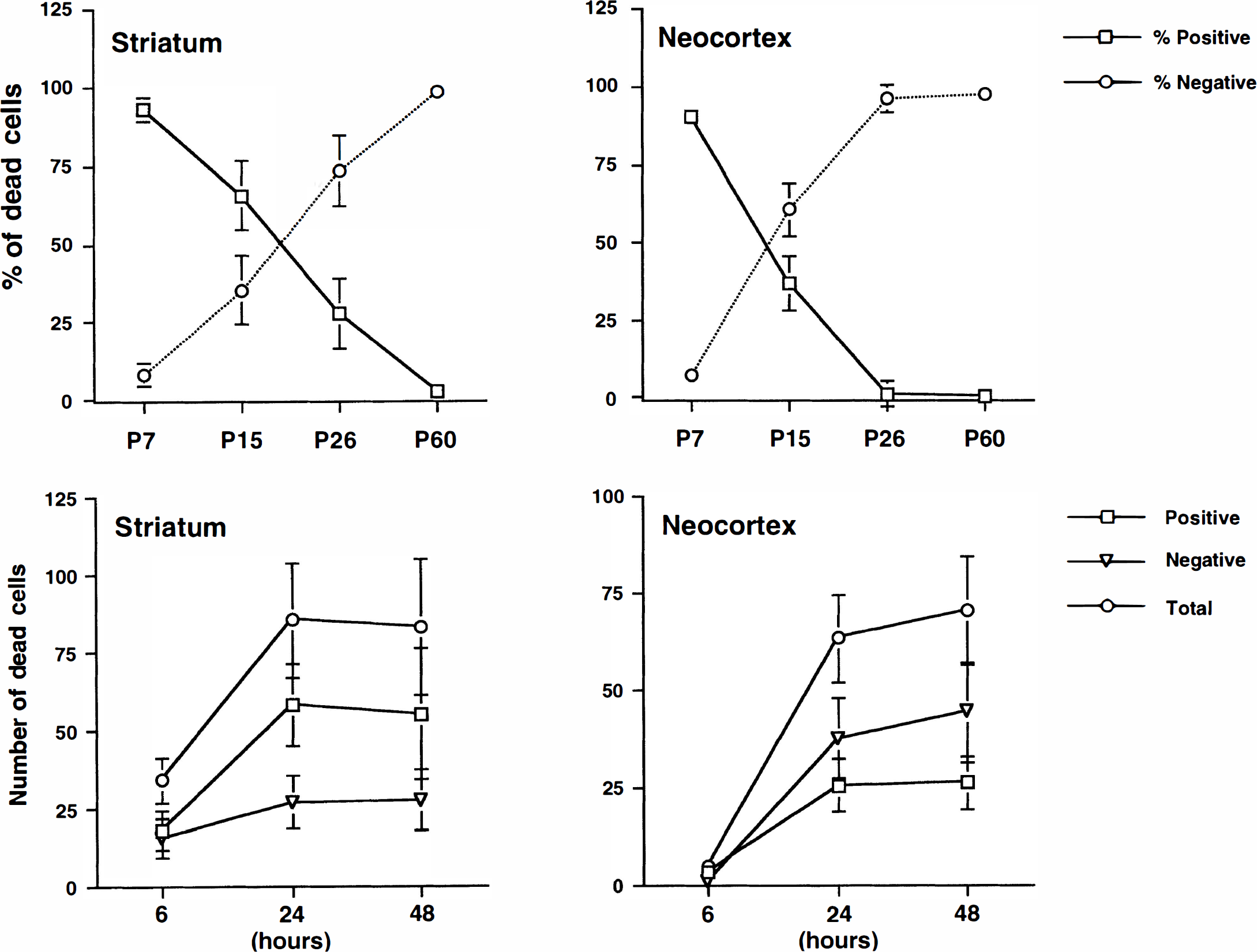
Upper panel shows active caspase-3 positive and negative neurons in the striatum and cortex of different age groups. Average numbers of active caspase-3 positive and negative and total dead cells at 48 hours after hypoxia–ischemia (HI) were counted in a field of 175 μm × 175 μm (60x objective field in confocal microscopy) on the brain sections of P7, P15, P26, and P60. Data are expressed as mean ± SD from four rats in each group. The differences between the P7 group and the other age groups are significant (P < 0.01; Fisher's PLSD test). Lower panel illustrates active caspase-3 positive and negative neurons in the striatum and cortex of different recovery periods after HI in P15 pups. Average numbers of active caspase-3 positive and negative and total dead cells were counted in a field of 175 μm × 175 μm (60x objective field in confocal microscopy) on the brain sections of 6, 24, and 48 hours after HI. Data are expressed as mean ± SD from four rats in each group. The differences between 6 hours and 24 or 48 hours are significant (P < 0.01; Fisher's PLSD test).
The time course of neuronal death, or induction, or both of active caspase-3 after HI were studied in the brain sections at 6, 24, and 48 hours after HI in P15 pups and P60 rats. In P15 pups, active caspase-3 positive neurons appeared as early as 6 hours and reached a plateau at 24 to 48 hours after HI in striatum (Fig. 2, lower panel). In contrast, the dead neurons after HI were hardly found in the cortical regions until 24 hours after HI and tended to increase further at 48 hours in P15 pups (Fig. 2, lower panel). In P60 rats, HI-damaged neurons could also be found at 6 hours in striatum and 24 hours in the cortex (data not shown). However, most of HI-damaged neurons in P60 rats were negative for active caspase-3 (Fig. 1A, upper panel).
Decreasing active caspase-3 in neurons with increasing animal age after HI was further confirmed by Western blot analyses. Levels of inactive caspase-3 (32 to 33 kDa), labeled with an antibody, gradually decreased from high levels in P7 pups to barely detectable levels in P60 rats in contralateral (Fig. 3, upper panel) and ipsilateral hemispheres (Fig. 3, middle panel). Interestingly, an unknown protein approximately 29 kDa labeled by the caspase-3 antibody gradually increased from undetectable levels in P7 pups to relative high levels in P60 (Fig. 3, arrows). The decrease of caspase-3 with increasing animal age in the brain tissues was further confirmed with another caspase-3 antibody from Santa Cruz (H-227) on Western blots. Concomitantly, induction of 17 to 19 kDa active caspase-3 after HI also declined from a relative high level in P7, to moderate levels in P15 and P26, to a barely detectable level in P60 rats in the ischemic brains (Fig. 3, lower panel).
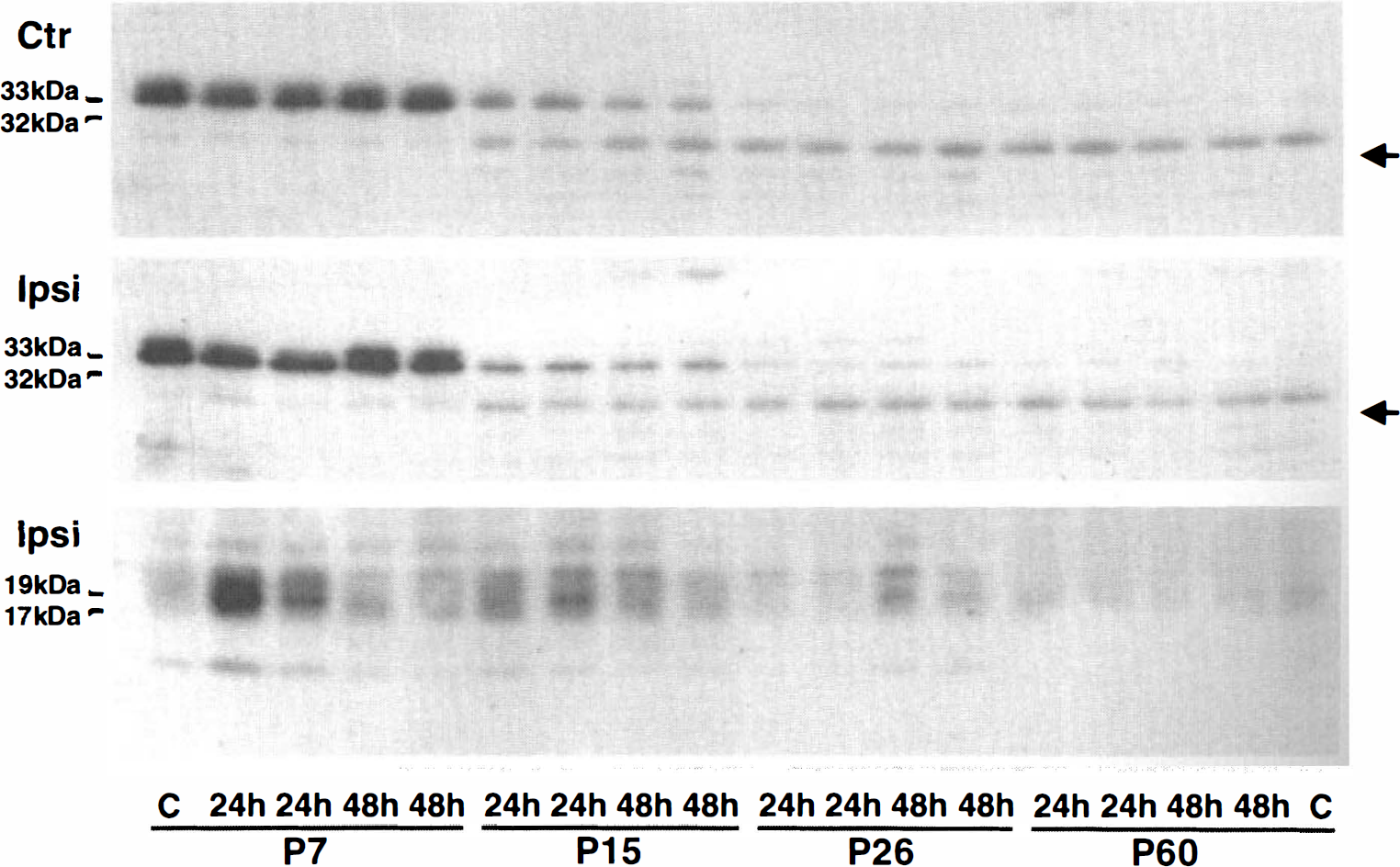
Western blots of total caspase-3 (32 to 33 kDa) and active caspase-3 (17 to 19 kDa) in brain homogenates after hypoxia–ischemia (HI). The brain homogenates were from the contralateral (Ctr, upper panel) and ipsilateral hemispheres (Ipsi, middle and lower panels) subjected to HI followed by 48 hours of recovery. Two samples from two individual rats in each experimental group were run on the same gel. Levels of 32 to 33 kDa caspase-3 in contralateral and ipsilateral hemispheres were decreased with increasing age of the animals. The active caspase-3 was detected only in the ipsilateral brain tissues and its level decreased with increasing animal age.
DISCUSSION
In the current study, the authors investigated the involvement of caspase-3 in the pathogenesis of HI-induced neuronal death in rats of different ages by confocal microscopy and biochemical analysis. The authors found that the active caspase-3 positive neurons after HI were decreasing with increasing animal age. Hypoxia–ischemia-induced neuronal death in P7 immature pups was almost entirely active caspase-3 positive. A mixture of active caspase-3 positive and negative neurons was found in P15 and P26 rats. In P60 rats, only a small portion of neurons after HI was positive for active caspase-3. These results demonstrate that the pathogenesis may shift from caspase-3-mediated to noncaspase-3-mediated neuronal death after HI during brain maturation.
The sensitivity of neurons to injuries varies with the animal's age. Thus, the authors adjusted the duration of HI in the different age groups to produce similar brain damage in rats of different ages according to the data presented in previous publications (Schurr and Rigor, 1987; Blumenfeld et al., 1992; Towfighi et al., 1997; Towfighi and Mauger, 1998). Consistent with these previous reports, 60 minutes of HI in P7 and P15 pups produced similar brain damage as did 30 minutes of HI in P26 and P60 animals. In the P60 group, the authors also used HI duration of 60 minutes and 20 minutes. Sixty minutes of HI caused severe mortality and large infarction, whereas 20 minutes of HI only induced selective neuronal death. All of these results are consistent with previous studies (Rice et al., 1981; Schurr and Rigor et al., 1987; Blumenfeld et al., 1992; Towfighi et al., 1997; Towfighi and Mauger, 1998).
The antiactive caspase-3 antibody used for confocal microscopic immunocytochemistry recognizes 17 to 19 kDa of active caspase-3 on Western blots. On brain sections, the active caspase-3 antibody only labels HI-damaged neurons with condensed and fragmented nuclei in young pups, but stains neither HI-damaged neurons in adult rats nor normal neurons from the contralateral hemisphere or from sham-operated controls in the different age groups. The active caspase-3 negative neurons usually have dispersed nuclear chromatin. Based on the current results, it is likely that activation of caspase-3 may contribute to HI-induced neuronal death in immature neurons, but would play a minor role in mature neurons, which agrees with several previous studies in adult ischemic animals (Deshpande et al., 1992; van Lookeren Campagne and Gill, 1996; Colbourne et al., 1999). Moderate insults may activate mechanisms of apoptosis, whereas dense and long-lasting insults are prone to cause necrosis (Nicotera and Lipton, 1999). In the current study, active caspase-3 positive neurons were rarely found in adult neurons after either severe 60 minutes, or mild 20 minutes, or 30 minutes HI. In addition, active caspase-3 positive neurons were also rarely found in the boarder region between damaged and nondamaged areas in adult animals. Furthermore, active caspase-3 positive neurons were rarely present in HI-damaged adult neurons at either 6 or 24 hours after HI. All of these results suggest that the negative staining of active caspase-3 in most adult neurons is not because the insult is too severe or too mild, or because the HI is too long.
It is interesting to note that 32 to 33 kDa caspase-3 in normal forebrain tissue decreases from high levels in P7 pups to barely detectable levels in P60 rats on Western blots. This result has been reproduced several times with two different caspase-3 antibodies. A recent report has shown that active caspase-3 and active caspase-8 are undetectable on Western blots after brain ischemia in a rat focal ischemia model, as is the caspase-3 activity (Velier et al., 1999). It has been demonstrated that naturally occurring neuronal death in the developing rat is primarily present from embryonic day 19 until postnatal day 8 with a peak occurring at birth (Waite et al., 1992), and that the dead neurons are TUNEL-positive (Maciejewska et al., 1998) and active caspase-positive (Siman et al., 1999). Thus, it seems likely that the caspase-3-mediated apoptotic machinery in immature neurons serves a purpose only during neuronal maturation. This cell death machinery may accidentally be turned on after HI to execute neuronal death in immature neurons. However, it may gradually fade out after neurons have matured. This possibility offers a plausible explanation for the shifting of pathogenesis from caspase-3-mediated neuronal death in immature neurons to noncaspase-3 mediated neuronal death in mature neurons after brain injury.