
Abstract
A short ischemic event (ischemic preconditioning (IPC)) can result in subsequent resistance to severe ischemic injury (ischemic tolerance (IT)). The expression and neuroprotective role of tumor necrosis factor (TNF-α) have been described in models of IPC and we have showed the participation of its processing enzyme, the TNF-α convertase enzyme (TACE) in this process. We have now decided to explore the expression and localization of TNF receptors (TNFR) as well as other signalling mechanisms involved in IT. A period of 10 mins of temporary middle cerebral artery occlusion (tMCAO) was used for focal IPC. To evaluate the ability of IPC to produce IT, permanent MCAO was performed 48 hours after IPC. Ischemic preconditioning produced a reduction in infarct volume, as we showed previously. Ischemic preconditioning caused upregulation of neuronal TNFR1 that was reduced by the selective TACE inhibitor BB1101. Intracerebral administration of TNFR1 antisense oligodeoxynucleotide, which caused a reduction in TNFR1 expression, inhibited the IPC-induced protective effect, showing that TNFR1 upregulation is implicated in IT. Moreover, treatment with BB1101, TNFR1 antisense and lactacystin—a specific proteasome inhibitor—blocked IPC-induced NF-κB. Immunohistochemical studies showed the expression of TACE and TNFR1 in neurons. In summary, these data show that IPC produces neuronal upregulation of TACE and TNFR1, and that the pathway TACE/TNF-α/TNFR1/NF-κB is involved in IT.
Introduction
A short ischemic event (ischemic preconditioning (IPC)) can result in a subsequent resistance to severe ischemic injury (ischemic tolerance (IT); Kitagawa et al, 1990). This phenomenon has been described in several organs, especially the heart and brain. Ischemic tolerance has also been shown in human clinical practice; indeed, less severe strokes have been described in patients with prior ipsilateral transient ischemic attacks (TIA) within a short period of time (Weih et al, 1999; Moncayo et al, 2000).
Several mechanisms of induction and maintenance of tolerance in the brain have been described (for a review, see Dirnagl et al, 2003), but this phenomenon is not fully elucidated. The activation of transcription factors such as nuclear factor-κB (NF-κB) has been implicated by several authors in the development of IT (Blondeau et al, 2001; Ginis et al, 2002). This factor plays a pivotal role in neuronal survival and is activated by various signals like proinflammatory cytokines, neurotrophic factors and neurotransmitters (for a review, see Mattson et al, 2000). One of these proinflammatory cytokines is tumor necrosis factor-α (TNF-α), which is expressed or released after brain ischemia (Liu et al, 1994). Tumor necrosis factor-α exerts its effects mainly on binding to different receptors, TNF receptor 1 (TNFR1; p55) and TNFR2 (p75), causing activation of complex signalling cascades that mediate different effects (for a review, see Baud and Karin, 2001). Although TNF-α contributes to ischemic brain damage, there are also multiple evidences supporting a neuroprotective role for this cytokine (for a review, see Shohami et al, 1999). Indeed, the expression of TNF-α and its neuroprotective role in in vivo and in vitro models of IPC have been described (Wang et al, 2000; Liu et al, 2000) and we have shown that high plasma levels of TNF-α are associated with human cerebrovascular IT (Castillo et al, 2003). In addition, although the upregulation of both TNF-α receptors after brain ischemia has been shown (Bruce et al, 1996; Botchkina et al, 1997), their role in IT is not known.
Tumor necrosis factor-α is shed in its soluble form by a disintegrin and metalloproteinase (ADAM) called TNF-α convertase (TACE/ADAM17). Some roles of TACE in CNS have been shown (for a review, see Moro et al, 2003). In this context, we have shown, using in vitro models, that TACE is upregulated after ischemic brain damage (Hurtado et al, 2001, 2002) and that the upregulation of TACE also occurs after in vivo IPC, where it is involved in brain tolerance (Cárdenas et al, 2002).
Therefore, we have now decided to investigate further mechanisms involved in the signalling triggered by the TACE/TNF-α pathway by using a rat model of transient middle cerebral artery occlusion (tMCAO) as IPC.
Methods
Animals
Adult male Fischer rats (240 to 260 g) were housed and cared for in accordance with the Committee of Animal Care at the Universidad Complutense of Madrid according to EU rules (DC86/609/CEE). Animals were allowed free access to food and water before and after surgery.
Focal Ischemic Preconditioning
Focal IPC was induced by a period of 10 mins of tMCAO, as described previously (Cárdenas et al, 2002). Cerebral blood flow was monitored by using laser Doppler flowmetry (Periflux System PF5010 Perimed, Stockholm, Sweden) over the parietal cortex before and during the 10 mins of tMCAO. Occlusion of the MCA caused a rapid and persistent decrease in laser Doppler blood flow (to 50% to 55% of pre-MCAO levels). A period of 10 mins of tMCAO was used because this short ischemic period results in no tissue injury but protects the tissue from later ischemia (Barone et al, 1998). Rats in which the MCA was exposed but not lifted served as sham-operated animals.
Permanent Focal Ischemia
To evaluate the ability of IPC (tMCAO) to produce IT, permanent MCAO (pMCAO) was performed 48 hours after sham or tMCAO surgery. We have used the procedure described above, but permanent occlusion was made in both CCA and MCA, as described previously (Brint et al, 1988; De Cristóbal et al 2001).
For permanent CCA occlusion a silk ligature was used, whereas MCA was occluded by applying an electrocoagulator tip (Select-Sutter Medizintechnik, Freiburg, Germany) just above the vessel on the wire hook; after transferring heat through the wire, the MCA was cauterized and severed.
Intracerebroventricular Injections
To block TNFR1 expression, intracerebral administration of TNFR1 antisense oligodeoxynucleotide (ODN) was made. The TNFR1 antisense-ODN sequence 5′-ACACGGTGTTCTGTTTCTCC-3′ was directed against a unique sequence of rat TNFR1. The mismatch ODN sequence, 5′-AC
The specific proteasome inhibitor lactacystin (100 μmol/L) was intracerebroventricularly administered in a volume of 4 μL at 2 μL/mins, 3 doses per day (0800, 1600 and 2400; Fornai et al, 2003). For infarct area determination, seven injections of lactacystin were administered from immediately after IPC (0800) until pMCAO was performed (0800, 48 hours after IPC), and infarct volume was measured 24 hours afterwards, as described below. For determination of protein expression levels (TNFR1 and p65), 4 injections of lactacystin were administered from immediately after IPC until 24 hours after, the time in which rats were killed and brain samples collected.
To neutralize TNF-α produced by TACE after IPC, intracerebroventricular injections of anti-TNF-α were made at 12, 1430, 17, 1930 and 22 hours after IPC. Each injection consisted of 5 μL of anti-TNF-α (10 μg≈60 pmol; Cárdenas et al, 2002).
Briefly, under anesthesia with 2% halothane in a mixture of 70% nitrogen/30% oxygen, an intracerebroventricular 26-gauge cannula (Plastics One, Roanoke, VA, USA) was inserted into the left lateral ventricle by using sterotaxic apparatus (Davis Kopf Instruments, Tujunga, CA, USA). The coordinates were as follows: anteroposterior, −0.8 mm; lateral, 1.0 mm; dorsoventral, 3.6 mm from the bregma, with the incisor bar placed at 3.3 mm below the interaural plane. Cannulas were anchored to the skull by stainless steel screws and dental cement. After a 5-day recovery period, the animals were anesthetized with halothane and underwent IPC, as described previously. Repeated injections were performed in conscious rats, lightly restrained, using a 33-gauge injection cannula, 1 mm longer than the guide cannula.
Experimental Groups
Several groups were used for determinations of TACE and TNFR expression and NF-κB translocation: (a) sham-operated rats, used as control group (SHAM); (b) tMCAO, used as IPC group (IPC); (c) tMCAO + BB-1101 (a selective TACE inhibitor) (IPC+BB); (d) tMCAO+TNFR1-antisense ODN (IPC+antisense); (e) tMCAO+mismatch ODN (IPC+mismatch); (f) tMCAO+lactacystin (a specific proteasome inhibitor; IPC+lactacystin) and (g) tMCAO+anti-TNF-α (IPC+anti-TNF). For protein expression levels, at least six animals were used in each group. Similarly, six animals per group were used for NF-κB translocation studies. BB-1101 was administered subcutaneously at 10 mg/kg/day after tMCAO for 2 days. BB-1101, a succinate-base hydroxamic acid compound, was used due to its ability to distinguish between other proteases that may cleave pro-TNF-α and TACE, because it is more potent against TACE (IC50 values around 0.05 to 0.1 μmol/L) than against others such as ADAM10 or α-secretase (IC50 values ranging from 3 up to >20 μmol/L) (Parvathy et al, 1998; Hooper et al, 2000). Indeed, BB1101 is a preferred inhibitor of metalloproteinases with sheddase activity (Ancuta et al, 1997; Middelhoven et al, 1997; Fiorucci et al, 1998; Lammich et al, 1999). The dose of BB-1101 was chosen according to our previous data showing an effective inhibition of TACE in brain (Cárdenas et al, 2002). The doses and routes of administration of the rest of the drugs were described above.
In addition, to evaluate the ability of IPC to produce IT, pMCAO was performed 48 hours after sham or tMCAO surgery and the infarct area was measured 24 hours after pMCAO. Therefore, eight additional groups were established: (a) SHAM+pMCAO (n=8), (b) IPC+pMCAO (n=8), (c) IPC+BB+pMCAO (n=8), (d) IPC+anti-TNF+pMCAO (n=6), (e) IPC+TNFR1-antisense ODN+pMCAO (n=6), (f) IPC+mismatch ODN+pMCAO (n=6), (g) SHAM+lactacystin+pMCAO (n=6), and (h) IPC+lactacystin+pMCAO (n=6). The microbial product lactacystin is a highly specific inhibitor of the 20 S proteasome (for a review, see Fenteany and Schreiber, 1998), which therefore inhibits NF-κB activation by preventing IκB degradation and subsequent NF-κB nuclear translocation (Grisham et al, 1999).
Finally, additional experiments were performed to analyze immunohistochemical localization of TACE and TNFR1. In this case, each group included three animals, from which several sections were obtained. After processing, each section was examined at least twice.
Infarct Area Determination
The brains were removed 24 hours after pMCAO, and series of 2 mm coronal brain slices were obtained (Brain Matrix, WPI, UK) and stained in 1% TTC (2,3,5-triphenyl-tetrazolium chloride, Merck) in 0.1 mol/L phosphate buffer, and infarct size was determined as described (Mackensen et al, 2001). Infarct volumes were measured by sampling stained sections with a digital camera (Nikon Coolpix 990, Nikon Corporation, Tokyo, Japan), and the image of each section was analyzed by an image analyzer (Scion Image for Windows 2000, Scion Corporation, Frederick, MD, USA). The digitalized image was displayed on a video monitor. With the observer masked to the experimental conditions, the contralateral hemisphere perimeter was overlapped on the ipsilateral hemisphere to exclude edema, and infarct borders in cortex, subcortex, and striatum were delineated using an operator-controlled cursor. The area of infarct, which was not stained, was determined by counting pixels contained within the outlined regions of interest and expressed in square millimeters. Infarct volumes (in mm3) were integrated from the infarct areas over the extent of the infarct calculated as an orthogonal projection.
Brain Samples
For determination of protein expression levels, rats were killed and brain samples were collected 24 and 48 hours after IPC. Brain areas corresponding to the infarct and surrounding area of the cortex were collected. For preparation of nuclear extracts, brains were collected 24 and 48 hours after IPC.
Protein Extraction
Protein extraction for Western blot analyses was performed as described previously (Cárdenas et al, 2002). Briefly, brain cortex from the ipsilateral hemisphere was removed and homogenized by sonication for 10 secs at 4°C in 4 volumes of homogenization buffer containing 320 mmol/L sucrose, 1 mmol/L
Preparation of Nuclear Extracts
Nuclear extracts were prepared as described previously (Cárdenas et al, 2000). Tissues (brain cortex) were homogenized with 300 μl of buffer A (10 mmol/L HEPES, pH 7.9; 1 mmol/L EDTA, 1 mmol/L EGTA, 10 mmol/L KCI, 1 mmol/L DTT, 0.5 mmol/L PMSF, 0.1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL TLCK, 5 mmol/L NaF, 1 mmol/L NaVO4, 0.5 mol/L sucrose, and 10 mmol/L Na2MoO4). After 15 mins, Nonidet P-40 was added to reach a 0.5% concentration. The tubes were gently vortexed for 15 secs and nuclei were collected by centrifugation at 8000 g for 5 mins. The pellets were resuspended in 100 μL of buffer A supplemented with 20% glycerol and 0.4 mol/L KCI, and gently shaken for 30 mins at 4°C. Nuclear protein extracts were obtained by centrifugation at 13,000 g for 5 mins, and aliquots of the supernatant were stored at −80°C. All steps of the fractionation were performed at 4°C.
Western Blot Analysis
Samples containing 20 μg of protein were loaded and the proteins size-separated in 12% SDS-polyacrylamide gel electrophoresis (90 mA). Proteins were blotted onto a PVDF membrane (Hybond™-P, Amersham Biosciences Europe GmbH, Freiburg, Germany) and incubated with specific primary antibodies against p65 (Santa Cruz; 1:1000), TNFR1 (Santa Cruz; 1:500) and TNFR2 (Santa Cruz; 1:250). Proteins recognized by the antibody were revealed by ECL™-kit following manufacturer's instructions (Amersham Biosciences Europe GmbH, Freiburg, Germany). The results reflect data from six to eight different blots from different animals, and are expressed as relative absorbance units obtained by densitometric analysis after normalization for β-actin and Sp1 levels, which were used as loading controls for TNFR1 and p65 proteins expression, respectively.
Double Immunofluorescence
Rats were anesthetized 24 hours after IPC with sodium pentobarbital and perfused through the left ventricle with 200 ml of 0.2 mol/L sodium phosphate buffer (PB) as a vascular rinse followed by 300 ml of fixative solution containing 4% paraformaldehyde in 0.1 mol/L phosphate buffer (PB), pH 7.4. The brains were removed, postfixed for 4 hours in the same solution of paraformaldehyde at room temperature, and then cryoprotected by immersion overnight at 4°C in 0.1 mol/L PB containing 30% sucrose. Brains were frozen and serial 8-μm-thick frontal sections were cut with a Leitz sledge microtome. Sections were acetone-permeabilized for 10 mins at 4°C and washed in PBS containing 3% bovine serum albumin and 0.1% Triton X-100 for 30 mins; then sections were incubated in the two primary antibodies for each double staining 1 hour at room temperature: anti-TACE (Prosci; 1:10 dilution) or anti-TNFR1 (Santa Cruz; 1:10 dilution) and a mouse antineuron nuclei (NeuN) antibody (Chemicon; 1:10 dilution) to identify neurons, anti-TACE or anti-TNFR1 and a mouse anti-GFAP antibody (Chemicon; 1:20 dilution) to identify astrocytes or anti-TACE or anti-TNFR1 and a fluorescein-labelled tomato lectin (from Lycopersicon esculentum; 1:150 dilution) to characterize microglia, macrophages and endothelium (Acarin et al, 1994). After washing in PBS, the sections were incubated in each respective secondary antibody for 1 hour. For TACE and TNFR1, Cy 3™-labelled goat anti-rabbit IgG was used (Amersham; 1:10 dilution; red colour with fluorescence maximum at 670 nm); for NeuN and GFAP, the sections were incubated with Cy 2™-labelled goat anti-mouse IgG (Amersham; 1:10 dilution; green colour with fluorescence maximum at 506 nm). Visualization was performed under a fluorescence microscope (Nikon Eclipse TE300, Nikon Corporation, Tokyo, Japan) using Plan Fluor × 20/0.45 or × 40/0.6 objectives, and phase optics, a B2A Nikon filter for FITC, and Cy2 fluorescence or a G2A Nikon filter for Cy3 fluorescence. The areas selected corresponded to regions in the proximity to the transiently occluded vessel, from both cortex and striatum. Each experiment was performed in duplicate and repeated three times. Image acquisition was peformed with a laser-scanning confocal imaging system (MRC1024, BioRad, Hempstead, UK).
Chemicals and Statistical Analyses
BB-1101(2S-allyl-N1-hydroxy-3R-isobutyl-N4-(1S-methylcarbamoyl-2-phenylethyl)-succinamide) was kindly supplied by British Biotech (Redford et al, 1997; Kupatt et al, 1999), TNFR1-antisense or mismatch ODNs were purchased from Sigma-Genosys (UK) and other chemicals were obtained from Sigma (Spain), or as indicated in the text. Results are expressed as mean±s.d. of the indicated number of experiments. Student's t-test or ANOVA followed by Dunnett's test (multiple comparison versus a control) were used. A value of P<0.05 was considered statistically significant.
Results
Effect of Ischemic Preconditioning on Tumor Necrosis Factor-α receptor expression
Western blot analysis revealed the presence of a TNFR1-immunopositive band (Figure 1) in brains from sham-operated animals, which was not different from unhandled animals (data not shown). IPC induced an increase in TNFR1 protein levels detected by immunoblotting at 24 hours (n=6; ∗P<0.05; Figure 1) which was maximal 48 hours after IPC (data not shown). Treatment with either BB-1101 or anti-TNF-α reduced IPC-induced upregulation of TNFR1 (n=6; #P<0.05; Figure 1). TNFR2-immunopositive band was not detected in any group studied (data not shown).
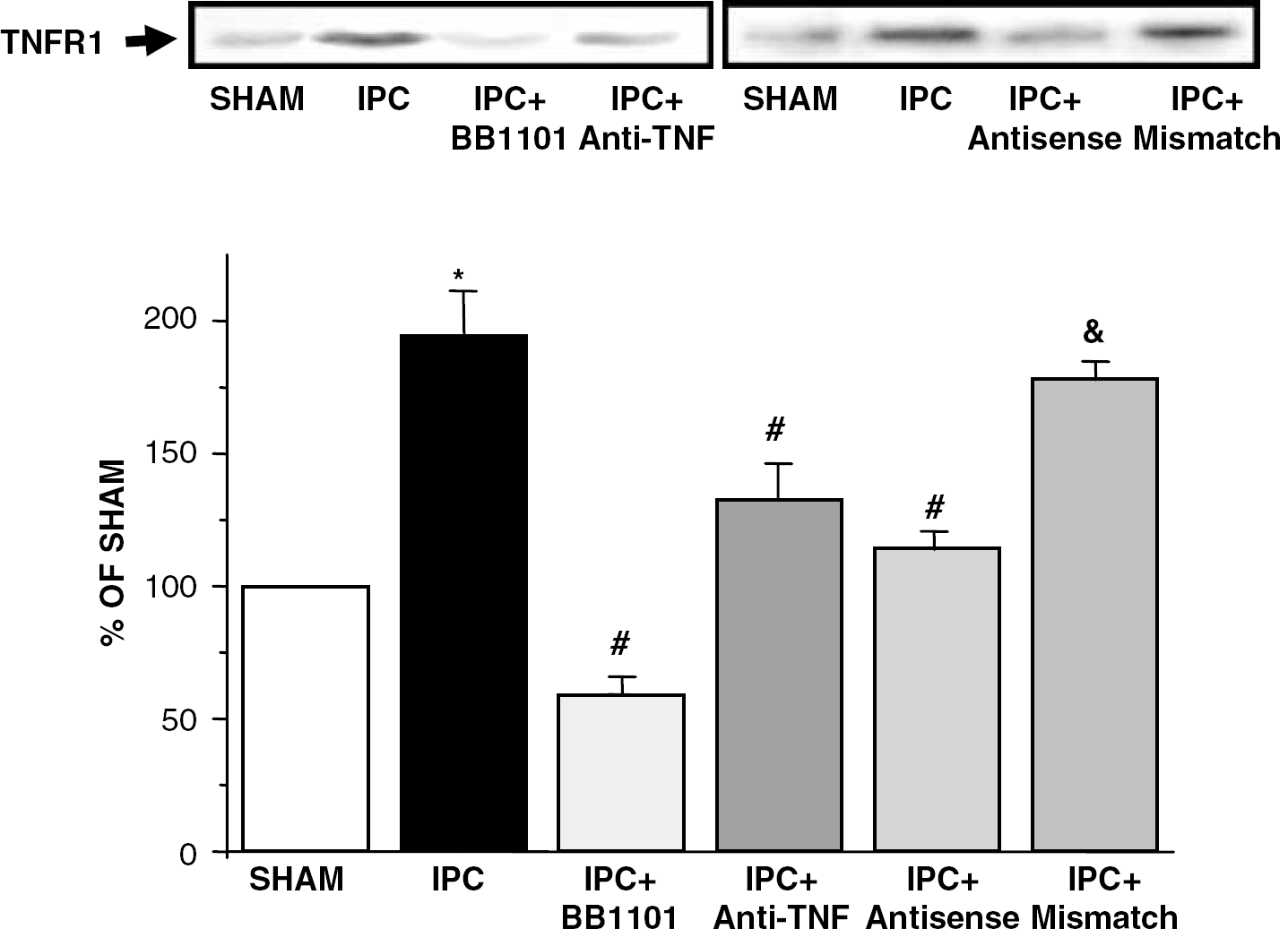
Western blot analysis of TNFR1 protein in brains from rats after IPC, sham-operation (SHAM), IPC treated with BB-1101 (IPC+BB1101), IPC treated with anti-TNF-α (IPC+anti-TNF), IPC treated with TNFR1 antisense-ODN (IPC+antisense) and IPC treated with TNFR1 mismatch-ODN (IPC+mismatch). The lower panel shows the laser densitometric analysis of bands after normalization with β-actin as loading control. Data are mean±s.d., n=6; ∗P<0.05 versus SHAM; #P<0.05 versus IPC; &P<0.05 versus IPC+antisense.
Effect of Ischemic Preconditioning on TACE and TNFR1 Immunoreactivities
TACE and TNFR1 localization were examined by double immunofluorescence staining. In sham-operated animals, double immunostaining showed very little or no TACE immunoreactivity in NeuN-positive cells (neurons, Figure 2A), GFAP-positive cells (astrocytes, Figure 2C) and Lycopersicon esculentum (tomato) lectin-positive cells (microglial cells and endothelium) (Figure 2E). Ischemic preconditioning produced the upregulation of TACE immunoreactivity in the cortical area surrounding transient occlusion at 24 hours, which appeared located mainly in neurons (NeuN-positive cells; Figure 2B). A few astrocytes (Figure 2D) and of microglial (L. esculentum lectin-positive cells not associated to vessels; Figure 2F) and endothelial cells (Figure 2F) showed TACE immunoreactivity after IPC. Ischemic preconditioning also caused an increase in the number of GFAP-stained cells, very likely due to astrogliosis in this area (Figure 2D).
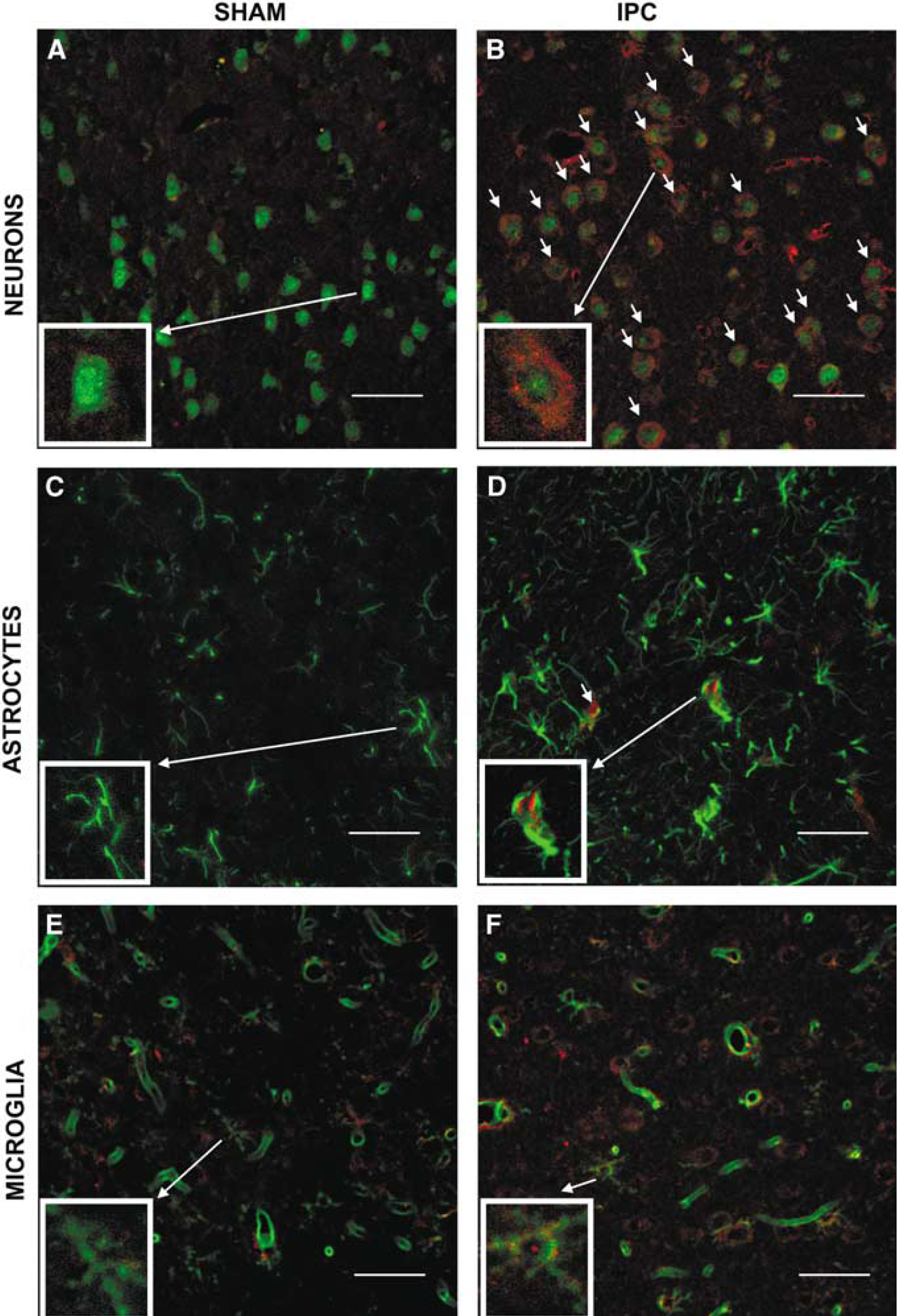
Cellular localization of TACE. Double immunofluorescence staining of brain sections from sham-operated (SHAM;
TNFR1 immunoreactivity was not detectable in preparations from sham-operated animals (Figures 3A, E and G). Ischemic preconditioning produced an upregulation of TNFR1 immunoreactivity, which was located in neuronal cells as shown by double immunofluorescence (Figure 3B). TNFR1 immunoreactivity was not detected either in astrocytes (Figure 3F) or microglial cells (Figure 3H) after IPC. Treatment with BB-1101 (Figure 3C) or the TNFR1 antisense ODN (Figure 3D) reduced IPC-induced upregulation of TNFR1 neuronal immunoreactivity.
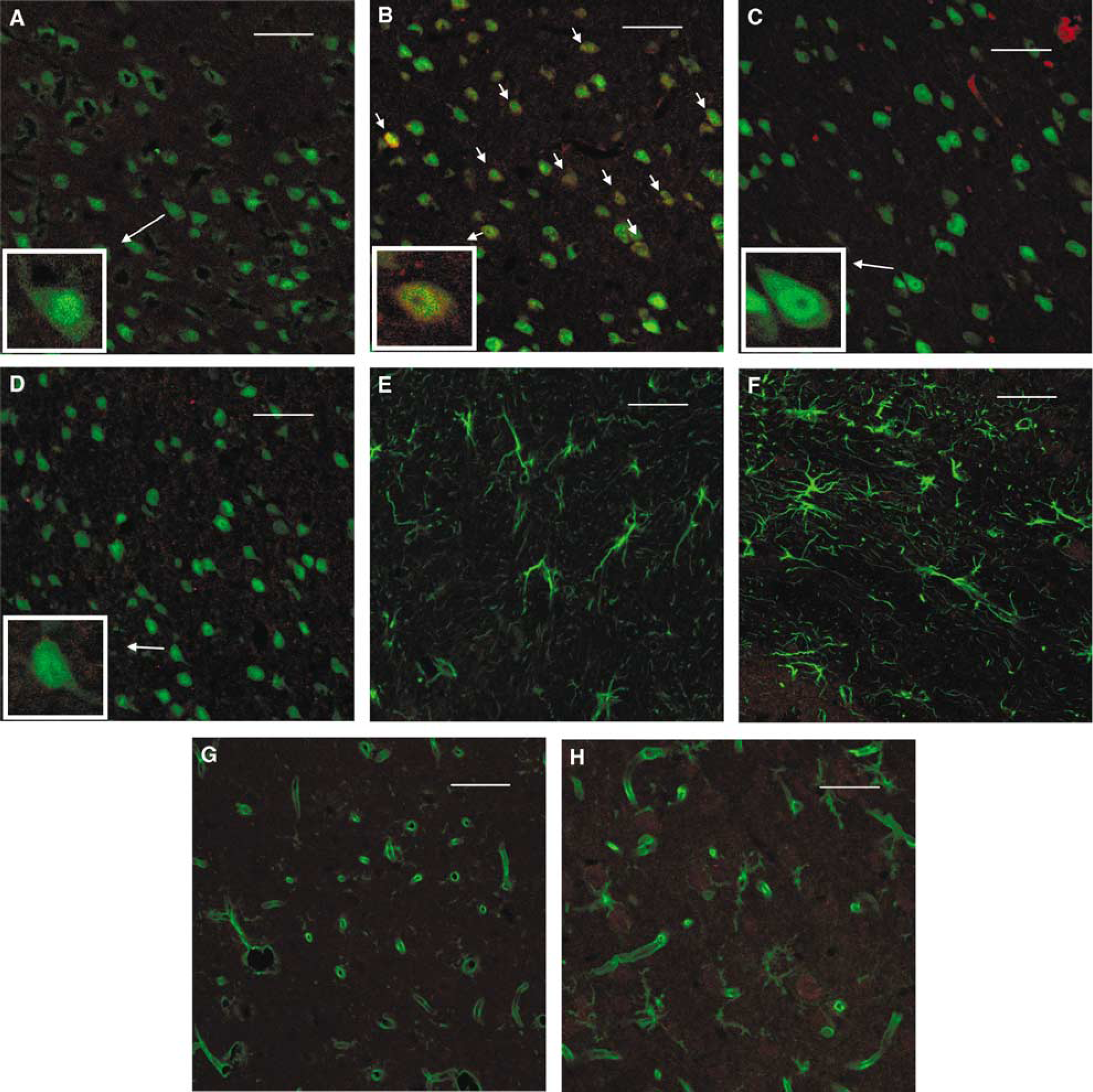
Cellular localization of TNFR1. Double immunofluorescence staining of TNFR1 (red) and the cellular markers (green) NeuN (neurons;
Effect of Ischemic Preconditioning on Nuclear Factor-κB activation
Ischemic preconditioning caused the nuclear translocation of the NF-κB monomer p65 in rat brain when measured 24 hours (n=6; ∗P<0.05; Figure 4) and 48 hours (data not shown) after temporary occlusion. Treatment with BB-1101, TNFR1 antisense-ODN blocked IPC-induced NF-κB activation (n=6; #P<0.05; Figure 4). TNFR1 mismatch-ODN did not modify nuclear translocation of p65 induced by IPC (Figure 4). In addition, lactacystin also blocked IPC-induced NF-κB activation (n=6; #P<0.05; Figure 4). Levels of p65 in sham animals at the times studied were not different from unhandled animals (data not shown).
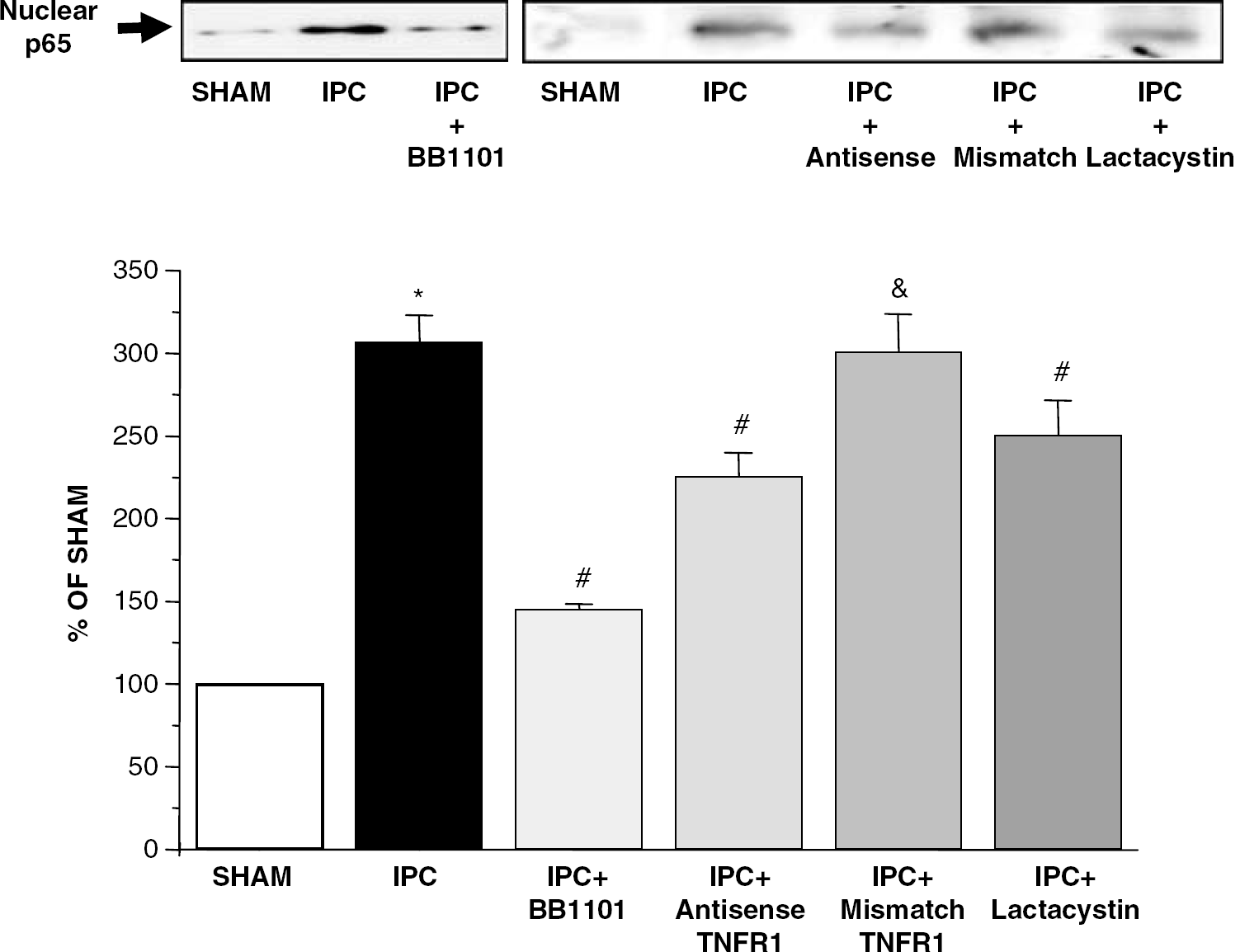
Nuclear translocation of NF-κB measured by Western blot analysis of p65 protein in nuclear extracts from brain rats after IPC, sham operation (SHAM), IPC treated with BB-1101 (IPC+BB1101), IPC treated with TNFR1 antisense-ODN (IPC+antisense), IPC treated with TNFR1 mismatch-ODN (IPC+mismatch) and IPC treated with lactacystin (IPC+lactacystin). The lower panel shows the laser densitometric analysis of bands after normalization with Sp1 as loading control. Data are mean±s.d., n=6; ∗P<0.05 versus SHAM; #P<0.05 versus IPC; &P<0.05 versus IPC+antisense.
Effect of Inhibition of TACE/Tumor Necrosis Factor-α/TNFR1 Pathway on Infarct Volume
Ischemic preconditioning produced a reduction in infarct volume measured at 24 hours after pMCAO when compared with the sham-operated animals group (SHAM+pMCAO: 243±27 mm3, 100%, IPC+pMCAO: 93±4 mm3, 72±2% of SHAM+pMCAO, n=8; P<0.05), as we have demonstrated previously (Cárdenas et al, 2002). We have also confirmed our previous data (Cárdenas et al, 2002) showing that treatment with BB-1101 as well as i.c.v. administration of anti-TNF-α inhibit IPC-induced decrease in infarct volume (IPC+BB+pMCAO: 219±10 mm3 and IPC+anti-TNF+pMCAO, 254±20 mm3; n=6 to 8; P<0.05 versus IPC+pMCAO).
The intracerebral administration of TNFR1 antisense-ODN but not TNFR1 mismatch-ODN also inhibited IPC-induced decrease in infarct volume (n=6; #P<0.05; Figure 5A). The efficiency of the ODN strategy was demonstrated by the fact that treatment with TNFR1 antisense-ODN, but not TNFR1 mismatch-ODN, caused a reduction in upregulation of TNFR1 induced by IPC, measured 24 hours after IPC (n=6; #P<0.05; Figure 1).
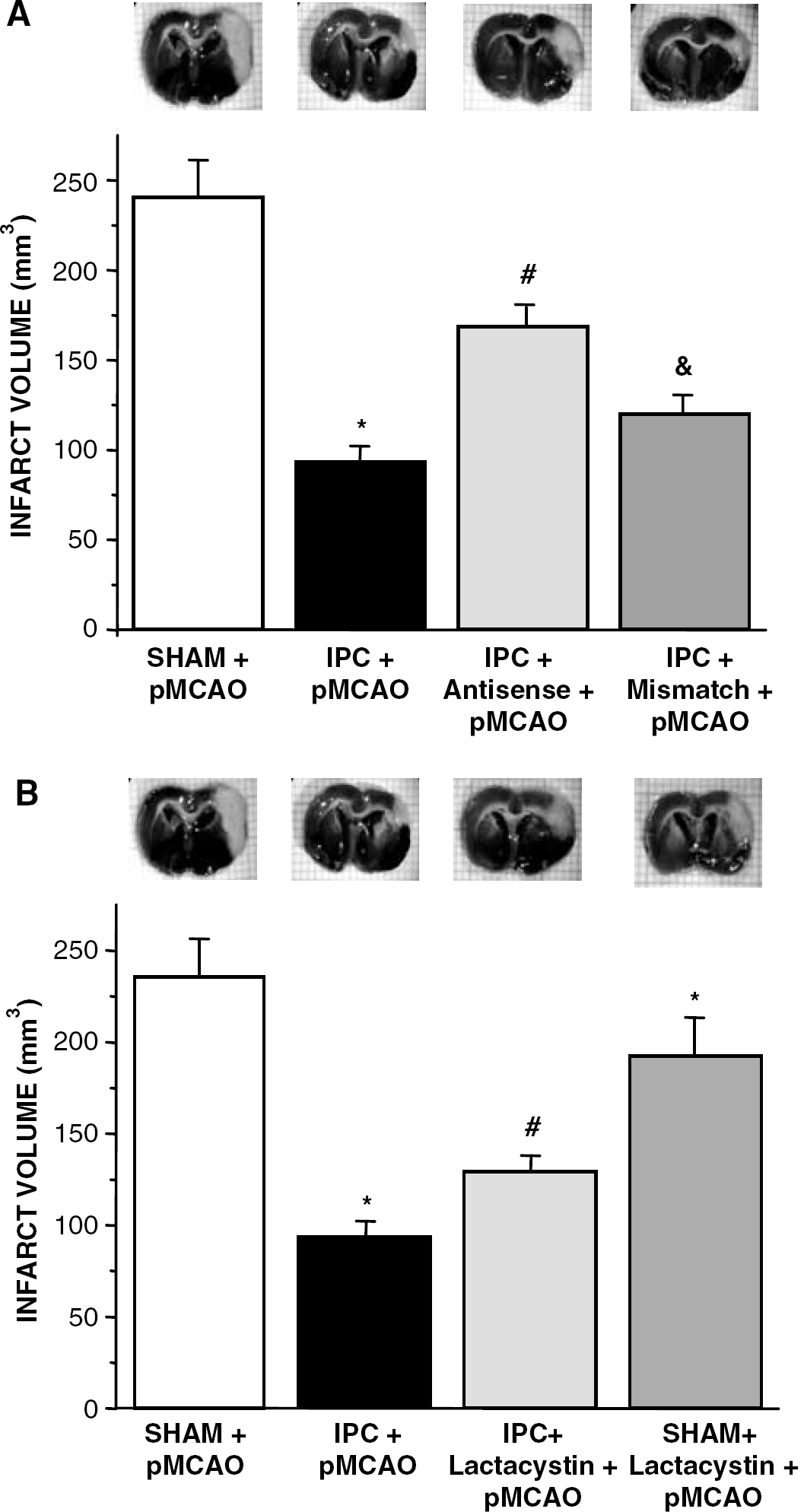
Infarct volume (mm3) measured 24 hours after permanent middle cerebral artery occlusion (pMCAO) in sham-operated (SHAM+pMCAO) or preconditioned rats (IPC+pMCAO) and in preconditioned rats treated with: TNFR1 antisense-ODN (IPC+antisense+pMCAO;
In addition, lactacystin also inhibited IPC-induced decrease in infarct volume (n=6; #P<0.05; Figure 5B). Lactacystin, by itself, produced a reduction in infarct volume when compared with the sham+pMCAO group (n=6; ∗P<0.05; Figure 5B). BB-1101 and TNFR1 antisense ODN did not affect infarct volume when compared with the sham+pMCAO group (data not shown).
Discussion
We have recently provided the first demonstration of the presence of TACE protein in CNS, of its upregulated expression after IPC and of its neuroprotective role, via TNF-α overproduction, in IT afforded by IPC (Hurtado et al, 2001, 2002; Cárdenas et al, 2002). In an attempt to elucidate the pathways involved, we have now shown that TNFR1 is upregulated after IPC, that there is a neuronal expression of TACE and TNFR1 after IPC and that TACE activity is responsible for TNFR1 upregulation and p65 nuclear translocation. These data indicate that TNFR1 upregulation mediates IT via NF-κB activation.
First, our data show low levels of TACE immunoreactivity in neurons, astrocytes, and microglial cells in control animals. Interestingly, IPC causes the upregulation of TACE, the localization of which is found mainly in neurons, most of which are TACE-immunoreactive after IPC. Some TACE immunostaining was also found in some microglial and astrocytes, although to a lesser extent. Of note, whereas the staining in neurons is clearly located in the membrane, this is not the case in astrocytes. In this context, using an in vitro model of IPC (Romera et al, 2004), we have recently demonstrated that IPC induces a different regulation of TACE in each cell type, with an increase in mature TACE and a decrease in its precursor, consistent with a regulation by processing, in neurons, and an increase in the expression of the precursor form, suggesting a regulation at the transcriptional level, in astrocytes. This is consistent with our new data, because the precursor form of TACE has been described to be located intracellularly, whereas the mature form is predominantly in the plasma membrane (Schlöndorff et al, 2000).
Furthermore, this is the first report showing an increased expression of TNFR1 expression after IPC. Analysis of TNFR1 protein by Western blot demonstrates its presence in brains from both control and IPC-exposed animals. More interestingly, upregulation of TNFR1 expression but not TNFR2, was detectable 24 hours after IPC. Previous reports have shown that TNFR1 immunoreactivity appears 4 to 6 hours after permanent focal ischemia (Botchkina et al, 1997) or 2 to 8 hours after temporary global ischemia (Sairanen et al, 2001). At the conditions used in this study, we have failed to detect TNFR2 protein even 48 hours after IPC (data not shown). In this context, it has been described that immunoreactivity of TNFR2 after ischemia is more delayed than that of TNFR1 and it is detected 24 hours after ischemia (Botchkina et al, 1997). In addition, our present finding showing that TNFR1 upregulation is reduced by the hydroxamate-based compound BB1101 and by anti-TNF-α antibody indicates that TACE activity might be responsible for the increased expression of its receptor after IPC, although other indirect mechanisms are not discarded. Hydroxamate-based compounds are inhibitors of TACE with a high selectivity, a fact that has been used to distinguish between TACE effect and that of other proteases (Parvathy et al, 1998; Hooper et al, 2000). We have previously demonstrated that upregulation of TACE after IPC causes the release of TNF-α, which is involved in the tolerance found in this model (Cárdenas et al, 2002). Our data suggest that TNF-α is responsible for an increased expression of its receptor in this setting.
We have also explored the cellular localization of TNFR1 after IPC. TNFR1 immunoreactivity in sham brains was not detected; however, after IPC, there was an upregulation of TNFR1 immunoreactivity, which was located in neurons. There is no previous information about cellular localization of TNFR1 immunoreactivity after IPC; however, it has been described that, after ischemia, increased TNFR1 immunoreactivity is located in neurons, activated macrophages and endothelial cells (Sairanen et al, 2001). Similar results have recently been described in human ischemic stroke (Dziewulska and Mossakowski, 2003). Because after IPC, increased TACE immunoreactivity appears mainly in neurons but also in some astrocytes and microglial cells, and increased TNF-α immunoreactivity after IPC is also localized in those cells (for a review, see Hallenbeck, 2002), we can suggest that both an autocrine and a paracrine mechanism may take place, having as the final target the neuronal expression of TNFR1.
It is well known that TNF-α, which is expressed or released after ischemia (Liu et al, 1994), contributes to the damaging effects of this condition (for a review, see Shohami et al, 1999). However, additional evidences have demonstrated a direct, neuroprotective role of TNF-α (for a review, see Shohami et al, 1999). Moreover, it has been shown that preconditioning produces an upregulation of TNF-α mRNA (Wang et al, 2000) and that TNF-α plays a protective role in this setting (Liu et al, 2000; Ginis et al, 2002); in this context, we have demonstrated that TACE-induced TNF-α release participates in the development of brain tolerance because treatment with BB-1101 and anti-TNF-α antibody inhibits the reduction of the infarct volume caused by IPC (Cárdenas et al, 2002).
Strong evidences pointed to NF-κB as the mediator of the protective actions of TNF-α (for a review, see Mattson et al, 2000), but until now it had not been well established. We have now demonstrated that the neuroprotective effect of TACE is parallel to the activation of NF-κB after the binding of TNF-α to its TNF receptor 1. Several data support this affirmation: first, TACE inhibition produced by BB-1101 prevents not only TNF-α release, neuroprotection and TNFR1 upregulation, but also NF-κB nuclear translocation. Second, treatment with TNFR1 antisense-ODN, which is able to reduce upregulation of TNFR1 induced by IPC, inhibits the nuclear translocation of p65 and the protective effect afforded by IPC, although, because the inhibition was partial in both cases, we cannot discard the fact that other mechanisms might also be implicated. The treatment with TNFR1 mismatch-ODN did not inhibit NF-κB activation and did not modify the infarct volume. Studies demonstrating NF-κB activation via TNFR1 (Adam et al, 1995; Liu et al, 1996) and other works describing NF-κB activation in TNF-α induction of IT (Ginis et al, 2002) are in agreement with our results. Finally, treatment with the specific proteasome inhibitor lactacystin, which not only produces the inhibition of p65 translocation but also partially blocks the protective effect induced by IPC, strongly supports our conclusion. Moreover, these data are in agreement with our previous results suggesting that the neuroprotective effect of TACE-induced TNF-α release using cortical cultures exposed to low glutamate concentration, a setting of mild neuronal damage that may correlate with IPC, was mediated via activation of NF-κB (Hurtado et al, 2002).
Several mechanisms have been postulated to account for the neuroprotective actions of NF-κB, such as inhibition of TNF-induced apoptosis, (Liu et al, 1996) by increasing the expression of antiapoptotic proteins such as Bcl-2, or decreasing the activation of proapoptotic caspase-3, because the presence of both processes has been demonstrated after IPC (Qi et al. 2001; Park et al. 2002). However, we cannot discard the fact that other possible actions may contribute to the neuroprotective actions of the TACE-TNF-α pathway in this setting. As in the case of TNF-α, NF-κB also mediates deleterious actions such as the induction of proinflammatory and inflammatory mediators, as occurs after ischemia (for a review, see Mattson et al, 2000). Indeed, the protective effect of lactacystin by itself is likely to be mediated by the inhibition of these noxious effects after pMCAO. Moreover, this protective effect of lactacystin on the sham+pMCAO group argue against proapoptotic actions of this compound, which may mediate the inhibition this compound causes on the tolerance induced by the IPC manoeuvre in this setting.
In summary, this is the first report showing (1) that TNFR1 protein is upregulated after IPC, (2) cellular localization of TACE and TNFR1 in this setting, and (3) that the pathway TACE/TNF-α/TNFR1/NF-κB is involved in IT (see Figure 6). We have recently reported an association between IT and elevation of TNF-α concentrations in humans with acute stroke (Castillo et al, 2003). Thus, elucidation of mechanisms that regulate the acquisition of brain tolerance could guide efforts to develop effective measures to protect the brain or reduce ischemic injury.
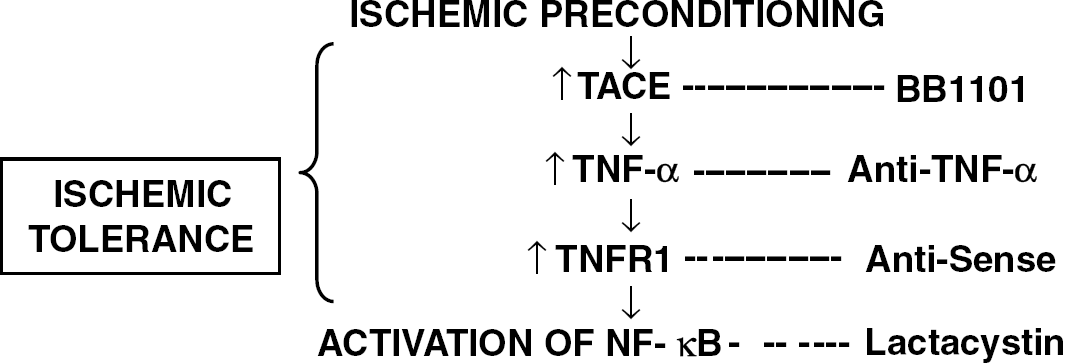
TACE/TNF-α pathway in IT.
Footnotes
Acknowledgements
This work was supported by grants from SAF2002-04487-C02-01 (IL), FIS-PI03/0314 (MAM), PGIDT99 PX120803B (JC) and CAM08.5/0001.1/2003 (PL). JMP, CR and OH are recipients of fellowships funded by Universidad Complutense, FIS and Comunidad de Madrid, respectively.