
Abstract
The role of the tumor necrosis factor (TNF)-α convertase (TACE/ADAM17) in the adult nervous system remains poorly understood. The authors have previously demonstrated that TACE is upregulated in rat forebrain slices exposed to oxygen–glucose deprivation (OGD). They have now used rat mixed cortical cultures exposed to OGD or glutamate to study (1) TACE expression and localization, and (2) the effects of TNF-α release on cell viability. OGD- or glutamate-caused TNF-α release, an effect that was blocked by the TACE inhibitor BB3103 (BB) (0.1–1 μmol/L; control: 1.67 ± 0.59; OGD: 6.59 ± 1.52; glutamate: 3.38 ± 0.66; OGD ± BB0.1: 3.23 ± 0.67; OGD ± BB1: 1.33 ± 0.22 pg/mL, n = 6, P < 0.05). Assay of TACE activity as well as Western blot showed that TACE expression is increased in OGD- or glutamate-exposed cells. In control cultures, TACE immunoreactivity was present in some microglial cells, whereas, after OGD or glutamate, TACE immunostaining appeared in most microglial cells and in some astrocytes. Conversely, BB3103 (0.1 μmol/L) caused apoptosis after glutamate exposure as shown by annexin and Hoechst 33342 staining and caspase-3 activity, an effect mimicked by the proteasome inhibitor MG-132 (caspase activity: glutamate: 5.1 ± 0.1; glutamate + BB: 7.8 ± 0.8; glutamate + MG: 11.9 ± 0.5 pmol · min−1 mg−1 protein, n = 4, P < 0.05), suggesting that translocation of the transcription factor NF-κB mediates TNF-α–induced antiapoptotic effect. Taken together, these data demonstrate that, in rat mixed neuronal–glial cortical cultures exposed to OGD or glutamate, (1) TACE/ADAM17 activity accounts for the majority of TNF-α shedding, (2) an increase in glial TACE expression contributes to the rise in TNF-α, and (3) TNF-α release in this setting inhibits apoptosis via activation of the transcription factor NF-κB.
Keywords
ADAMs are a family of proteins containing a disintegrin and metalloproteinase domain, thus mediating both adhesive interactions and proteolysis. They are therefore expected to possess diverse implications in neurobiology as in neuronal development, but their role in CNS has been scarcely explored (for review, see Yong et al., 2001).
Both physiologic and pathologic roles for the protease activity of tumor necrosis factor α (TNF-α) converting enzyme (TACE/ADAM17) have been indicated (for review, see Killar et al., 1999) and its gene expression has been demonstrated in the adult CNS (Kärkkäinen et al., 2000). The purification and identification of TACE/ADAM17 as a disintegrin and metalloproteinase marked the first instance in which a proteolytically active ADAM was paired with a bona fide substrate (Black et al., 1997; Moss et al., 1997). As regards the CNS, shedding of the pleiotropic cytokine TNF-α could have implications in neuroinflammatory conditions in which an increase in TNF-α levels has been described, such as cerebral ischemia, multiple sclerosis, or trauma (Feuerstein et al., 1998).
In a first approach to elucidate the role of TACE/ADAM17, we recently reported that this enzyme is upregulated in rat forebrain slices exposed to oxygen–glucose deprivation (OGD), a paradigm of cerebral ischemia (Hurtado et al., 2001). Moreover, we found that one of the functions of TNF-α released in this condition was to participate in the expression of the inducible isoform of nitric oxide synthase (iNOS), an enzyme associated with cell damage and inflammation. We have also found that TACE is increased in animal models of psychological stress, where it again participates in TNF-α–mediated iNOS-expression (Madrigal et al., 2002). Although this evidence points to a deleterious effect of the TACE/TNF-α pathway, both neurotoxic and neuroprotective roles of TNF-α have been demonstrated. Indeed, divergent signaling pathways downstream of TNFR1 (p55), the most ubiquitously expressed TNF receptor, lead to either survival or cell death (Natoli et al., 1998). TNF-α may induce apoptosis via TNFR1-death domains and adapter proteins that lead to caspase activation, whereas it can increase cell survival via activation of NF-κB-dependent genes (Askenazi and Dixit, 1998). Indeed, there is much evidence demonstrating a TNF-α–induced neuroprotective role (Cheng et al., 1994; Houzen et al., 1997; Mattson et al., 1995; Shinpo et al., 1999; Sullivan et al., 1999; Tarkowski et al., 1999) and antiapoptotic actions on neural cells (Barger et al., 1995; Mattson et al., 1997; Tamatani et al., 1999; Diem et al., 2001). Interestingly, TNF-α–mediated expression of NF-κB-dependent genes such as iNOS in glial cells may also mediate apoptosis on target neurones (Ogura et al., 1997; Chung et al., 1999; Heneka et al., 1999; Tezel and Wax, 2000; Combs et al., 2001). To further explore TACE/TNF-α pathway in CNS pathology, we have decided to use rat mixed cortical cultures exposed to OGD or glutamate, to investigate (1) the expression and localization of TACE, and (2) some of the effects of TNF-α release on apoptosis after glutamate exposure.
MATERIALS AND METHODS
Primary rat mixed cortical cultures
All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense (following DC 86/609/EU). Primary cultures of cortical cells were performed as described before (Fernández-Tomé and Segal, 1987; Moro et al., 1998): brains were removed from fetal Wistar rats at embryonic day 18, and the cortical area was dissected. Cells were mechanically dissociated in incubation medium consisting of 80% Eagle's minimum essential medium containing 33-mmol/L glucose, 2-mmol/L glutamine, 16 mg/L gentamicin, 10% horse serum and 10% fetal calf serum (growth medium). The dissociated cells were plated at a density of 3 × 10 cells per square centimeter in polylysine-precoated 6-, 12-, or 24-multiwell plates. Plates were kept in a 37°C incubator in a humidified atmosphere containing 95% air/5% CO2. Medium was replaced every 3 to 4 days to fresh growth medium lacking fetal calf serum.
Percentage of neurons in the rat mixed cortical cultures was determined by flow cytometry. Cells were detached by trypsinization [0.025% trypsin, 0.02% EDTA in phosphate-buffered saline (PBS)], washed once in PBS, and then fixed for 30 minutes in a solution containing 4% p-formaldehyde in 0.1 mol/L phosphate buffer, pH 7.4 at room temperature. Then, cells were spun down at 13,000 rpm in a microcentrifuge (Hettich, Germany), and pellet was resuspended in PBS containing 3% bovine serum albumin and 0.2% Triton X-100 for 30 minutes. Cells were washed and incubated 2 hours at room temperature in a monoclonal anti-MAP2 antibody (Chemicon, Temecula, CA, U.S.A.; 1:200 dilution). After washing in PBS, cells were incubated in Cy2-labeled anti-mouse immunoglobulin G (Amersham Pharmacia Biotec, Piscataway, NJ, U.S.A.; 1:300 dilution) for 1 hour. Cells were then analyzed in a FAC-Scan flow cytometer (Becton-Dickinson, Mountain View, CA, U.S.A.) connected to a MacIntosh Quadra II computer system to collect fluorescence from Cy2. For all studies, 10,000 cells were acquired per sample and statistical analysis was done. The mean relative fluorescence intensity of the population under study was 37.59 ± 1.43, and its median relative fluorescence intensity was 11.64 ± 1.25 (n = 4). Data were recorded and analyzed using the Cell Quest software program. Flow cytometer was checked daily with fluorescent beads to detect daily variations, in the measurement. Studies were performed at in vitro days 9 to 10, the time at which the cultures consisted of 60 ± 10% neurons.
Exposure of rat cortical cultures to oxygen–glucose deprivation or to glutamate
Oxygen–glucose deprivation was performed as described before (Goldberg and Choi, 1993) with modifications (De Cristóbal et al., 2002). Culture medium was replaced by a solution containing (in millimoles per liter): NaCl (130), KCl (5.4), CaCl2 (1.8), NaHCO3 (26), MgSO4 (0.8), NaH2PO4 (1.18), and 2% horse serum bubbled with 95% N2/5% CO2 for OGD cells (OGD solution). OGD cells were transferred to an anaerobic chamber (Billups-Rothenberg Inc., Del Mar, CA, U.S.A.) containing a gas mixture of 95% N2/5% CO2 and humidified at 37°C, and maintained at a constant pressure of 0.15 bar. The time of exposure to OGD was 150 minutes, a time at which we have observed that there exists glutamate release but no necrosis is found (De Cristóbal et al., 2002).
In some experiments, either a succinate-based hydroxamic acid compound, BB3103 (0.1–1 μmol/L; British Biotech, Oxford, U.K.), which inhibits metalloproteinases that mediate ectodomain shedding (Ancuta et al., 1997; Middelhoven et al., 1997; Lammich et al., 1999), TNF-α (1–10 pg/mL, Pepro Tech EC Ltd., U.K.) or the N-methyl-
In a different set of experiments, cells were exposed to 10 μmol/L glutamate in control solution lacking Mg2+ for 5 minutes, time after which this solution was replaced by GGPS and cultures returned to the incubator. Simulated reperfusion was performed as already indicated, either in the absence or the presence of BB3103, neutralizing concentrations of anti–TNF-α (2 μg/mL), or the proteasome inhibitor MG-132 (75 μmol/L; carbobenzoxy-
Several apoptotic parameters were evaluated 45 minutes to 3 hours after the treatment without changing the medium unless indicated.
Tumor necrosis factor-α determination
Soluble TNF-α released from cells to the incubation solution was determined by a rat TNF-α immunoassay (Quantikine M, R&D Systems, Minneapolis, MN, U.S.A.) in samples collected at the end of the experiment.
Cleavage of a fluorescein-labeled propeptide containing the cleavage site in protumor necrosis factor-α by control and glutamate-subjected cortical cultures
To study whether the increase in TNF-α levels after glutamate exposure derives from TACE upregulation and/or from an increase in the levels of the substrate pro–TNF-α, the cleavage of saturating concentrations of a 12-residue peptide spanning the Ala -Val site in pro–TNF was studied. TACE activity in control and glutamate-exposed cultured cells was then monitored as the ability to cleave a fluorescein-labeled propeptide fluorescein-SPLAQAVRSSSR-Lys-biotin (propeptide) containing the cleavage site in pro–TNF-α as described (Hurtado et al., 2001) with some modifications. The fluorophoric propeptide (final concentration 0.025 mg/mL, 18 μmol/L) was included in the culture medium of control and glutamate-exposed cortical cultures, during the “reperfusion” period. The unhydrolyzed propeptide and the biotin-linked, nonfluorescent peptide product VRSSSR-Lys-biotin were removed using an avidin resin (avidin immobilized on cross-linked 6% beaded agarose). The fluorescence of the peptide product fluorescein-SPLAQA was measured in a fluorescence microplate reader (Fluoroskan FL Ascent, Labsystems, Helsinki, Finland), with filters of λexc = 485 nm and λem = 510 nm. At the end of the experiment, cells were lysed with perchloric acid, and the amount of protein was calculated. The activity of TACE was calculated from the difference between the fluorescence produced from samples and samples containing 100 μmol/L 1,10-phenanthroline, and expressed as relative fluorescence units per milligram of protein.
Determination of matrix metalloproteinase activity by cleavage of a fluorogenic MMP substrate by control and glutamate-subjected cortical cultures
To check matrix metalloproteinase (MMP) activity in the model used in the present study, both control and glutamate-exposed cultures were incubated with a fluorogenic MMP substrate (Calbiochem, Schwalbach, Germany) in equimolar concentrations (0.020 mg/mL, 18 μmol/L) and the same conditions as described earlier herein for the TACE substrate. Activity was also calculated as already mentioned.
Determination of TACE/ADAM 17 and ADAM10 by Western blot
Cells were homogenized by sonication at 4°C in 5 volumes of buffer containing 320-mmol/L sucrose, 1-mmol/L
Double fluorescence immunostaining for glial fibrillary acidic protein (GFAP)/TACE and for biotin conjugate of Lycopersicon esculentum agglutinin (tomato lectin)/TACE
Two hours after the end of OGD or glutamate exposure, cells were fixed by immersion for 30 minutes in a solution containing 4% p-formaldehyde in 0.1 mol/L phosphate buffer, pH 7.4 at room temperature. Cells were washed and then permeabilized in PBS containing 3% bovine serum albumin and 0.2% Triton X-100 for 30 minutes and then incubated in the two primary antibodies for each double staining 2 hours at room temperature: a specific polyclonal anti-TACE (ProSci Inc.; 1:300) and a monoclonal anti–GFAP antibody (Chemicon, Temecula, CA, U.S.A.; 1:100 dilution) to identify astrocytes, or anti-TACE and a fluorescein-labeled agglutinin from Lycopersicon esculentum; (Sigma, Madrid, Spain; 1:150 dilution) to characterize microglia and macrophages (Acarin et al., 1994, 1996; Velasco et al., 1995). After washing in PBS, cells were incubated in each respective secondary antibody for 1 hour: for TACE, Cy 2-labeled goat anti-rabbit immunoglobulin G was used (Amersham Pharmacia Biotec; 1:300 dilution); for GFAP, the sections were incubated in a Cy 3-labeled goat anti-mouse immunoglobulin G (Amersham Pharmacia Biotec; 1:300 dilution). Visualization was performed under a fluorescence microscope (Nikon Eclipse TE300, Nikon Corporation, Tokyo, Japan) using Plan Fluor 20x/0.45 or 40x/0.6 objectives, and phase optics, a B2A Nikon filter for fluorescein isothiocyanate and Cy2 fluorescence or a G2A Nikon filter for Cy3 fluorescence. Each experiment was performed in duplicate and repeated three times. Image acquisition was carried out with a laser scanning confocal imaging system (MRC1024, BioRad, Hempstead, U.K.).
Determination of caspase-3 activity
Caspase-3 activity was determined as an indicator of apoptosis. Caspase-3 activity was measured in a fluorometric assay by measuring the extent of cleavage of the fluorescent peptide substrate with a commercial kit (Molecular Probes, Eugene, OR, U.S.A.) following manufacturer recommendations. The fluorescence of the rhodamine 110-labeled caspase-3 product was determined in a fluorescence microplate reader (Fluoroskan Ascent FL, Labsystems).
Determination of phosphatidylserine-exposing cells
Apoptotic cells were determined as phosphatidylserine-exposing cells by labeling with fluorescein isothiocyanate-annexin V using a commercial kit (Molecular Probes). The counting was performed under a fluorescence microscope (Nikon Eclipse TE300) using a Plan Fluor 20x/0.45 objective and a B2A Nikon filter for fluorescein isothiocyanate. A counting grid was placed in the microscope ocular to count cell numbers from four random fields for each staining. Each experiment was performed in duplicate and repeated three times. Data are expressed as percent of total cells/field ± SD.
Determination of condensed chromatin of apoptotic cells with Hoechst 33342
Apoptotic cells were determined by staining with the blue fluorescent Hoechst 33342 dye, which stains the condensed chromatin of apoptotic cells more brightly than the chromatin of normal cells. Staining was performed using a commercial kit (Molecular Probes, Eugene, OR, U.S.A.). The counting was performed as described earlier, using a Plan Fluor 40x/0.60 objective and a UV-2A Nikon filter for Hoechst 33342. Each experiment was performed in duplicate and was repeated three times. Data are expressed as percent of total cells/field ± SD.
Protein determination
Protein contents were determined using bicinchoninic acid (Hill and Straka, 1988).
Chemicals and statistical analyses
TNF-α and anti–TNF-α were obtained from Pepro Tech EC Ltd., BB3103 was supplied by British Biotech, propeptide substrate was synthesized by Dr. David Andreu (Peptide Synthesis Unit, Universidad de Barcelona, Spain), and antibodies and other chemicals were from Sigma (Spain) or as indicated in the previous sections. Results are expressed as mean ± SD of the indicated number of experiments, and statistical comparisons were made using a Newman-Keuls test.
RESULTS
Release of TNF-α from rat cortical cultures exposed to OGD or glutamate: Effect of the TACE inhibitor BB3103
When rat cortical cultures were exposed to OGD, TNF-α levels in the culture medium at the end of the OGD period were not significantly different from control (1.5 ± 1.5 pg/mL in control and 3.2 ± 3.0 pg/mL in OGD-exposed cells, P > 0.05, n = 6). However, after 150 minutes of “reperfusion,” TNF-α levels in OGD-exposed cells were significantly higher than those in control cells (Fig. 1, P < 0.05, n = 6). The N-methyl-
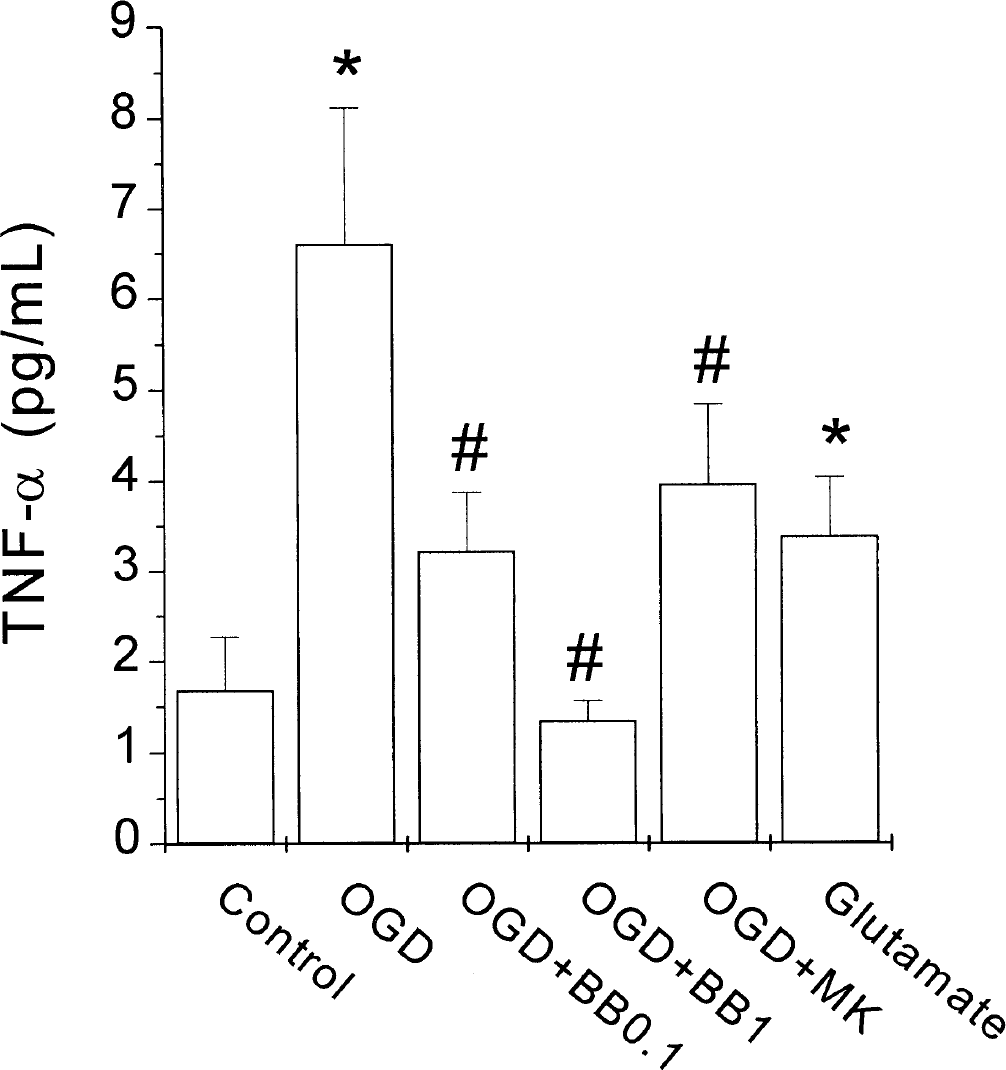
Effect of oxygen–glucose deprivation (OGD) or glutamate on tumor necrosis factor-α (TNF-α) release from rat cortical cultures. Effect of the TNF-α convertase inhibitor BB3103 (0.1 to 1 μmol/L, BB). TNF-α levels in the incubation solution of rat cortical cultures at the end of the reperfusion period were determined by enzyme-linked immunosorbent assay (see Materials and Methods). MK, MK-801 (an N-methyl-
TACE/ADAM17 and ADAM10 protein levels in control, glutamate and OGD-exposed cortical cultures
Western blot analysis revealed the presence of a TACE-immunopositive band (Fig. 2A); exposure to either OGD or glutamate and a subsequent 120-minute “reperfusion” period caused a significant increase in the amount of TACE protein detected by immunoblotting (Fig. 2A, n = 4, P < 0.05). Because ADAM10 has been reported to possess the ability to cleave pro–TNF-α in the same site as ADAM17, we analyzed its levels in our samples. An ADAM10-immunopositive band was shown to be present in control cortical cultures, but its levels were decreased after exposure to OGD or glutamate (Fig. 2B).
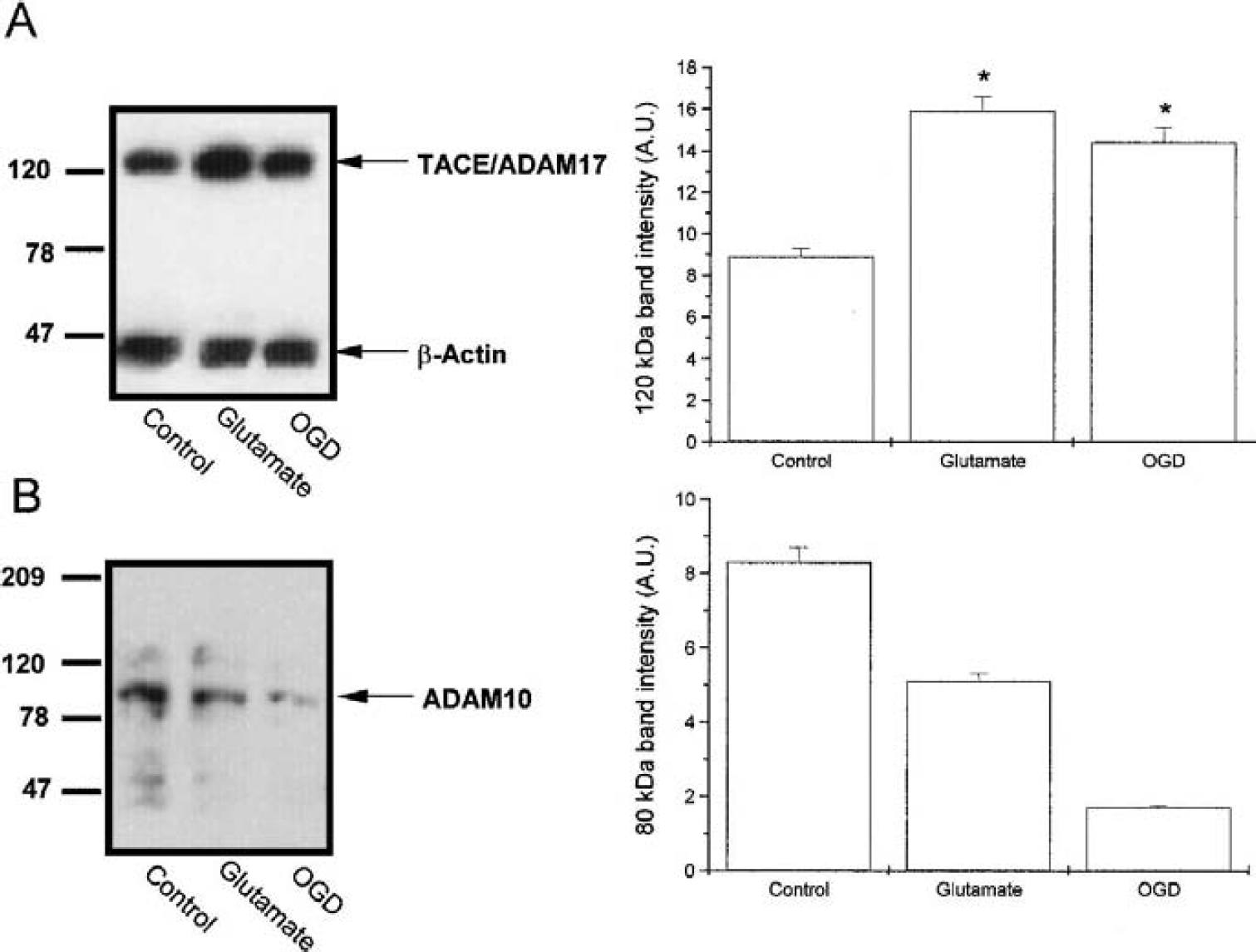
Effect of oxygen–glucose deprivation (OGD) or glutamate on the levels of tumor necrosis factor-α convertase (TACE)/ADAM17 and ADAM10 proteins in rat cortical cultures. Western blot analysis of TACE
Cleavage of fluorescein-labeled peptide containing the cleavage site in pro–TNF-α by control and glutamate-subjected cortical cultures: Effect of BB3103 and the protein synthesis inhibitor cycloheximide
When TACE activity was assayed in cortical cultures by incubation with the fluorophoric propeptide containing the cleavage site in pro–TNF-α, there was a time-dependent increase in the specific fluorescence of peptide product in the solution bathing the cultures (Fig. 3), which was significantly higher in cells exposed to glutamate (Fig. 3). Both the TACE inhibitor BB3103 (0.1 μmol/L) and the protein synthesis inhibitor cycloheximide (10 μmol/L) inhibited peptide cleavage (n = 4, P < 0.05; Fig. 3).
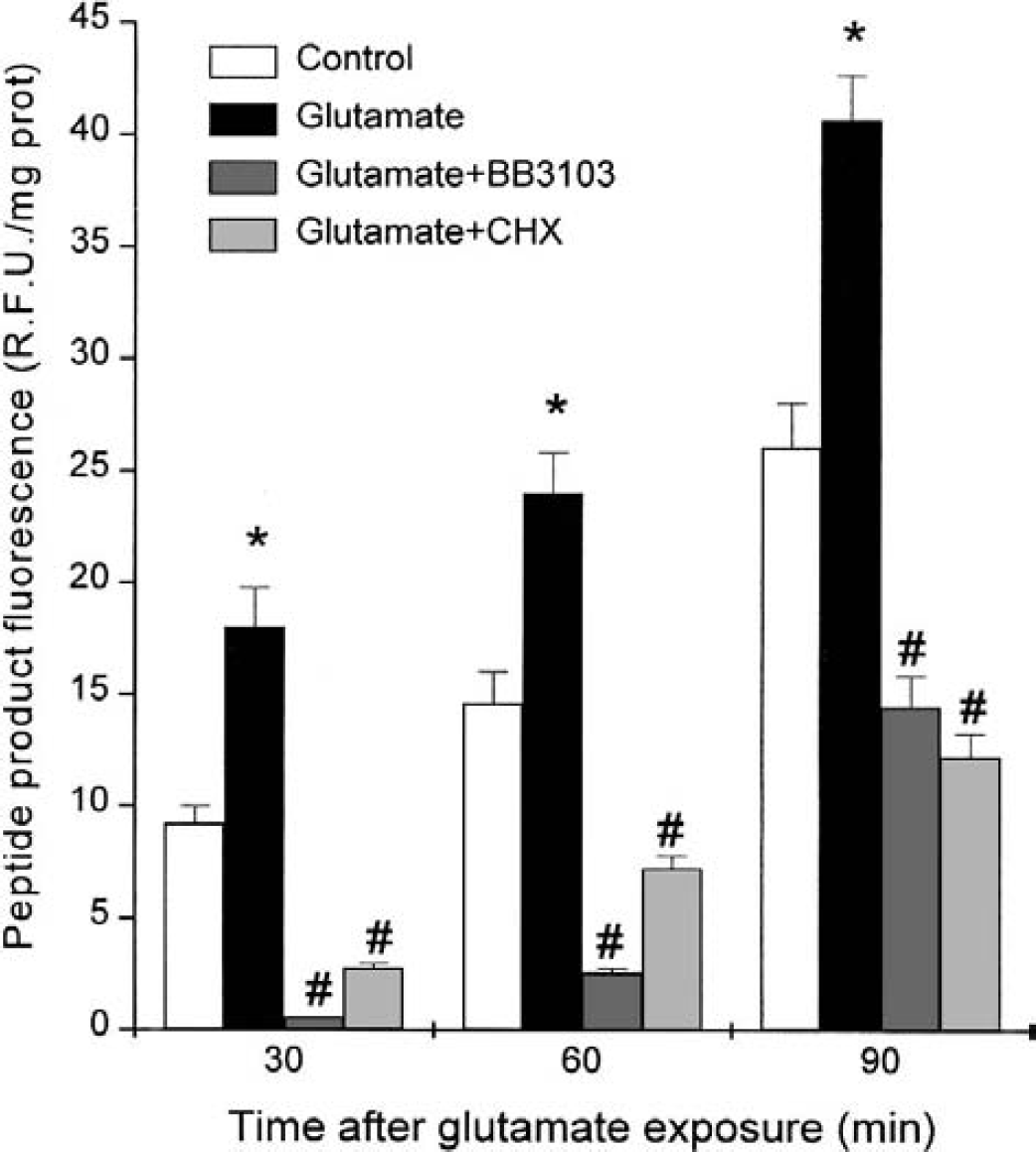
Tumor necrosis factor (TNF)-α convertase (TACE) activity in control and glutamate-treated cortical cultures during the reperfusion period. Effect of the TACE inhibitor BB3103 (BB, 0.1 μmol/L) and of the protein synthesis inhibitor cycloheximide (CHX, 10 μmol/L). TACE activity was determined as the ability to cleave a fluorescent peptide containing the cleavage site in pro-TNF. Activity was quantified from the fluorescence of the peptide product fluorescein-SPLQA in the incubating solution of cells, which was collected at different times. The data are mean ± SD, n = 4, *P < 0.05 versus control, #P < 0.05 versus glutamate.
Matrix metalloprotease activity in control and glutamate-exposed cultures
To check the participation of metalloproteases such as the MMPs after glutamate exposure, MMP activity was assayed in cortical cultures by incubation with a genic peptide, which acts as MMP substrate (Calbiochem). After 90 minutes of incubation, the MMP activity found in control cultures (11.3 ± 2.4 relative fluorescence units/mg protein, n = 4) was not significantly different from that found in glutamate-exposed cells (9.4 ± 0.5 R.F.U./mg protein, n = 4, P > 0.05).
Cellular localization of TACE in mixed cortical cultures
TACE/ADAM17 localization was examined in rat cortical cultures by double immunofluorescence staining. Control cultures showed that TACE-immunostaining was restrained to some L. esculentum agglutinin positive cells (30 ± 3% of agglutinin-stained cells), corresponding to microglial cells/macrophages (Fig. 4A). Interestingly, after exposure to glutamate, most macrophage/microglial cells showed TACE immunoreactivity (Fig. 4B, 90 ± 10%), which typically appears at the cell membrane. Moreover, after glutamate exposure, TACE immunofluorescence was found also in some GFAP-positive cells (12 ± 2% of GFAP-stained cells; Fig. 4D) that correspond to astrocytes, which were not TACE positive before the treatment with glutamate (control: 2 ± 2%; Fig. 4C). The pattern of TACE immunoreactivity after OGD was similar to that found after glutamate (data not shown).
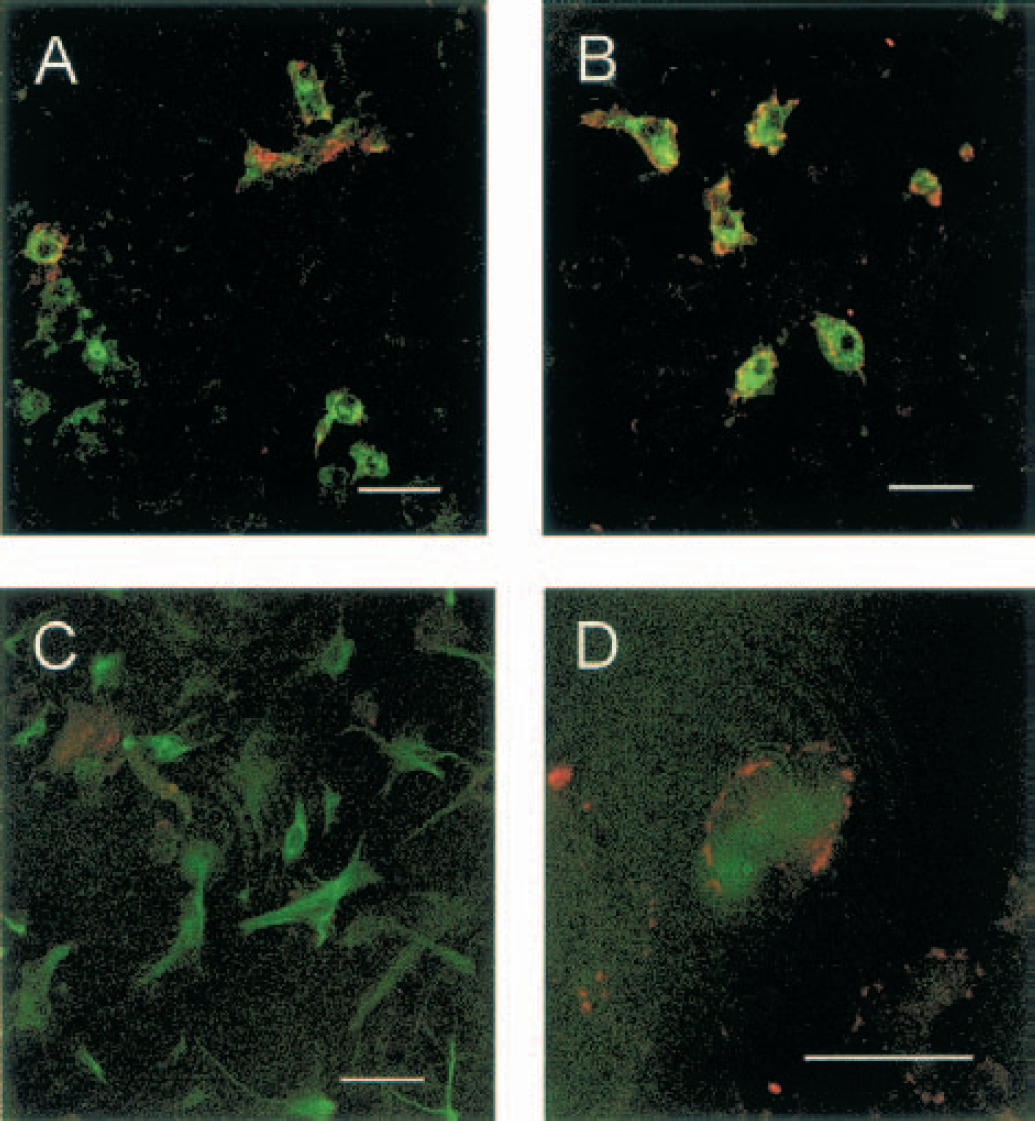
Double immunostaining of control and glutamate-exposed rat cortical cultures.
Apoptotic parameters in mixed cortical cultures after incubation with glutamate: Effect of BB3103
Annexin staining for detection of phosphatidylserine membrane exposure, activity of caspase-3, and Hoechst 33342 staining of condensed chromatin nuclei were determined as parameters of apoptosis after treatment of the mixed cortical cultures with glutamate 10 μmol/L for 10 minutes. Glutamate did not significantly affect either annexin staining (Fig. 5A), Hoechst 33342 staining (Fig. 5B), or caspase-3 activity when measured 45 minutes (Fig. 5C) or 150 minutes after the treatment (Fig. 5C). However, the exposure to glutamate in the presence of BB3103 (0.1 μmol/L) caused an increase in both annexin staining (Fig. 5A), Hoechst 33342 staining of condensed chromatin nuclei (Fig. 5B), and caspase-3 activity (Fig. 5C) at both times studied. The effect of BB3103 was mimicked by neutralizing concentrations of anti–TNF-α (2 fig/mL; 7.9 ± 0.8 pmol · min−1 mg−1 prot, n = 3, 45 minutes after treatment). In addition, BB3103-induced effect was inhibited in the presence of exogenously added TNF-α (10 pg/mL, Fig. 5C).
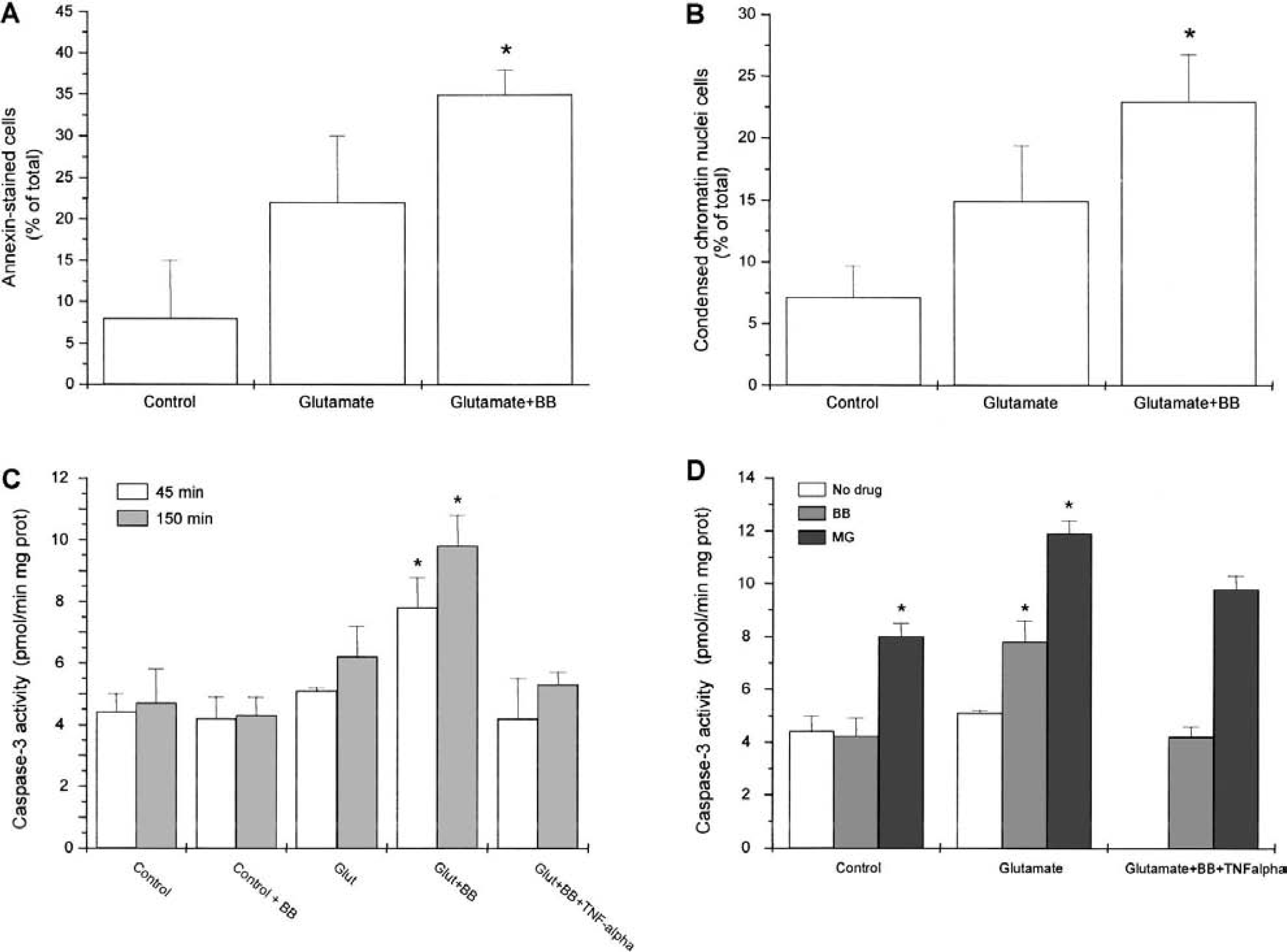
Apoptotic parameters in control and glutamate-exposed cortical cultures, and effect of tumor necrosis factor (TNF)-α convertase inhibitor BB3103 (BB). Annexin staining
Pharmacologic characterization of the antiapoptotic pathway induced by TACE/TNF-α
The inhibitor of NF-κB activation MG-132 (75 μmol/L) increased caspase-3 activity in control cells. In addition, after glutamate exposure, MG132 increased caspase-3 activity when compared with their respective controls (Fig. 5D). To check that endogenous TNF-α is responsible for the antiapoptotic action unmasked in the presence of BB3103, the experiment was repeated adding exogenous TNF-α once endogenous production had been blocked by BB3103 (0.1 μmol/L). In these conditions, MG-132 reversed the inhibitory action of TNF-α on caspase-3 activity after exposure to glutamate in the presence of BB3103 (Fig. 5D).
DISCUSSION
We have recently provided the first demonstration of the presence of TACE/ADAM17 protein in CNS and of its increased protein and mRNA expression in rat forebrain exposed to OGD (Hurtado et al., 2001), or of its activation after immobilization stress in rat brain cortex (Madrigal et al., 2002). We have now explored TACE/ADAM17 expression and cellular localization in rat neuronal–glial cortical cultures exposed to OGD or glutamate and searched for a role of TACE in this model. Our results indicate that both OGD and glutamate upregulate TACE/ADAM17, and that the resulting TNF-α overproduction may exert antiapoptotic effects in some circumstances.
First, our results show that OGD causes the release of soluble TNF-α from cortical cultures, an effect that is inhibited by low concentrations of BB3103, suggesting that this release results predominantly from pro–TNF-α shedding by TACE/ADAM17. Indeed, the hydroxamatebased compound BB3103 is a preferred inhibitor of metalloproteinases with sheddase activity (Ancuta et al., 1997; Middelhoven et al., 1997; Fiorucci et al., 1998; Lammich et al., 1999), which shows an IC50 of approximately 0.1 μmol/L to inhibit OGD-induced TNF-α production in rat cortical cultures as well as in rat forebrain slices (Hurtado et al., 2001). Although other proteases have been suggested to possess the ability to cleave pro–TNF-α (Lammich et al., 1999), hydroxamatebased compounds have been used to distinguish between them and TACE because they are more potent against TACE (IC50 values approximately 0.05 to 0.1 μmol/L) than against others such as ADAM10 or α-secretase (IC50 values ranging from 3 to less than 20 μmol/L) (Parvathy et al., 1998; Hooper et al., 2000).
Exposure to OGD causes the release of the excitatory amino acid glutamate from cortical cultures (Goldberg and Choi, 1993; De Cristóbal et al., 2002). We have now found that OGD-induced TNF-α release is inhibited by the N-methyl-
Analysis of TACE protein by Western blot demonstrates its presence in both control and OGD- or glutamate-exposed cortical cultures; moreover, TACE expression is increased after OGD or glutamate, in agreement with our recent results in rat forebrain slices (Hurtado et al., 2001). Although pharmacologic inhibition of TNF-α production with low concentrations of BB3103 excludes the involvement of ADAM10 in the processing of this cytokine, we have further confirmed these data by showing that, although ADAM10 is present in rat cortical cultures, its levels are even decreased after exposure to glutamate or OGD. This is an unexpected result, and its meaning merits further study in the context of cerebral ischemia.
A question arising from these findings concerns the effect of the increased expression of TACE on TNF-α release. To elucidate this point without interferences from de novo pro–TNF-α synthesis, we have studied the cleavage by cortical cultures of saturating concentrations of a fluorescein-labeled propeptide spanning the pro-TNF Ala76-Val77 cleavage site in pro–TNF-α. Our results showing that propeptide cleavage by glutamate-subjected cells is inhibited both by cycloheximide and BB3103 strongly suggest that both increased expression and activity of TACE/ADAM17 are responsible for the increase in TNF-α release. Other metalloproteases such as MMPs do not seem to be implicated, because the poor MMP activity found in the cultures was not affected by exposure to glutamate.
An important study pointed out by these findings concerns the cellular localization of TACE. The mixed cortical cultures used in the present work contain both astrocytes and microglial cells apart from cortical neurons as the major cellular components. Therefore, the presence of TACE was explored by double-immunostaining techniques using anti-TACE in combination with specific cellular markers. Our results show that, in control cultures, TACE staining appears at the cell membrane in some microglial cells. More interestingly, after exposure to OGD or glutamate, most microglial cells showed TACE immunoreactivity. Furthermore, some astrocytes now appeared as TACE positive. These data suggest that these two paradigms of excitotoxicity are able to trigger the expression of TACE in glial cells. These results may explain the reported increased TNF-α production after stimulation of AMPA-kainate glutamate receptors by microglial cells (Noda et al., 2000) as well as by astrocytes exposed to glucose deprivation and oxygen reduction (Tezel and Wax, 2000). These evidences suggest that activation of glutamate receptors on glial cells induces upregulation of TACE/ADAM17 in these cells. TACE/ADAM17 promoter region contains multiple AP2 or Sp1 transcription factor binding sites (Mizui et al., 1999), which may be responsible for this effect. To the best of our knowledge, this is the first demonstration of the localization of TACE in cortical cultures and its regulation by increased expression in glial cells. During the preparation of this article, TACE immunostaining has been described in astrocytes and endothelial cells from adult human CNS (Goddard et al., 2001).
Our findings on the constitutive expression of TACE/ADAM17 in glial cells and on its increased expression after OGD or glutamate raise the question as to what its role is. We have shown that TACE plays a major role in the shedding of TNF-α after OGD. This cytokine is expressed or released after ischemia (Liu et al., 1994; Buttini et al., 1996; De Bock et al., 1996), where it contributes to the damaging effects of this condition (Dawson et al., 1996; Barone et al., 1997; Meistrell et al., 1997; Nawashiro et al., 1997). It has also been described to exert proapoptotic actions (Sipe et al., 1996; Viviani et al., 1998; Downen et al., 1999; Sortino et al., 1999) on neural cells. Some of these proapoptotic actions on neurons are mediated indirectly via glial iNOS expression (Ogura et al., 1997; Chung et al., 1999; Heneka et al., 1999; Tezel and Wax, 2000; Combs et al., 2001). Conversely, there are also multiple additional evidences that demonstrate a direct, neuroprotective role of TNF-α (Cheng et al., 1994; Houzen et al., 1997; Mattson et al., 1995; Shinpo et al., 1999; Sullivan et al., 1999; Tarkowski et al., 1999) and antiapoptotic actions of this cytokine in neural cells (Barger et al., 1995; Mattson et al., 1997; Tamatani et al., 1999; Diem et al., 2001). These controversial evidences suggest that injury extent as well as the amount of TNF-α released may influence cellular fate. In the present work, we decided to focus on the effects of TACE/TNF-α pathway on the apoptotic parameters induced by the exposure of mixed cortical cultures to glutamate. Although glutamate, at the concentration used, was not able per se to induce significant apoptosis, when glutamate treatment was performed in the presence of BB3103, apoptosis was triggered, suggesting that glutamate-induced endogenous TNF-α production was masking a concomitant induction of apoptosis. This was corroborated by the inhibition of apoptosis caused by addition of exogenous TNF-α, and by the results showing that neutralizing concentrations of anti–TNF-α mimic the effect of BB3103. These antiapoptotic actions of TNF-α were found both 45 and 150 minutes after the end of the OGD, suggesting that both activation of TACE enzymatic activity as well as increased TACE expression play an important role in this antiapoptotic effect. These evidences show a neuroprotective effect of TNF-α in a setting of mild neuronal damage, which may find a clinical correlate in situations such as ischemic preconditioning. It remains to be studied whether endogenous TNF-α production may have opposite effects on viability in a different scenario, such as a situation of a higher production of TNF-α or a longer exposure to this cytokine, which may lead to the induction of inflammatory prooxidant enzymes such as iNOS or COX-2.
Strong evidence points to NF-κB as the mediator of the antiapoptotic actions of TNF-α (for a review, see Mattson et al., 2000). NF-κB is a ubiquitously expressed transcription factor that is held in an inactive form in the cytosol by interaction with a member of the IκB family (Baldwin, 1996; Verma and Stevenson, 1997; Ghosh et al., 1998; Karin, 1999). NF-κB is activated by phosphorylation and subsequent degradation of IκB, which results in translocation of NF-κB to the nucleus, where it induces transcription of target genes. In the paradigm of neural damage used in the present report, the neuroprotective effect of TNF-α is mediated via activation of NF-κB, as the proteosome inhibitor MG132 is able to revert it. Interestingly, MG132 induced apoptosis in control cells, suggesting that this pathway is necessary for cell survival.
In summary, this is the first report showing (1) TACE activity and protein and its localization and upregulation in CNS, (2) its major role in TNF-α shedding, and (3) its functional implication after OGD or glutamate. These findings give new insights into the molecular mechanisms underlying a range of conditions from ischemic preconditioning up to neuropathologic disorders where TNF-α is implicated, such as degenerative diseases and stroke. Other effects derived from TACE expression and upregulation in CNS remain to be studied.
Footnotes
Acknowledgments:
The authors thank Dr. David Andreu and Dr. Luis Rivas for helpful discussion in the design of TACE substrate peptide, and British Biotech for the generous gift of BB3103.