
Abstract
A short ischemic event (ischemic preconditioning [IPC]) can result in a subsequent resistance to severe ischemic injury (ischemic tolerance [IT]). Although tumor necrosis factor-α (TNF-α) contributes to the brain damage, its expression and neuroprotective role in models of IPC have also been described. However, the role of TNF-α convertase (TACE) in IPC and IT is not known. Using in vitro models, the authors previously demonstrated that TACE is upregulated after ischemic brain damage. In the present study, the authors used a rat model of transient middle cerebral artery occlusion as IPC to investigate TACE expression, its involvement in TNF-α release, and its role in IT. Western blot analysis showed that TACE expression is increased after IPC. Ischemic preconditioning caused TNF-α release, an effect that was blocked by the selective TACE inhibitor BB-1101 (10 mg · kg−1 · day−1; SHAM, 1,050 ± 180; IPC, 1,870 ± 290; IPC + BB, 1,320 ± 260 ng/mg; n = 4, P < 0.05). Finally, IPC produced a reduction in infarct volume, which was inhibited by treatment with BB-1101 and with anti–TNF-α (10 μg/5 doses; SHAM + permanent middle cerebral artery occlusion [pMCAO], 335 ± 20; IPC + pMCAO, 244 ± 14; IPC + BB + pMCAO, 300 ± 6; IPC + anti-TNF + pMCAO, 348 ± 22 mm3; n = 6–10, P < 0.05). Taken together, these data demonstrate that TACE is upregulated after IPC, plays a major role in TNF-α shedding in IPC, and has a neuroprotective role in IT.
In the last few years, it has been demonstrated that a short ischemic event (ischemic preconditioning [IPC]) can result in a subsequent resistance to severe ischemic injury (ischemic tolerance [IT]; Kitagawa et al., 1990). This phenomenon has been described in several organs, especially the heart and brain. Although certain pathophysiologic issues may be similar, the mechanisms of induction and maintenance of tolerance in the brain are distinct from those described in the heart. Ischemic tolerance has also been demonstrated in human clinical practice, since less severe strokes have been described in patients with prior ipsilateral transient ischemic attacks within a short period of time (Blanco et al., 1999: Moncayo et al., 2000; Weih et al., 1999).
There are two temporally and mechanistically distinct types of protection afforded by IPC: acute and delayed tolerance. It is well known that the effects of delayed tolerance require new protein synthesis and are sustained for days to weeks. The precise mechanism is not known, but de novo synthesis of proteins, promoting neuronal survival, includes heat-shock proteins (Kirino et al., 1991), Bcl-2 (Shimazaki et al., 1994), and superoxide dismutase (MnSOD) (Toyoda et al., 1997). In addition, the activation of transcription factors such as nuclear factor-κ (NF-κ) has recently been implicated by several authors in the development of IT (Blondeau et al., 2001; Ginis et al., 2002). This factor plays a pivotal role in neuronal survival and is activated by various signals, such as proinflammatory cytokines, neurotrophic factors, and neurotransmitters (Mattson et al., 2000).
Tumor necrosis factor-α (TNF-α) is one of these proinflammatory cytokines, and is expressed or released after brain ischemia (Liu et al., 1994). Although TNF-α contributes to the ischemic brain damage found in this condition, there is evidence supporting a neuroprotective role for this cytokine (Shohami et al., 1999). Furthermore, the expression of TNF-α and its neuroprotective role in in vivo and in vitro models of IPC have also been described (Liu et al., 2000; Wang et al., 2000).
Tumor necrosis factor-α is shed in its soluble form by a membrane-anchored zinc protease, identified as ADAM (A Disintegrin And Metalloproteinase), called TNF-α convertase (TACE/ADAM17). The role of this protease in the adult nervous system is poorly known. We have recently demonstrated, using in vitro models, that TACE is upregulated after ischemic brain damage and that the increase in TACE expression contributes to a rise in TNF-α and a subsequent neuroprotective effect after excitotoxic stimuli (Hurtado et al., 2001, 2002). The role for TACE in IT, however, has not been described.
We investigated the expression of TACE protein, its involvement in TNF-α release, and its role in IT using a rat model of transient middle cerebral artery occlusion (tMCAO) as IPC.
MATERIALS AND METHODS
Animals
Adult male Fischer rats (240 to 260 g) were housed and cared for in accordance with the Committee of Animal Care at the Universidad Complutense of Madrid according to E.U. rules (DC86/609/CEE). All animals had free access to food and water before and after surgery.
Focal ischemic preconditioning
A period of 10 minutes of tMCAO was used for focal IPC according to the method previously described by Barone et al. (1998). Animals were anesthetized with 2% halothane in a mixture of 70% nitrogen and 30% oxygen, and body temperature was maintained at 37°C using a heating pad throughout the surgical procedure and during postsurgical recovery. We used a surgical preparation consisting of a tandem occlusion of the distal middle cerebral artery (MCA) and the ipsilateral common carotid artery (CCA). The ventral neck and the area behind the left eye were shaved and a povidone–iodine solution was applied to sterilize the skin. For transient CCA occlusion, a midline ventral cervical incision was made and the CCA was isolated and occluded with an aneurysm clip. For tMCAO, a 1-cm incision perpendicular to the line connecting the lateral canthus of the left eye and the external auditory canal was made to expose and retract the temporalis muscle. A 2-mm burr hole was drilled (Drill Foredom, Stoelting, Wood Dale, IL, U.S.A.) 2 or 3 mm rostral to the fusion of the zygomatic arch with the squamosal bone. Heat injury to the cerebral cortex was prevented by continuously dripping 0.9% saline onto the skull during drilling. The MCA was exposed by cutting and retracting the dura, and was located by means of a dissecting binocular microscope (Nikon SMZ-1; Nikon Corporation, Tokyo, Japan). The tip of a stainless steel wire hook, bent at 90°, was placed under the MCA with a micromanipulator (M3301; World Precision Instruments, U.K.) and used to lift the artery away from the brain surface to temporarily occlude blood flow as described previously (Barone et al., 1998). A period of 10 minutes of tMCAO was used because this short ischemic period results in no tissue injury but protects the tissue from later ischemia (Barone et al., 1998). Rats in which the MCA was exposed but not lifted served as sham-operated controls.
Permanent focal ischemia
To evaluate the ability of IPC (tMCAO) to produce IT, permanent MCAO (pMCAO) was performed 48 hours after sham or tMCAO surgery. We have used the procedure described previously, but permanent occlusion was made in both CCA and MCA as described elsewhere (Brint et al., 1988; De Cristóbal et al., 2001).
For permanent CCA occlusion a silk ligature was used, whereas MCA was occluded by applying an electrocoagulator tip (Geiger Model-100; Stoelting) just above the vessel on the wire hook. Heat was transferred through the wire, and the MCA was cauterized and severed.
Intracerebroventricular injection of anti–tumor necrosis factor-α
In order to neutralize TNF-α produced by TACE after IPC, intracerebroventricular injections of anti–TNF-α were made at 18, 21, 24, 27, and 30 hours after IPC.
Briefly, using pentobarbital (50 mg/kg; IP) anesthesia, an intracerebroventricular 26-G cannula (Plastics One, Roanoke, VA, U.S.A.) was inserted into the left lateral ventricle by using a stereotaxic apparatus (Davis Kopf Instruments, Tujunga, CA, U.S.A.) and the following coordinates: anteroposterior, −0.8 mm; lateral, 1.0 mm; dorsoventral, 3.6 mm from the bregma, with the incisor bar placed at 3.3 mm below the interaural plane. Cannulas were anchored to the skull by stainless steal screws and dental cement. After a 5-day recovery period, the animals were anesthetized with halothane and underwent IPC as described previously. Repeated injections were performed in conscious, lightly restrained rats using a 33-G injection cannula, 1 mm longer than the guide cannula. Each injection consisted of 5 μL anti–TNF-α (10 (μg ã 60 pmol) or vehicle (sterile phosphate-buffered saline) infused into the ventricle with a microinfusion pump (HA 22; Harvard Apparatus, Holliston, MA, U.S.A.) over 2 minutes.
Forty-eight hours after IPC (18 hours after the last injection), pMCAO was performed and infarct volume was measured 24 hours later.
Experimental groups
Three groups were used for determinations of TNF-α release: (1) a control group of sham-operated rats (SHAM; n = 4); (2) an IPC group of rats that underwent tMCAO (IPC; n = 4); and (3) a group of rats in which tMCAO + BB-1101 (an selective TACE inhibitor) was used to determine the involvement of TACE in TNF-α release after IPC (IPC + BB; n = 4). BB-1101 was administered subcutaneously at 10 mg · kg−1 · day−1 after tMCAO for 2 days. The dose of BB-1101 was chosen according to our previous data showing an effective inhibition of TACE in brain (Madrigal et al., 2002). In addition, to evaluate the ability of IPC to produce IT, pMCAO was performed 48 hours after sham or tMCAO surgery and the infarct area was measured 24 and 72 hours after pMCAO. Therefore, the four groups were established as: (1) SHAM + pMCAO (n = 10); (2) IPC + pMCAO (n = 10); (3) IPC + BB + pMCAO (n = 10); and (4) IPC + anti-TNF + pMCAO (n = 6).
TACE protein expression
TACE protein levels were determined 0, 12, and 24 hours after IPC as previously described (Hurtado et al., 2002). Briefly, brain cortices from both ipsilateral and contralateral hemispheres were removed at those times and homogenized by sonication for 10 seconds at 4°C in four volumes of homogenization buffer containing 320 mmol/L sucrose, 1 mmol/L DL-dithiothreitol, 10 μg/mL leupeptin, 10 μg/mL soybean trypsin inhibitor, 2 μg/mL aprotinin, 0.2% Nonidet P40 (Roche, Barcelona, Spain), 100 μmol/L 1,10-phenanthroline, and 50 mmol/L Tris brought to pH 7.0 at 20°C with HCl. 1,10-Phenanthroline was included to avoid removal of the cytoplasmic domain after cell lysis by a metalloprotease, very likely TACE itself (Schlóndorff et al., 2000). Homogenate containing 20 μg protein was loaded, and the proteins were size-separated by 10% SDS-polyacrylamide gel electrophoresis (90 mA). The proteins were blotted onto a PVDF membrane (Millipore, Madrid, Spain) and incubated with a specific polyclonal anti-TACE (Chemicon, Temecula, CA, U.S.A.; 1:1000 dilution; raised against a peptide corresponding to amino acids 807–823 of human TACE; Black et al., 1997). Proteins recognized by the antibody were revealed by ECL-kit (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) following the manufacturer's instructions. SDS-PAGE was performed in two different gels each containing n = 2 per group, making a total of n = 4.
Tumor necrosis factor-α levels
To determine tissue levels of soluble TNF-α, brain cortices from both ipsilateral and contralateral hemispheres were removed 24 hours after IPC and homogenized by sonication for 10 seconds at 4°C in four volumes of homogenization buffer. The homogenates were centrifuged (13,000 g, 10 minutes, 4°C) and supernatants were used for determinations using a commercially available ELISA kit (Quantikine; R&D Systems, Minneapolis, MN, U.S.A.) following the manufacturer's instructions. Tumor necrosis factor-α ELISA was performed in at least n = 4 per group.
Determining infarct area
The brains were removed 24 and 72 hours after pMCAO, and a series of 2-mm coronal slices were obtained (Brain Matrix; World Precision Instrument, U.K.) and stained in 1% TTC (2,3,5-triphenyl-tetrazolium chloride, Sigma) in 0.1 mol/L phosphate buffer. The infarcted area, which is not stained, was quantified by image analysis (Scion Image for Windows 2000, Scion Corporation, Frederick, MD, U.S.A.).
Chemicals and statistical analyses
BB-1101(2S-allyl-N1-hydroxy-3R-isobutyl-N4-(1S-methylcarbamoyl-2-phenylethyl)-succinamide) was kindly supplied by British Biotech (Kupatt et al., 1999; Redford et al., 1997), anti–TNF-α was supplied by R&D Systems (Vitro; Madrid, Spain), and other chemicals were obtained from Sigma (Spain) or as indicated in the text. Results are expressed as mean ± SD of the indicated number of experiments, and statistical comparisons were made using a Newman-Keuls test.
RESULTS
Effect of ischemic preconditioning on TACE expression
Western blot analysis revealed the presence of a TACE-immunopositive band (Fig. 1A) in brains from sham-operated animals. IPC induced a time-dependent increase in TACE protein levels detected by immunoblotting (at 12 to 24 hours) that was maximal 24 after IPC (n = 4; *P < 0.05; Fig. 1A).
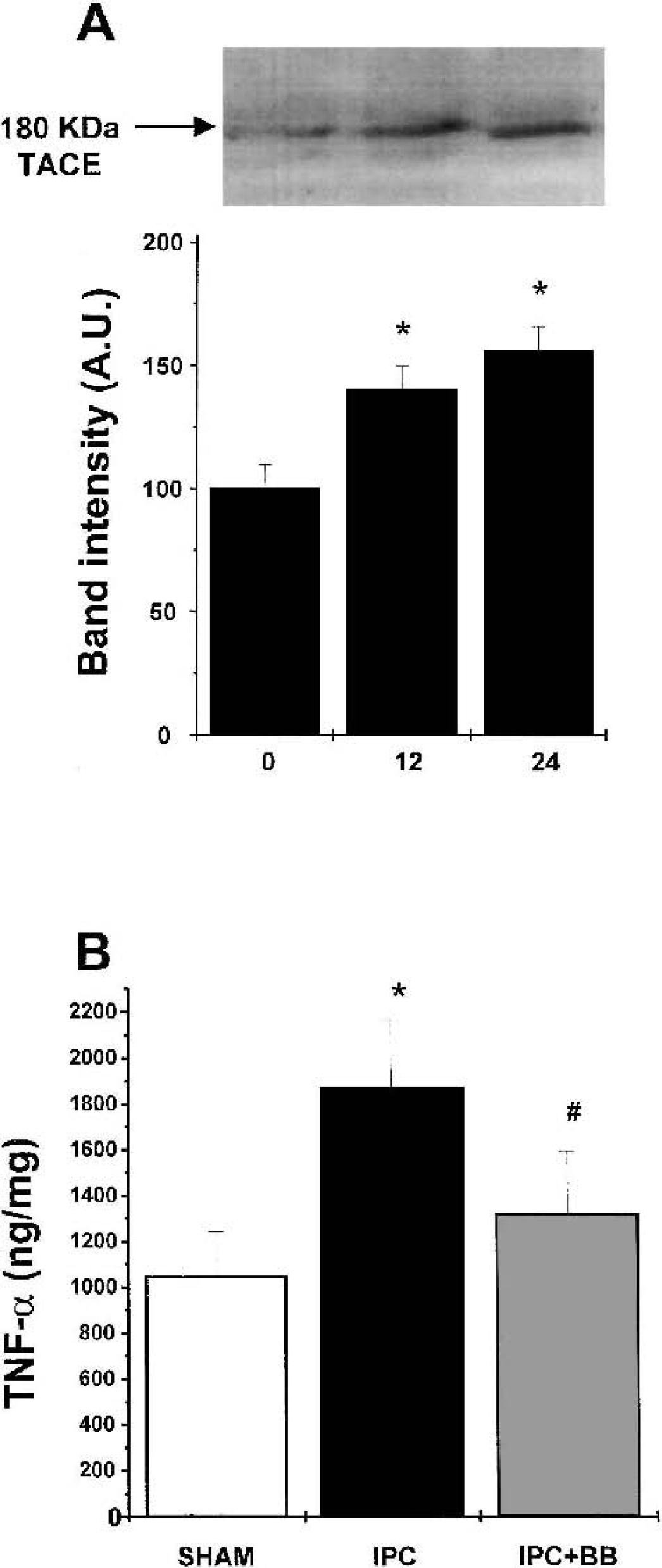
Western blot analysis of TNF-α convertase (TACE) protein in brain homogenates 0, 12, and 24 hours after ischemic preconditioning (IPC). Lower panel shows the laser densitometric analysis of bands.
Effect of ischemic preconditioning on tumor necrosis factor-α levels: Effect of inhibition of TACE
Ischemic preconditioning caused an increase in TNF-α tissue levels at 24 hours (1,870 ± 290 ng/mg; n = 4) when compared with the sham-operated group (1,050 ± 180 ng/mg; n = 4; *P < 0.05; Fig. 1B).
Treatment with BB-1101, a selective inhibitor of TACE activity, inhibited the TNF-α increase induced by IPC (1,320 ± 260 vs. 1870 ± 290 ng/mg in IPC + BB and IPC, respectively; n = 4; #P < 0.05; Fig. 1B).
Effect of ischemic preconditioning on infarct volume: Effect of inhibition of TACE
Ischemic preconditioning produced a reduction in infarct volume measured at 24 hours after pMCAO (IPC + pMCAO, 244 ± 14 mm3, 73 ± 4% of SHAM + pMCAO value; n = 10) when compared with the sham-operated animals (SHAM + pMCAO, 335 ± 20 mm3, 100%; n = 10; *P < 0.05; Fig. 2). Treatment with BB-1101 as well as intracerebroventricular administration of anti–TNF-α inhibited the IPC-induced decrease in infarct volume measured 24 hours after pMCAO (IPC + BB + pMCAO, 300 ± 6 mm3; n = 10; #P < 0.05; and IPC + anti-TNF + pMCAO, 348 ± 22 mm3; n = 6; #P < 0.05; Fig. 2). Administration of BB-1101 alone did not affect infarct volume after pMCAO (SHAM + BB + pMCAO, 309 ± 15 mm3; n = 10; P > 0.05). In addition, intracerebroventricular administration of vehicle did not produce any effect (data not shown).
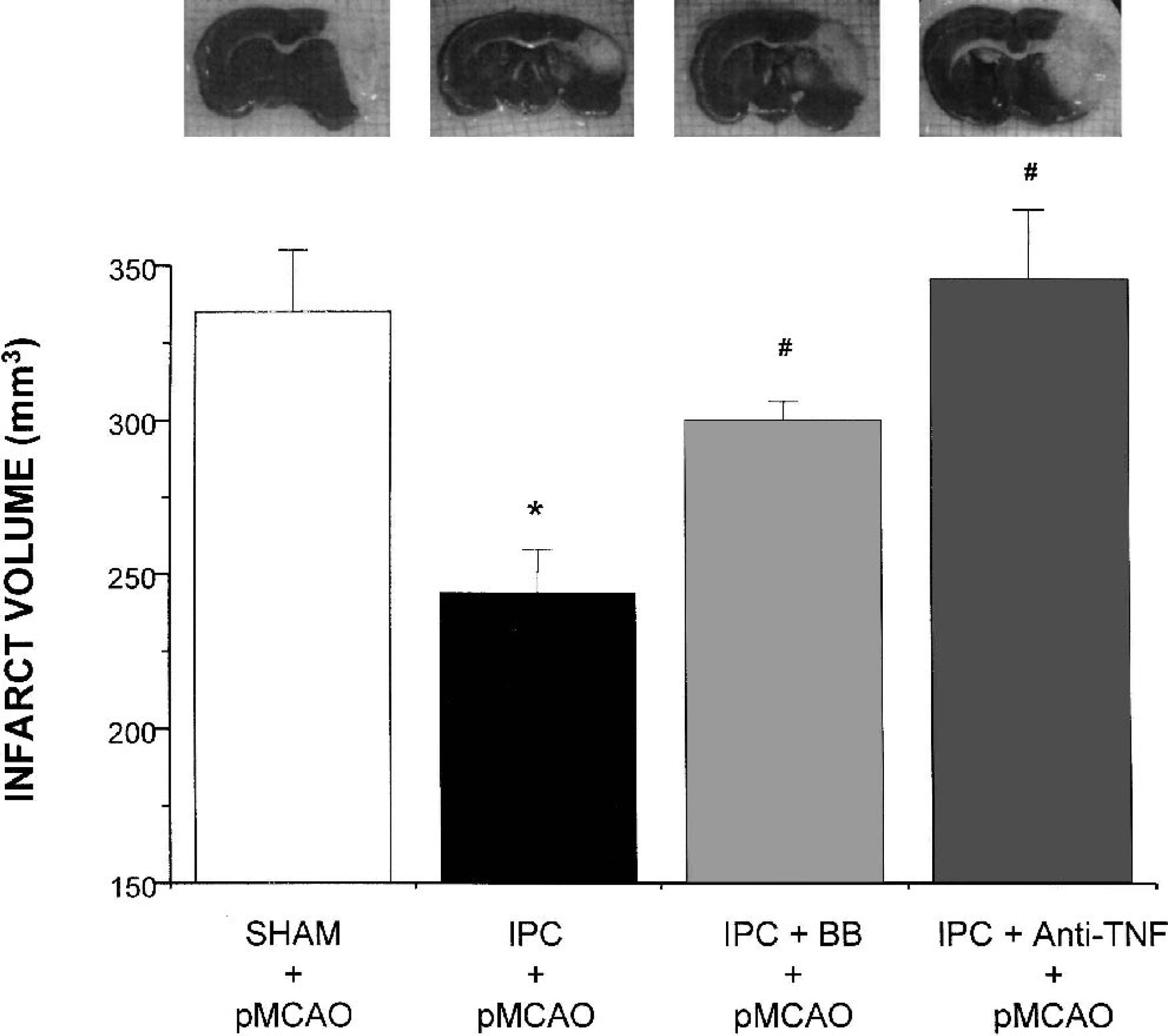
Infarct volume (mm3) measured 24 hours after permanent middle cerebral artery occlusion (pMCAO) in sham-operated (SHAM + pMCAO), preconditioned (IPC + pMCAO), preconditioned rats treated with BB-1101 (IPC + BB + pMCAO) and preconditioned rats treated with anti–TNF-α (IPC + Anti-TNF + pMCAO). Inset: photographs of representative animals. Data are mean ± SD, n = 6 to 10; *P < 0.05 versus SHAM + pMCAO; #P < 0.05 versus IPC + pMCAO. TNF-α, tumor necrosis factor-α.
Although IPC caused a larger reduction in infarct volume when measured 72 hours after pMCAO (61% ± 6% of SHAM + pMCAO), BB1101 induced inhibition of the protective effect of IPC at both times studied (89% ± 2% and 81% ± 5% of SHAM + pMCAO values at 24 and 72 hours after pMCAO, respectively).
DISCUSSION
We recently provided the first demonstration of the presence of TACE protein in CNS and of its upregulated expression after ischemia or excitotoxicity using in vitro models (Hurtado et al., 2001, 2002). In the present study, we have demonstrated that TACE is upregulated after IPC, that TACE plays a major role in TNF-α shedding in IPC, and that TACE-induced TNF-α overproduction is responsible, at least in part, for the IT afforded by IPC.
First, analysis of TACE protein by Western blot analysis demonstrates its presence in brains from both control and IPC-exposed animals. These results are in agreement with the demonstration of TACE mRNA in both rat and mouse adult brain (Kärkkäinen et al., 2000) and with our previous data demonstrating the presence of TACE protein in the CNS (Hurtado et al., 2001). Moreover, TACE expression is increased after IPC, also in agreement with our previous data showing increased TACE protein and mRNA expression in in vitro models of brain ischemia (Hurtado et al., 2001, 2002), or of its activation after immobilization stress in rat brain cortex (Madrigal et al., 2002).
Our findings of the constitutive expression of TACE and of its increased expression after IPC raise the question of its role in TNF-α release. Our results show that IPC causes the release of soluble TNF-α, an effect that is inhibited by the hydroxamate-based compound BB1101, suggesting that this release results predominantly from pro–TNF-α shedding by TACE/ADAM17. Although other proteases have been suggested to possess the ability to cleave pro–TNF-α (Lammich et al., 1999), hydroxamate-based compounds are more potent against TACE, a fact that has been used to distinguish between the TACE effect and that of the other proteases (Hooper et al., 2000; Parvathy et al., 1998). Regarding the regulation of TNF-α release, although our data clearly indicate that TACE upregulation plays a role in this setting, we cannot discard the possibility that increased pro–TNF-α synthesis (Wang et al., 2000) or increased TACE catalytic activity is involved in this process.
It is well known that TNF-α, which is expressed or released after ischemia (Liu et al., 1994), contributes to the damaging effects of this condition; additional evidence, however, has demonstrated a direct, neuroprotective role of TNF-α (Shohami et al., 1999). We have now shown that TACE-induced TNF-α release participates in the development of brain tolerance, since addition of BB1101, which has already been determined to reduce TNF-α release, is able to inhibit the reduction of the infarct volume caused by IPC. Consistent with this finding, the anti–TNF-α antibody mimicked this inhibitory effect of BB-1101, strongly supporting this conclusion. In addition, although IPC-induced reduction in infarct volume 72 hours after pMCAO was even larger than that found at 24 hours (39% reduction), in agreement with previous results (Mullins et al., 2001) suggesting that reparative processes become active between 24 and 72 hours after secondary MCAO in preconditioned animals, the effect produced by BB1101 was maintained at this later time.
Our data are in agreement with previous work showing that TNF-α pretreatment induces protective effects against focal ischemia (Nawashiro et al., 1997) and that augmented expression of TNF-α mRNA is found after preconditioning (Wang et al., 2000), both findings suggesting a role of TNF-α in the development of brain tolerance. Other authors have also shown the protective role of TNF-α in IPC (Ginis et al., 2002; Liu et al., 2000).
Strong evidence points to NF-κ as the mediator of the protective actions of TNF-α (Mattson et al., 2000). We have demonstrated that the neuroprotective effect of TACE-induced TNF-α release in a setting of mild neuronal damage using cortical cultures exposed to glutamate, which may correlate with IPC, is mediated via activation of NF-κ (Hurtado et al., 2002). Furthermore, other authors have also described NF-κ activation in
TNF-α induction of IT (Ginis et al., 2002). Nuclear factor-κ would increase the expression of antiapoptotic proteins such as Bcl-2 or decrease the activation of proapoptotic caspase-3 since the presence of both processes have been demonstrated after IPC (Qi et al. 2001; Park et al. 2002). In addition, we cannot discard the possibility that other actions, such as a modification of blood flow change, may contribute to the neuroprotective actions of the TACE–TNF-α pathway in this setting.
In summary, this is the first report showing the upregulation of TACE protein after IPC, its major role in TNF-α shedding in this setting, and its neuroprotective role in IT. We have recently reported an association between IT and elevation of cerebrospinal fluid TNF-α concentrations in humans with acute stroke (Castillo et al., 2002). Thus, elucidation of mechanisms that regulate the acquisition of brain tolerance could guide efforts to develop effective measures or safe pharmacological preconditioning agents to protect the brain or reduce ischemic injury.