
Abstract
Neurogenesis in the brain continues throughout life and is promoted by brain insults including ischemia. There is no critical conclusion, however, about whether proliferated cells acquire neuronal function after ischemia. Transient global ischemia was produced by a four-vessel occlusion procedure in rats (n = 54). To label proliferative cells, rats were administrated with a single dose of 5-bromo-2’-deoxyuridine (BrdU) at 4, 6, 8, 10, 13, or 15 days after ischemia. Increases in BrdU-positive cells were detected in the hippocampal dentate gyrus at 5, 7, and 9 days after ischemia. To determine the phenotype of BrdU-positive cells, BrdU was administrated twice daily for 3 consecutive days during 6 to 8 days after ischemia. A basic helix–loop–helix transcription factor NeuroD at 7 and 14 days and an immature migrating neuronal marker doublecortin at 14 days after ischemia were expressed transiently in proliferative cells. These proliferative cells after ischemia differentiated to the phenotype of neuron at 28 days after ischemia. Furthermore, BrdU-positive neurons showed phosphorylation of extracellular signal-regulated kinase (ERK) by intracerebroventricular injection of N-methyl-D-aspartate (NMDA) at 28 and 56 days after ischemia as seen in surrounding mature neurons. The number of BrdU-positive neurons, which responded to NMDA stimulation, increased with time after ischemia and was greater than that of sham-operated animals. The present study provides evidence for in vivo ERK phosphorylation in response to NMDA stimulation of BrdU-positive neurons in the adult hippocampus after transient forebrain ischemia.
Keywords
Accumulating evidence indicates that neurogenesis in the central nervous system continues throughout life in restricted regions, including the subventricular zone of the lateral ventricles and the subgranular zone (SGZ) of the hippocampal dentate gyrus (Eriksson et al., 1998; Gould et al., 1998). In addition, focal and global ischemia increase neurogenesis in the subventricular zone and SGZ (Arvidsson et al., 2002; Jin et al., 2001; Liu et al., 1998; Yagita et al., 2001). Neurogenesis induced by brain ischemia implies that damaged neuronal function may be repaired by replacing lost neurons with BrdU-positive neurons in the adult brain. This ability could offer hope for the development of restorative therapies for neurologic disorders or brain injuries. To achieve such a goal, a good understanding of neurogenesis in the adult brain is required.
Recently, it was reported that newly generated neurons, which were labeled with green fluorescent protein, in the adult hippocampal dentate gyrus were able to show electrophysiologic function recorded in acute hippocampal slices (van Praag et al., 2002; Liu et al., 2003). These findings suggested that physiologic neurogenesis in the adult brain could generate functional mature neurons. However, the functional properties of BrdU-positive neurons induced by brain insults remain to be determined, although there are a number of factors influencing neuronal function of newly generated neurons, which could depend not only on the growth factors around them, but also on the nature and intensity of ischemic insult, the origin of newly generated neurons, and their final destination and formation of neuronal network with surrounding mature neurons.
Mature hippocampal neurons mainly use glutamate for excitatory neurotransmission, and abundantly express the N-methyl-D-aspartate (NMDA) receptor, an ionotropic glutamate receptor. A synaptic Ras-GTPase activating protein SynGAP is associated with NMDA receptor complex via postsynaptic density 95 (Kim et al., 1998). The Ras-GTP activating activity is inhibited by CaMKII, which is activated by Ca2+ influx through the NMDA receptor. Consequently, activated Ras signaling at excitatory synapses stimulates the mitogen-activated protein (MAP) kinase/ERK cascade (Chen et al., 1998). In fact, stimulation of the NMDA receptor activates intracellular signal transduction cascades including the MAP kinase/ERK cascade (Bading and Greenberg, 1991; Xia et al., 1996). It has been suggested that NMDA receptor-mediated activation of ERK cascade may play a pivotal role in synaptic plasticity underlying learning processes (Atkins et al., 1998; English and Sweatt, 1997). Therefore, we focused on ERK phosphorylation after NMDA receptor stimulation to determine a biochemical response of neurons, although the NMDA receptor could activate other signal transduction pathways.
In the present study, we investigated cell proliferation and differentiation in the adult hippocampus after transient forebrain ischemia. Furthermore, we have hypothesized that BrdU-positive cells that show phenotypes of mature neurons after transient global ischemia acquire intracellular signal transduction cascades. To determine whether BrdU-positive cells induced by ischemia have characteristics of neurons, we administered NMDA to the lateral ventricle to activate ERK. As an in vivo response to excitatory input through the NMDA receptor, we assessed ERK phosphorylation of BrdU-positive cells after ischemia. The findings demonstrated that the number of proliferative cells increased transiently after ischemia. Furthermore, the number of BrdU-positive cells, which showed the phenotype of mature neuron and ERK phosphorylation, increased with time after ischemia as compared with sham-operated animals.
MATERIALS AND METHODS
Animal model
Adult male Wistar rats (8 weeks old, Charles River Japan, Atsugi, Japan) had free access to food and water according to the Guidelines of Experimental Animal Care issued by the Prime Minister's Office of Japan. The experimental protocol was approved by the Committee of Animal Care and Use of Tokyo University of Pharmacy and Life Science. Transient forebrain ischemia was produced by four-vessel occlusion procedure of rats for 15 minutes and reperfusion as previously described (Pulsinelli and Brierley, 1979; Takagi et al., 2003). Rats were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally). Both second cervical vertebras were exposed, and visible vertebral arteries were permanently electrocauterized. Twenty-four hours after electrocauterization, anesthesia was induced with 3% enflurane and maintained with 1.5% enflurane in a mixture of oxygen/nitrous oxide (25/75%). Bitemporal subdermal electroencephalogram needle electrodes were placed in reference to a frontal subdermal electrode. After establishing a baseline electroencephalogram level, both common carotid arteries were exposed and occluded with aneurysm clips for 15 minutes, at which time the clips were removed and the rat was allowed to recover. Rectal temperature was continuously monitored during ischemia and was maintained at 37.0°C to 37.5°C with a heating pad. Only rats that showed a completely flat electroencephalogram and a loss of consciousness were chosen in the present study. Each set of animals received the same degree of surgical preparation and the same recovery paradigms, and the loss of neurons in the hippocampal CA1 region was confirmed in all occluded animals in this study. To minimize insult variability that could result from surgical procedures, animals without neuronal cell loss in the hippocampal CA1 region were excluded. Sham-operated animals received exactly the same surgical procedure without arterial occlusion.
Bromodeoxyuridine labeling
To label proliferative cells, the thymidine analog 5-bromo-2’-deoxyuridine (BrdU, Sigma-Aldrich, St. Louis, MO, U.S.A.) was administered intraperitoneally. Two injection paradigms (short-term and long-term) were used. In the short-term experiment, rats were administered a single dose of BrdU (50 mg/kg) at 4, 6, 8, 10, 13, or 15 days after ischemia (n = 5 at each time point and for control group). Twenty-four hours after the administration, rats were perfused transcardially with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4, PB) under deep anesthesia. This experiment reveals the number of cells that incorporated BrdU during a 24-hour period and is an index of the rate of generation of cells at a specific time point after ischemia. In the long-term experiment, BrdU (50 mg/kg) was administered to rats twice daily for 3 consecutive days during 6 to 8 days after ischemia, and rats were perfused transcardially with 4% paraformaldehyde in 0.1 mol/L PB at 14, 28, or 56 days after ischemia. This experiment discloses the phenotype, survival, and migration pattern of BrdU-positive cells.
Microinjection of N-methyl-D-aspartate
To determine the in vivo intracellular response of BrdU-positive neurons, rats received intracerebroventricular administration of NMDA (Sigma-Aldrich), an agonist of the NMDA receptor at 14, 28, and 56 days after ischemia (n = 4 at each time point). Rats were anesthetized with pentobarbital and placed on a stereotaxic apparatus. After making two burr holes on the bilateral parietal skull at 1.0 mm posterior and 1.8 mm lateral from bregma, 30-gauge needles were inserted into both ventricles at a depth of 4.0 mm from the cortical surface. NMDA (25 μg/kg) diluted to 2 mg/mL by saline was infused at the rate of 0.5 μL/min using a microsyringe pump (EP-60, EICOM, Kyoto, Japan). The same volume of saline was injected into both ventricles as a control for NMDA injection (n = 3). At 30 minutes after the start of administration, rats were perfused with 4% paraformaldehyde in 0.1 mol/L PB containing 0.1 mol/L sodium pyrophosphate and 1 mol/L NaF to prevent dephosphorylation. In additional experiments, a NMDA receptor antagonist, dizocilpine malate (MK-801, 1 mg/kg, Sigma-Aldrich) was injected intraperitoneally 30 minutes before NMDA stimulation (n = 3).
Immunohistochemistry
After the perfusion with paraformaldehyde, the brains were quickly removed, cut into approximately 5-mm-thick coronal slabs, and postfixed overnight with 4% paraformaldehyde in 0.1 mol/L PB containing 0.1 mol/L sodium pyrophosphate. Coronal slabs were embedded in paraffin and cut serially at 10 μm with a microtome. For immunohistochemical detection of BrdU-labeled nuclei, coronal sections placed on poly-L-lysine-coated slides were incubated for 30 minutes with 2N HCl at 37°C for DNA denaturation. After blocking, sections were incubated overnight with mouse monoclonal anti-BrdU antibody (1:200; Sigma-Aldrich) at 4°C, and then incubated with biotinylated horse anti-mouse immunoglobulin G (IgG) (1:200; Vector Laboratories, Burlingame, CA, U.S.A.) for 2 hours at room temperature. They were washed with phosphate-buffered saline (pH 7.4, PBS) and further incubated in streptavidin–biotin–peroxidase solution (Vector Laboratories) for 2 hours. The peroxidase reaction was carried out by incubating with diaminobenzidine and hydrogen peroxide (Vector Laboratories).
Double-immunostaining was performed with immunofluorescence. To retrieve antigen, sections were boiled by a microwave in 10 mmol/L citrate buffer for 5 minutes. DNA denaturation was done as mentioned above. After blocking, sections were incubated overnight with rat monoclonal anti-BrdU antibody (1:200, Oxford Biotechnology, Oxford, UK) at 4°C and then incubated for 2 hours with biotinylated goat anti-rat IgG (1:200, Vector Laboratories) at room temperature. They were then incubated with streptavidin–fluorescein (1:100, Amersham, Buckinghamshire, UK) for 90 minutes. Thereafter, sections were incubated with mouse anti-NeuN (1:100, Chemicon, Temecula, CA, U.S.A.), mouse anti-microtubule-associated protein 2ab (1:250, Sigma-Aldrich), mouse anti-CNPase (1:200, Sigma-Aldrich), goat anti-doublecortin (DCX) (1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA, U.S.A.), rabbit anti-GFAP (DAKO, Carpinteria, CA, U.S.A.), or goat anti-NeuroD antibody (1:200, Santa Cruz) for 2 hours. This was followed by incubation for 90 minutes with Cy3-labeled goat anti-mouse IgG (1:50, Amersham), TRITC-conjugated swine anti-rabbit IgG (1:200, DAKO), or Cy3-conjugated donkey anti-goat IgG antibody (1:200, Jackson Immunoresearch, West Grove, PA, U.S.A.). For triple staining, sections were incubated overnight with rat monoclonal anti-BrdU antibody at 4°C and then incubated for 1 hour with fluorescein isothiocyanate–conjugated donkey anti-rat IgG antibody (1:400, Jackson Immunoresearch) at room temperature. Sections were further incubated with a mixture of rabbit anti-pERK (1:200, phosphop44/42 MAP Kinase [Thr202/Tyr204], Cell Signaling Technology, Beverly, MA, U.S.A.) and mouse anti-NeuN antibodies (1:100) for 2 hours at room temperature. These sections were then incubated with Cy3-conjugated donkey anti-rabbit IgG (1:1,000, Jackson Immunoresearch) and 7-amino-4-methylcoumarin-3-acetic acid–conjugated donkey anti-mouse IgG antibodies (1:400, Jackson Immunoresearch) for 1 hour. For parvalbumin-pERK-BrdU triple staining, the sections, which were reacted with anti-BrdU and fluorescein isothiocyanate–conjugated anti-rat IgG antibodies, were further incubated overnight with mouse anti-parvalbumin antibody (1:200, Sigma-Aldrich) at 4°C and then incubated for 1 hour with 7-amino-4-methylcoumarin-3-acetic acid–conjugated donkey anti-mouse IgG (1:400) at room temperature. This was followed by incubation with rabbit anti-pERK antibody (1:200) for 2 hours at room temperature and then with Cy3-conjugated donkey anti-rabbit IgG antibody (1:1,000) for 1 hour. Fluorescence was detected using an Olympus fluorescence microscopy (BX-51) or a Bio-Rad MRC 1024 confocal imaging system equipped with a krypton–argon laser and a Nikon Diapot microscope. Images were processed by Adobe Photoshop (Adobe Systems, Mountain View, CA, U.S.A.). The microscopic observations were performed by a person unaware of the study group.
Cell counting
5-Bromo-2’-deoxyuridine–labeled cells in the hippocampal dentate gyrus were counted in five to seven sections that were spaced 200 μm apart and corresponded to coronal coordinates from bregma −2.80 to −4.52 mm. All BrdU-positive cells located in both SGZ and granule cell layer (GCL) were counted regardless of size or shape under x400 magnification (Olympus BX-51). Results were expressed as the average number of cells per square millimeter in the areas that consist of both SGZ and GCL. In counting of double-staining sections, all BrdU-labeled cells, which were located in the SGZ and GCL, were first examined for colocalization with NeuN in eight sections that were spaced 150 μm apart and corresponded to coronal coordinates of bregma from −3.14 to −4.52 mm. The number of immunoreactive cells stained with anti-NeuN antibody, which were colocalized with BrdU-labeled cells (yellow fluorescence image), were counted. Double labeling was confirmed with a Bio-Rad MRC 1024 confocal imaging system. In counting of triple-staining sections, triple-labeled images in both SGZ and GCL were imported into Photoshop under x400 or x1,000 magnification (Olympus BX-51), and the number of BrdU-positive cells that colocalized with NeuN and pERK (white fluorescence image) was counted on a computer monitor. Double labeling with pERK and BrdU was confirmed with a Bio-Rad MRC 1024 confocal imaging system. Results were expressed as the average number of double- or triple-labeled cells per square millimeter in the areas where consist of both SGZ and GCL. Statistical analysis for BrdU-labeled cells in the hippocampal dentate gyrus was performed using analysis of variance followed by Scheffe's post-hoc test. Statistical analysis for BrdU-NeuN-labeled and BrdU-NeuN-pERK-labeled cells was performed using analysis of variance followed by Fisher's PLSD as post-hoc test. P values less than 0.05 were considered significant.
RESULTS
Cell proliferation and differentiation
We initially determined cell proliferation after transient forebrain ischemia in the hippocampal dentate gyrus (n = 5 at each time point). There were BrdU-labeled cells in the hippocampal SGZ of control animals (n = 5, Fig. 1A). The number of BrdU-labeled cells in the SGZ was 54.1 ± 26.9 count/mm2 dentate gyrus (Fig. 1C). Increases in BrdU incorporation were detected at 5 days after ischemia (Fig. 1C). Maximally, a 6-fold increase in the number of BrdU-labeled cells compared with control animals was seen at 7 days after ischemia (Fig. 1B and 1C). The significant increase was continued up to 9 days after ischemia (Fig. 1C). BrdU-labeled cells after ischemia were located exclusively in the SGZ. These cells were shaped irregularly and were smaller than the surrounding mature granule cells (Fig. 1B).
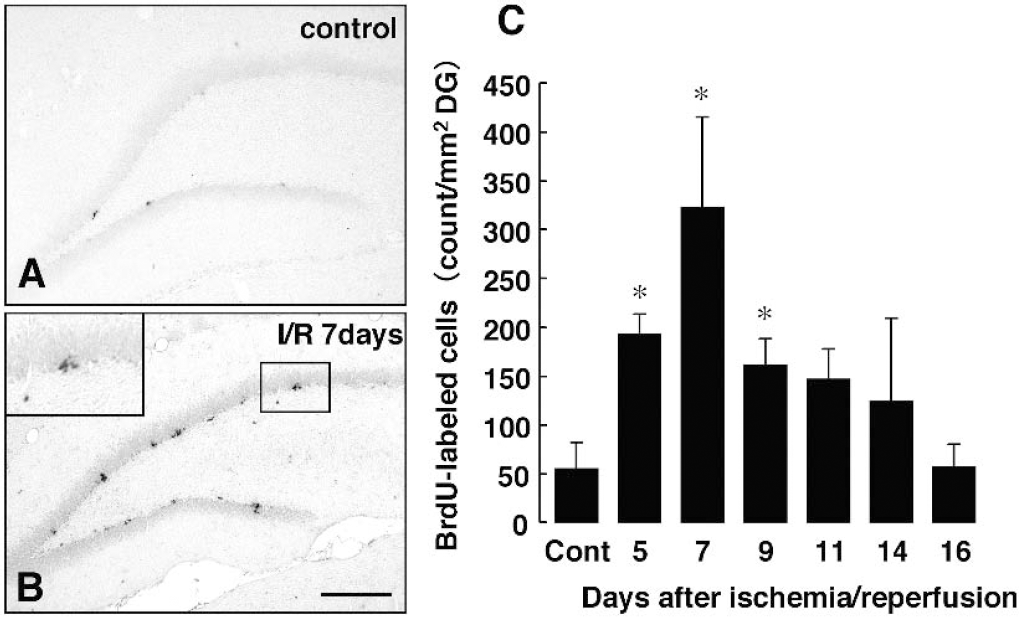
Transient forebrain ischemia-induced cell proliferation in adult dentate gyrus. Rats received a single dose of BrdU (50 mg/kg) 24 hours before fixation.
It is known that basic helix–loop–helix (bHLH) transcription factors control differentiation of cells in the embryonic brain. We examined whether BrdU-positive cells after ischemia expressed a bHLH factor, NeuroD, which was present in differentiating neurons. Seven (Fig. 2A, 2B, and 2C) and 14 (Fig. 2D, 2E, and 2F) days after ischemia (n = 3 at each time point), the BrdU-labeled cells in the SGZ expressed NeuroD (Fig. 2C and 2F). There were also NeuroD-labeled cells without BrdU immunoreactivity in the SGZ. These cells might be immature neurons, which were generated in the period without BrdU. In contrast, NeuroD was not expressed in the BrdU-labeled cells at 28 days after ischemia (n = 3, Fig. 2G, 2H, and 2I). The results obtained at each time point were similar in a group.
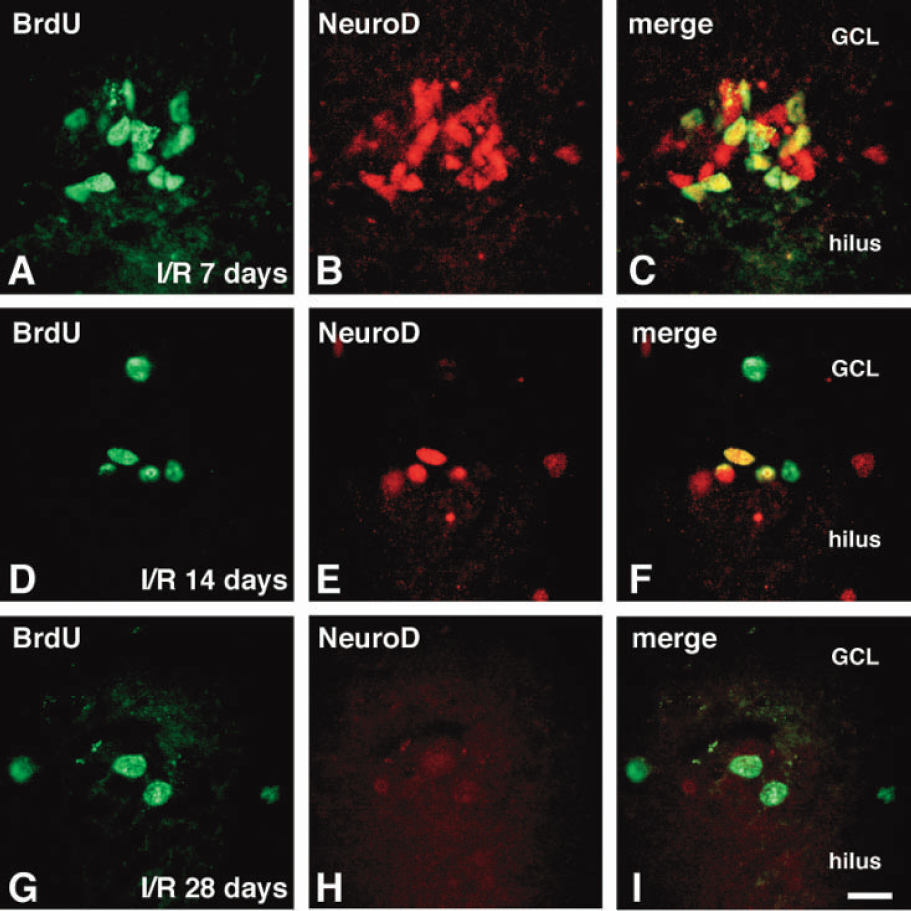
5-Bromo-2’-deoxyuridine (BrdU)-positive cells after ischemia expressed a basic helix–loop–helix transcription factor NeuroD. Images of double staining with BrdU (green) and NeuroD (red) in the sections from rats at 7 (A, B and C), 14
To determine whether BrdU-positive cells in the SGZ differentiate to mature neurons, we examined the expression of neuron, astrocyte, or oligodendrocyte markers in BrdU-labeled cells at 14, 28, or 56 days after ischemia. Fourteen days after ischemia, BrdU-labeled cells migrated from clusters and distributed evenly along the SGZ (n = 5, Fig. 3A). At this time point, BrdU-labeled cells expressed DCX, a marker of migrating immature neurons (n = 3, Fig. 3B). Twenty-eight days after ischemia, BrdU-labeled cells existed in the GCL (n = 5, Fig. 3C) and did not express DCX (n = 3, Fig. 3D). Morphology of BrdU-labeled cells in the dentate gyrus changed from irregular to round, like those of the mature granule neurons (Fig. 3C and 3D). BrdU-labeled cells in the GCL at 28 days after ischemia expressed both NeuN (n = 3, Fig. 3E) and MAP2 (n = 3, Fig. 3F). On the other hand, there were no BrdU-labeled cells expressing GFAP (n = 3, Fig. 3G) or CNPase (n = 3, Fig. 3H) in the GCL. The numbers of BrdU-labeled cells located in the SGZ and GCL at 14, 28, and 56 days after ischemia were 230.5 ± 52.0, 183.5 ± 67.3, and 206.5 ± 54.7 count/mm2 dentate gyrus, respectively (n = 4 at each time point, Fig. 4A). There was no significant difference among the cells 14, 28, and 56 days after ischemia. The proportions of NeuN-BrdU-labeled cells at 14, 28, and 56 days after ischemia were 65.0 ± 2.2, 94.8 ± 1.3, and 98.0 ± 0.2% of total BrdU-labeled cells, respectively (n = 4 at each time point, Fig. 4B). NeuN-BrdU-labeled cells were increased with time after ischemia (Fig. 4B).
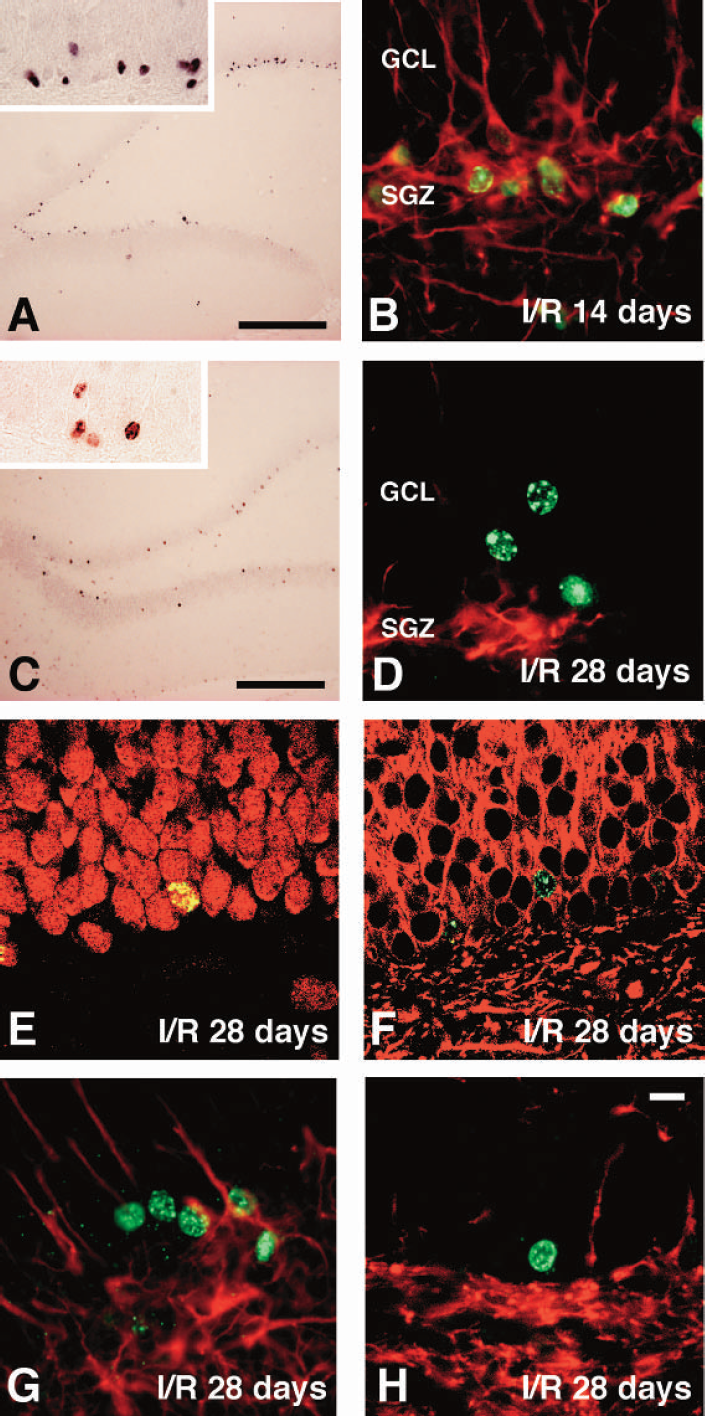
5-Bromo-2’-deoxyuridine (BrdU)-positive cells after ischemia expressed immature and mature neuronal markers.
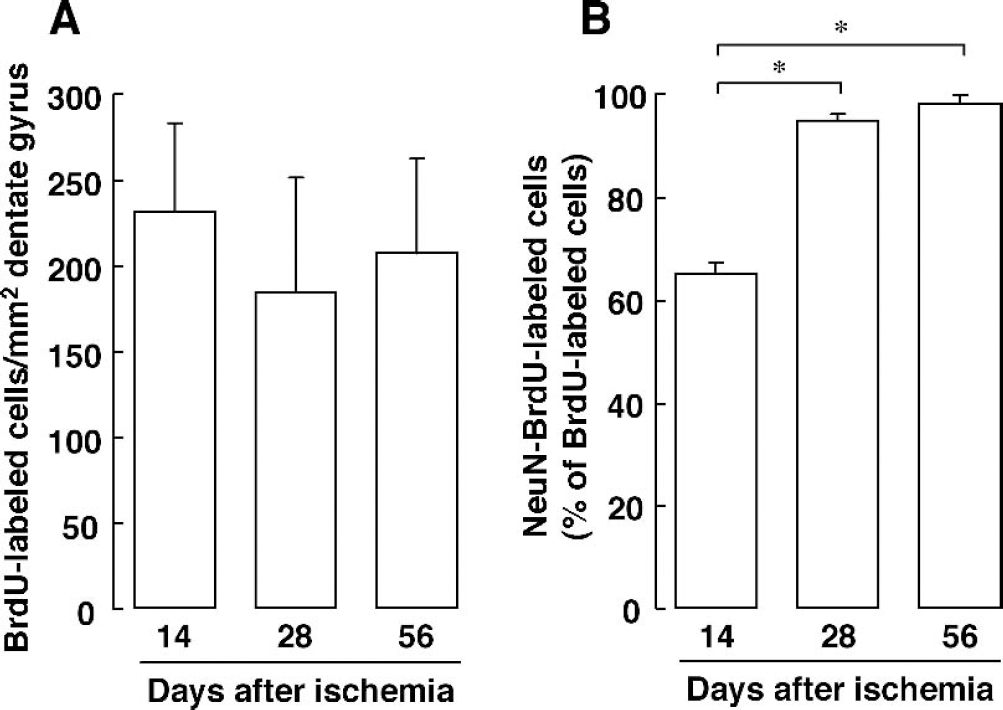
Cell proliferation and differentiation in the dentate gyrus after transient forebrain ischemia. BrdU was administered twice daily from 6 to 8 days after ischemia.
To determine whether programmed cell death occurs in BrdU-positive cells after ischemia, terminal deoxynucleotidyl transferase-mediated 2’-deoxyuridine 5’-triphosphate-biotin nick-end labeling (TUNEL) staining was performed at 7 and 14 days after ischemia. In our animal model, however, no TUNEL-labeled cells were detected in the SGZ and GCL, although there were TUNEL-labeled cells in the CA1 region (n = 3 at each time point, data not shown).
Phosphorylation of ERK in BrdU-positive neuron
Next, we examined whether differentiated neurons after ischemia acquired functional features of mature neurons. In hippocampal neurons, glutamate receptors including the NMDA receptor exist abundantly. The ability of BrdU-positive neurons to respond to excitatory input through the NMDA receptor would be a feature of mature neurons (Jelitai et al., 2002). We ascertained in the nonoperated control rats that injection of NMDA (25 μg/kg), an agonist of the NMDA receptor, into the bilateral ventricles induced the phosphorylation of ERK in the granule cells at 30 minutes after the injection (n = 3, data not shown). Then, NMDA was injected at 14, 28, and 56 days after ischemia (n = 4 at each time point). Triple staining with antibodies against BrdU, NeuN, and phosphorylated ERK (pERK) revealed that less than 7% of BrdU-labeled cells were positive to pERK at 14 days after ischemia (data not shown). Twenty-eight days after ischemia, when the mature neuronal marker NeuN was expressed in 95% (173.6 ± 63.2 count/mm2 dentate gyrus) of total BrdU-labeled cells (Fig. 4B and 6A), 34.2% of total NeuN-BrdU-labeled neurons were positive for pERK (Figs. 5D and 6B). Injection of saline into both ventricles as a control for NMDA injection did not affect immunoreactivity of pERK at each time point after ischemia. The numbers of pERK-NeuN-BrdU-labeled cells at 28 days after ischemia were 63.8 ± 38.2 count/mm2 dentate gyrus and increased significantly compared to those (7.9 ± 5.1 count/mm2 dentate gyrus) of sham-operated animals (Fig. 6B). Fifty-six days after ischemia, pERK-positive BrdU-positive neurons increased significantly up to 61.3% of total NeuN-BrdU-labeled neurons (Figs. 5H and 6B). The number of pERK-NeuN-BrdU-labeled cells at 56 days after ischemia was 139.5 ± 18.4 count/mm2 dentate gyrus (Fig. 6B). Although pERK-positive BrdU-positive neurons in the sham-operated animals increased at 56 days after the operation, the number in the ischemic animals increased significantly compared to that in the sham-operated animals (Fig. 6B). The phosphorylation of ERK was inhibited completely by preadministration of MK-801, an NMDA receptor antagonist (Figs. 5I, 5J, 5K, and 5L).
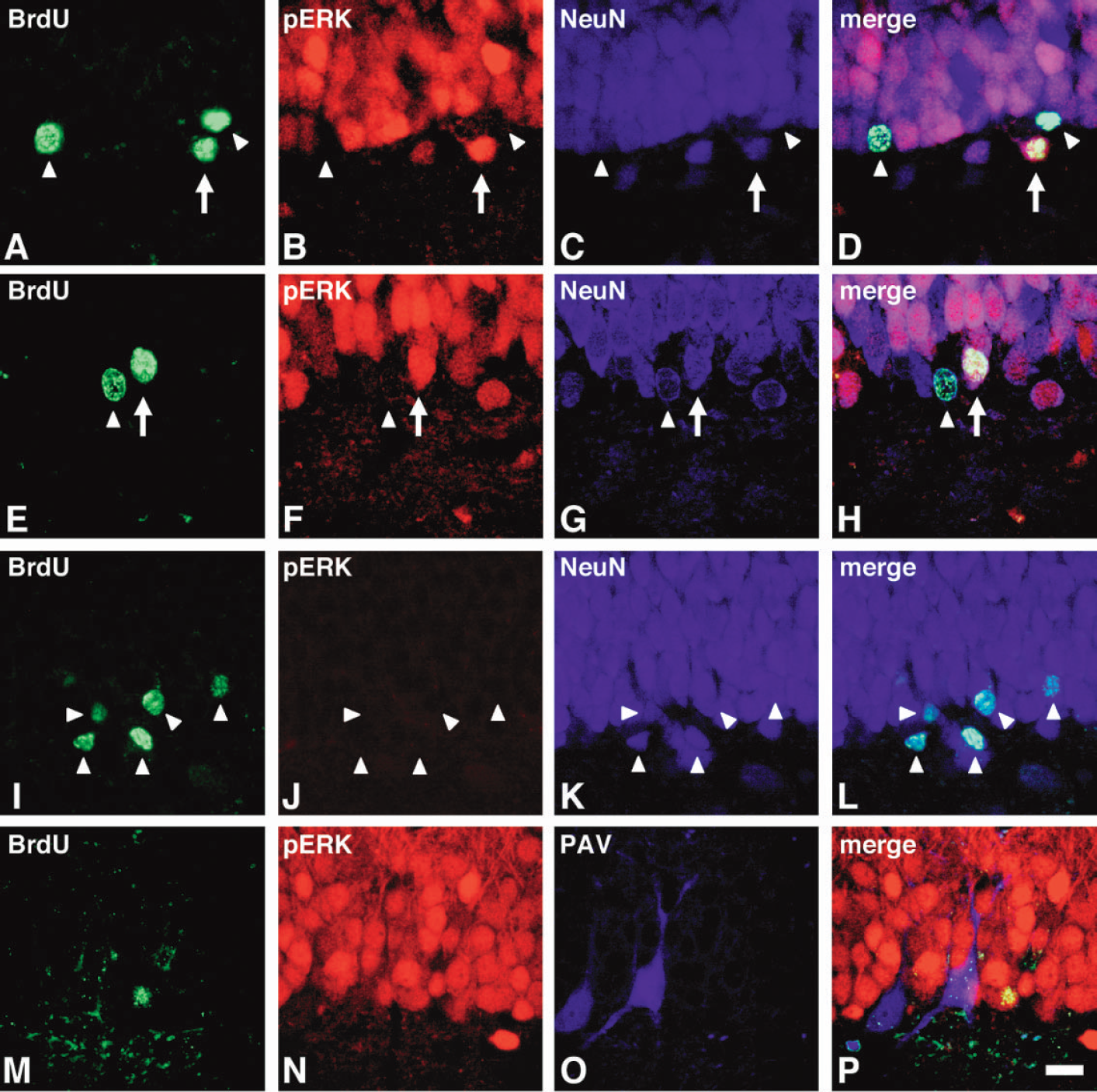
5-Bromo-2’-deoxyuridine (BrdU)-positive neurons after ischemia acquired the function to respond to NMDA stimulation. (A–L) Triple staining with BrdU (green), pERK (red), and NeuN (blue) at 30 minutes after NMDA injection in the sections from rat at 28
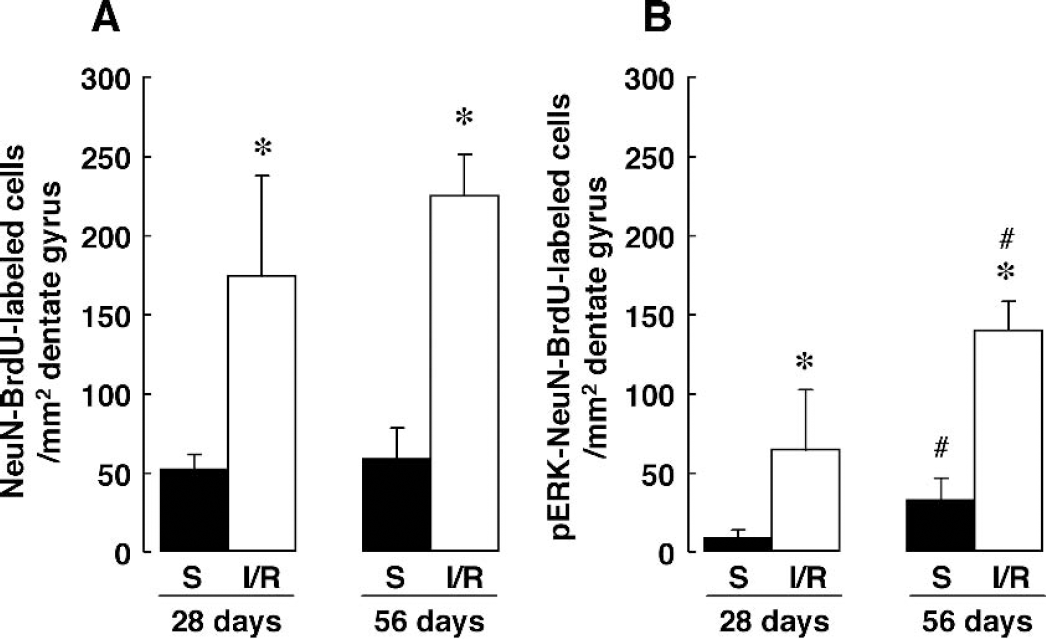
The numbers of NeuN-BrdU-labeled cells
In the present study, about 40% of NeuN-BrdU-labeled cells located in the GCL and SGZ were negative for pERK induced by NMDA injection at 56 days after ischemia (Fig. 6B). Recently, it was reported that BrdU-positive cells in the adult SGZ differentiated to both excitatory and inhibitory neurons (Liu et al., 2003). We examined expression of parvalbumin, a marker of the GABAergic neuron, after ischemia. Although there were a few parvalbumin-labeled cells in the SGZ and GCL, no BrdU-labeled cell expressed parvalbumin at 28 (data not shown) and 56 (Figs. 5M, 7N, 7O, and 7P) days after ischemia.
DISCUSSION
In the present study, we demonstrated that transient forebrain ischemia accelerated cell proliferation in the SGZ and that the proliferated cells migrated and differentiated to phenotypes of neurons with time after ischemia. These results were comparable to previous studies in terms of ischemia-induced cell proliferation and differentiation (Liu et al., 1998; Yagita et al., 2001). It is known that neural progenitors and BrdU-positive cells are eliminated by spontaneous apoptosis during development (Oppenheim, 1991; Oppenheim et al., 1995; Oppenheim et al., 2000). However, in the present study, no TUNEL-labeled cells were detected in the SGZ and GCL after ischemia. It has been suggested that 80% or more of the newly generated neurons in the striatum died during 2 to 6 weeks after transient focal ischemia (Arvidsson et al., 2002). Therefore, apoptotic BrdU-positive cells in the hippocampus might be detected in the long-term periods after transient global ischemia. Future studies will be required to clarify whether the number of BrdU-positive neurons in the SGZ and GCL after transient global ischemia depends on apoptosis cell death. Neurogenesis takes place predominantly in the developing embryonic brain. Therefore, it is possible that neurogenesis in the adult brain is controlled by the mechanism that is associated with embryonic neurogenesis. In the development process, NeuroD, a bHLH transcription factor, is required for differentiation of the hippocampal granule cells and cerebellar granule cells (Miyata et al., 1999; Schwab et al., 2000; Pleasure et al., 2000). NeuroD mRNA was also expressed in mature differentiated neurons (Lee, 1997; Elliott et al., 2001). In adult injured spinal cord, neither the expression of the neurogenic bHLH factors including NeuroD nor subsequent generation of new neurons could be detected (Yamamoto et al., 2001). Furthermore, forced expression of Neurogenin 2, a neurogenic bHLH factor, significantly enhanced neurogenesis in adult neural progenitors (Yamamoto et al., 2001). Our findings showed that BrdU-labeled cells expressed NeuroD protein 7 and 14 days, but not 28 days, after ischemia. Although the biologic basis of ischemia-induced neurogenesis remains to be understood, the transient expression of a neurogenic bHLH factor NeuroD in proliferative cells may contribute to an adult neurogenesis after ischemia. A number of transcription factors are expressed in BrdU-positive cells and regulate neurogenesis in the brain. Therefore, further studies will be needed to clarify roles of transcription factors in adult neurogenesis after ischemia.
In other studies, BrdU-positive neurons had electrophysiologic activity in acute hippocampal slices and formed functional synapses in culture (van Praag et al., 2002; Liu et al., 2003; Vicario-Abejon et al., 2000; Song et al., 2002). Another feature of mature neurons is the presence of intracellular signal transduction cascades. We focused on NMDA receptor–mediated intracellular signaling, because the NMDA receptor is expressed in the hippocampus abundantly and plays a key role in synaptic plasticity or learning and memory function in the brain. We evaluated in vivo biochemical response of BrdU-positive neurons by examining the activation of ERK after NMDA receptor stimulation. The number of BrdU-positive neurons that responded to NMDA increased with time, from 34% of BrdU-NeuN labeled cells at 28 days and reaching 61% by 56 days after ischemia. ERK phosphorylated-BrdU-positive neurons increased with time in the sham-operated rats. However, the number of BrdU-positive neurons that responded to NMDA in the ischemic rats increased to a greater extent than in the sham-operated rats. Our finding suggested that BrdU-positive neurons acquired components for the intracellular signal transduction cascade, linking the NMDA receptor to phosphorylation of ERK in the nucleus, and ischemic insults enhanced this effect. This is evidence for the in vivo biochemical response of BrdU-positive neurons in the adult hippocampus after transient forebrain ischemia. In the present study, some BrdU-positive neurons (BrdU-NeuN double-positive cells) after ischemia did not phosphorylate ERK in response to NMDA stimulation. Immunostaining using antibody against the GABAergic neuronal marker revealed that GABAergic inhibitory neuron was neither colocalized with BrdU nor stained with anti-pERK antibody in response to NMDA stimulation. Therefore, pERK-negative-BrdU-positive neurons after NMDA injection might not be GABAergic inhibitory neurons. An alternative reason might be that some BrdU-positive neurons did not become fully mature because of the imperfect expression of components for intracellular signaling pathways. Alternatively, it is also conceivable that phosphorylation of ERK is not a specific marker of NMDA receptor activation, or even mature neuronal function in general. It remains to be determined whether ERK phosphorylation in BrdU-positive neurons is mediated directly by the NMDA receptor or indirectly through other cells. Further studies are needed to clarify the physiologic consequences of the presence of BrdU-positive neurons in the ischemic brain.
Footnotes
Abbreviations used
Acknowledgments:
We thank Dr. James W. Gurd (University of Toronto at Scarborough) for helpful comments on the manuscript.