
Abstract
The signal transducers and activators of transcription (STATs) were found to be essential for cardioprotection. However, their role in preconditioning (PC) neuroprotection remains undefined. Previously, our studies showed that PC mediated a signaling cascade that involves activation of epsilon protein kinase C (εPKC), extracellular signal-regulated kinase (ERK1/2), and cyclooxygenase-2 (COX-2) pathways. However, the intermediate pathway by which ERK1/2 activates COX-2 was not defined. In this study, we investigated whether the PC-induced signaling pathway requires phosphorylation of STAT isoforms for COX-2 expression. To mimic PC or lethal ischemia, mixed cortical neuron/astrocyte cell cultures were subjected to 1 and/or 4 h of oxygen—glucose deprivation (OGD), respectively. The results indicated serine phosphorylation of STAT3 after PC or εPKC activation. Inhibition of either εPKC or ERK1/2 activation abolished PC-induced serine phosphorylation of STAT3. Additionally, inhibition of STAT3 prevented PC-induced COX-2 expression and neuroprotection against OGD. Therefore, our findings suggest that PC signaling cascade involves STAT3 activation after εPKC and ERK1/2 activation. Finally, we show that STAT3 activation mediates COX-2 expression and ischemic tolerance.
Keywords
Introduction
Ischemic preconditioning (PC) is an endogenous protective mechanism invoked by a brief, sublethal ischemic insult. Ischemic PC initiates a number of cytoprotective signaling pathways elicited by transcriptional activation of new protective genes and protein synthesis. Previously, our laboratory showed that activation of epsilon protein kinase C (εPKC) plays a key role in the PC signaling cascade in the organotypic hippocampal slice cultures by activating the extracellular signal-regulated kinase (ERK1/2) pathway (Lange-Asschenfeldt et al, 2004; Raval et al, 2003). Furthermore, we showed that oxygen—glucose deprivation (OGD)/εPKC-mediated PC induced cyclooxygenase-2 (COX-2) expression in neuronal cultures (Kim et al, 2007); however, the transcriptional signaling that modulates COX-2 expression after ischemic tolerance remains undefined.
Cyclooxygenase-2 is the rate-limiting enzyme for prostaglandin synthase, catalyzing the conversion of arachidonic acid to prostaglandin H2 (Smith et al, 1996). Recently, other groups have shown that COX-2 expression is a key mediator of the late phase of PC in both heart and brain (Gendron et al, 2005; Shinmura et al, 2000). Both cortical spreading depression
The signal transducers and activators of transcription (STATs) family consists of seven members, 1, 2, 3, 4, 5A, 5B, and 6 (Darnell, 1997). The STAT activation has a role in cell proliferation, apoptosis, and immune response (Karamouzis et al, 2007; Wittig and Groner, 2005; Zhang et al, 2007). For example, STAT1 has been shown to induce apoptosis (Stephanou et al, 2000). In contrast, STAT3 protects cardiomyocytes against ischemic injury (Hilfiker-Kleiner et al, 2004). Another STAT isoform was found to be neuroprotective against cerebral ischemia. STAT5 phosphorylation and CA1 neuronal survival after cerebral ischemia were observed after erythropoietin treatment (Zhang et al, 2007).
In general, the STAT activation involves dimerization, which is mediated by tyrosine phosphorylation (Zhong et al, 2005). Depending on the cell type or stimulus, STAT activation requires a second phosphorylation at a serine 727 residue as well as tyrosine phosphorylation (Shen et al, 2004). Interestingly, in the heart, PC caused serine phosphorylation of STAT1 and STAT3 via the εPKC → Raf-1 → MEK-1/2 → ERK1/2 signaling cascade, which resulted in COX-2 expression (Xuan et al, 2005). These studies suggested that the phosphorylation of STATs at the serine residue is an essential component of the COX-2-mediated induction pathway where
Materials and methods
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and were approved by the Animal Care and Use Committee of the University of Miami. According to these guidelines, efforts were made to minimize the number of animals and their suffering.
Preparation of Mixed Cortical Neuron/Astrocyte Cell Cultures
For mixed cortical neuron/astrocyte cell cultures, first, astrocytes were prepared from neonatal rat as described previously (Kim et al, 2002). Sprague—Dawley neonatal (1 to 2 days old) rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg/pup). Animals were decapitated and the brains were quickly removed. The cerebral cortices of the embryos were isolated and the dissociated astrocytes were plated with minimum essential medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum, 10% equine serum, 2 mmol/L glutamine, and 1 μg/mL epidermal growth factor (Sigma, St Louis, MO, USA) at 3 cortical hemispheres/24-well plates. After 10 days, 19-day pregnant Sprague-Dawley rats were anesthetized by halothane and embryos were quickly removed and decapitated to isolate cortical neurons. The cerebral cortices of the embryos were isolated and the dissociated cortical neurons were plated on a confluent monolayer of astrocytes cultured for 10 days to generate coculture with minimum essential medium containing 5% fetal bovine serum, 5% equine serum, and 2 mmol/L glutamine. The mixed cortical neuron/astrocyte cells were used after 10 to 11 days
Scheme of OGD Injury
To mimic ischemic injury, we subjected cells to OGD for 4 h. To simulate PC, cell cultures were subjected to OGD for a short period of 1 h, 48 h before OGD. For OGD, cell cultures were washed twice with glucose-free Hank's balanced salt solution (pH 7.4) of the following constitution: 1.26 mmol/L CaCl2·2H2O, 5.37 mmol/L KCl, 0.44 mmol/L KH2PO4, 0.49 mmol/L MgCl2, 0.41 mmol/L MgSO4 · 7H2O, 136.9 mmol/L NaCl2, 4.17 mmol/L NaHCO3, 0.34 mmol/L Na2HPO4 · 7H2O, and 10 mmol/L HEPES (Sigma). The cell cultures were then transferred to an anaerobic chamber (PROOX model 110, BioSpherix, Ltd, Redfield, NY, USA), which was placed in a water-jacketed incubator gassed with 95% N2/5% CO2 at 37°C. The chamber was sealed and incubated for either 1 h (PC) or 4 h (ischemic insult; OGD). After OGD, the cell cultures were transferred to their respective normal culture media and placed back into the incubator.
Assessment of Cell Death of Mixed Cortical Neuron/Astrocyte Cell Cultures
To determine cell death, cytotoxicity was measured by lactate dehydrogenase (LDH) released for 48 h into culture medium (Koh and Choi, 1987). Maximal neuronal LDH release was measured in the neuronal cultures exposed to
Western Blot Analysis
Cells were lysed in a lysis buffer containing 1% Nonidet P-40, 20 mmol/L Tris (pH 8.0), 137 mmol/L NaCl, 0.5 mmol/L EDTA, 10% glycerol, 10 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, 1 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L vanadate, and 1 mmol/L phenylmethylsulfonyl fluoride (Raval et al, 2003). Equal amounts of proteins were subjected to 8% to 12% SDS—polyacrylamide gel electrophoresis and the separated proteins were electrophoretically transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). The blot was blocked with 5% non-fat dried milk, incubated overnight at 4°C with COX-2 (1:1,000; Cayman Chemicals, Ann Arbor, MI, USA), β-actin (1:5,000; Sigma), εPKC, lamin B (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-serine 727-STAT3, p-tyrosine 705-STAT3, STAT3, p-serine 727-STAT1, STAT1, pERK1/2, and ERK1/2 (1:1,000; Cell Signaling Technology, Danvers, MA, USA) antibodies. Then, incubation was followed by horseradish peroxidase-conjugated specific secondary antibody for 1 h at room temperature. The immunoreactive bands were revealed by ECL western blotting detection reagents (Amersham, Buckinghamshire, England). Western blot images were digitized at 8-bit precision by means of a CCD camera (8 to 12 bit; Xillix Technologies Corporation, Vancouver, BC, Canada) equipped with a 55 mm Micro-Nikkor lens (Nikon, Tokyo, Japan). The camera was interfaced to an advanced image analysis system (MCID Model M2, Imaging Research Inc., St Catherines, ON, Canada). The digitized immunoblots were subjected to densitometric analysis using MCID software.
Preparation of Nuclear Extracts
To visualize nuclear translocation of p-serine 727-STAT3, nuclear extracts of neurons were prepared by using procedures described previously (Wang et al, 2007a). Cells were collected and washed twice with ice-cold phosphate-buffered saline (PBS). Cells were then lysed in lysis buffer containing 1% Nonidet P-40, 20 mmol/L Tris (pH 8.0) 137 mmol/L NaCl, 0.5 mmol/L EDTA, 10% glycerol, 10 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, 1 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L vanadate, and 1 mmol/L phenylmethylsulfonyl fluoride for 15 mins on ice. Cells were vortexed vigorously for 15 secs and then centrifuged at 14,000 r.p.m. for 10 mins. The supernatant was used as the cytosolic extract. The nuclear pellet was resuspended in buffer B (10 mmol/L HEPES, pH 8, 2 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol, 1 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L vanadate, and 1 mmol/L phenylmethylsulfonyl fluoride) and centrifuged at 14,000 r.p.m. for 10 mins. The resulting pellet was incubated with the extraction buffer (20 mmol/L HEPES, pH 8, 400 mmol/L NaCl, 0.5 mmol/L EDTA, 2 mmol/L MgCl2, 1 mmol/L dithiothreitol, 1 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L vanadate, and 1 mmol/L phenylmethylsulfonyl fluoride) for 30 mins on ice. After centrifugation at 15,000 r.p.m. for 15 mins, the supernatant was used as the nuclear extract. Protein samples (15 to 20 μg) of nuclear extracts were fractionated on an 8% SDS-polyacrylamide gel and transferred to a PVDF membrane. The membrane was then incubated with diluted p-serine 727-STAT3 antibody (1:1,000; Cell Signaling Technology). Immunoreactive proteins on the membrane were visualized by using ECL protocol as described above. To verify the purity of the nuclear fraction, the membrane was stripped and reblotted with polyclonal anti-lamin B, a nuclear marker (1:500; Santa Cruz Biotechnology). Lamin B was present in the nuclear fraction and was not detected in the cytosolic extract.
Immunocytochemistry
Mixed cortical neuron/astrocyte cell cultures were grown onto cover glass in 24-well plates and cultured
Immunoprecipitation
Cells were lysed in a lysis buffer containing 1% Nonidet P-40, 20 mmol/L Tris (pH 8.0), 137 mmol/L NaCl, 0.5 mmol/L EDTA, 10% glycerol, 10 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, 1 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L vanadate, and 1 mmol/L phenylmethylsulfonyl fluoride (Raval et al, 2003). Cell lysates (500 μg) were incubated with 1 to 2 μg of appropriate antibody at 4°C overnight and precipitated with protein A-Sepharose beads (Sigma) for 2 h at 4°C. The immunoprecipitated proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Bio-Rad Laboratories). Western blot analysis was performed with εPKC (1:500; Santa Cruz Biotechnology), p-serine 727-STAT3, and pERK1/2 (1:1,000; Cell Signaling Technology) antibodies.
Statistical Analysis
Data were expressed as the mean ± s.e.m. An analysis of variance followed by Dunnett's multiple comparison tests was used for statistical comparison. The results from the densitometric analysis were analyzed by a two-tailed Student's
Experimental Design
The mixed cortical neuron/astrocyte cell cultures were divided into five major groups as follows (Figure 1).
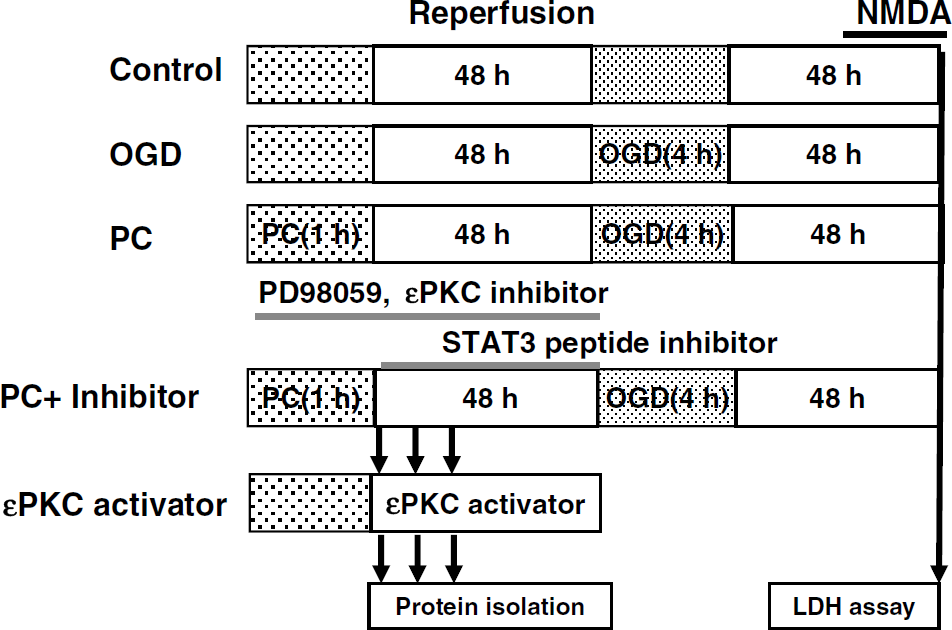
Experimental design. Control; OGD: cell cultures were subjected for 4 h of OGD; PC: cell cultures were subjected for 1 h of OGD and 48 h of reperfusion and then 4 h of OGD was induced; PC + inhibitor: either MAPK-K inhibitor (PD98059, μ10 mmol/L) or εPKC-specific inhibitory peptide (εV1–2, 100 nmol/L) or STAT3 inhibitory peptide (PpYLKTK, 100 nmol/L, 1 and 5 μmol/L) was administered to cell cultures during PC and 48 h of reperfusion and then 4 h of OGD was induced; εPKC activator: εPKC-specific activator peptide (ψεRACK, 100 nmol/L) was administered to cell cultures for 2 h without PC.
Control: cell cultures maintained in normal culture conditions were used as controls. The cell death was measured by the LDH assay. For western blot experiments, cells were lysed and the lysate was used as the control sample.
Oxygen—glucose deprivation: cell cultures were subjected to sham OGD PC with glycemic Hank's balanced salt solution, and 48 h later, 4 h OGD was induced. At 48 h after OGD, cell death was measured by the LDH assay.
Preconditioning: cell cultures were subjected to sublethal OGD (1 h OGD), and 48 h later, 4 h OGD was induced followed by the LDH assay. Cells were lysed with lysis buffer at the indicated times for western blot analysis.
Preconditioning + inhibitor treatment: cell cultures were treated with PD98059 (an mitogen-activated protein kinase kinase (MAPK-K) inhibitor, 10 μmol/L; Sigma) or an εPKC-specific inhibitory peptide (εV1–2, 100 nmol/L; KAI Pharmaceuticals Inc., South San Francisco, CA, USA) during 1 h of PC and 48 h of reperfusion. The dosage and duration of inhibitor (PD98059 and εPKC-specific inhibitory peptide) treatment were defined based on our previous study (Kim et al, 2007). A STAT3-specific inhibitory peptide (100 nmol/L, 1 and 5 μmol/L; EMD Chemicals Inc., Gibbstown, NJ, USA) was administered to cell cultures during 48 h of reperfusion after 1 h of PC just before OGD. At 48 h after OGD, cell death was measured by the LDH assay. Cells were lysed with lysis buffer at the indicated times for western blot analysis.
εPKC activation with an εPKC-specific activating peptide: cell cultures were treated with an εPKC-specific activating peptide for 2 h after washing with culture media two times without PC (ψεRACK at 100 nmol/L; KAI Pharmaceuticals Inc.). Cell samples were collected at the indicated durations (0, 15, 30, 60, and 120 mins after treatment) for western blot analysis.
Results
Preconditioning Induces Phosphorylation of STAT3 but not STAT1
In a previous study, we showed that PC upregulated COX-2 via εPKC and ERK1/2 activation and COX-2 played a crucial role in PC-induced neuroprotection (Kim et al, 2007). In this study, we investigated whether STAT1 and STAT3 are downstream pathways of serine/threonine kinase εPKC/MEK/ERK1/2 after PC and whether they could induce COX-2 expression.
First, to investigate whether PC (1 h OGD) induces activation of STATs, cortical neuron/astrocyte cell cultures were preconditioned for 1 h and cells were collected at 0 min, 15 mins, 30 mins, 1 h, and 2 h of reperfusion after PC. Western blot analysis revealed that PC induced serine phosphorylation of STAT3 in the cytosolic fraction at 15 and 30 mins and persisted for 2 h of reperfusion after PC (
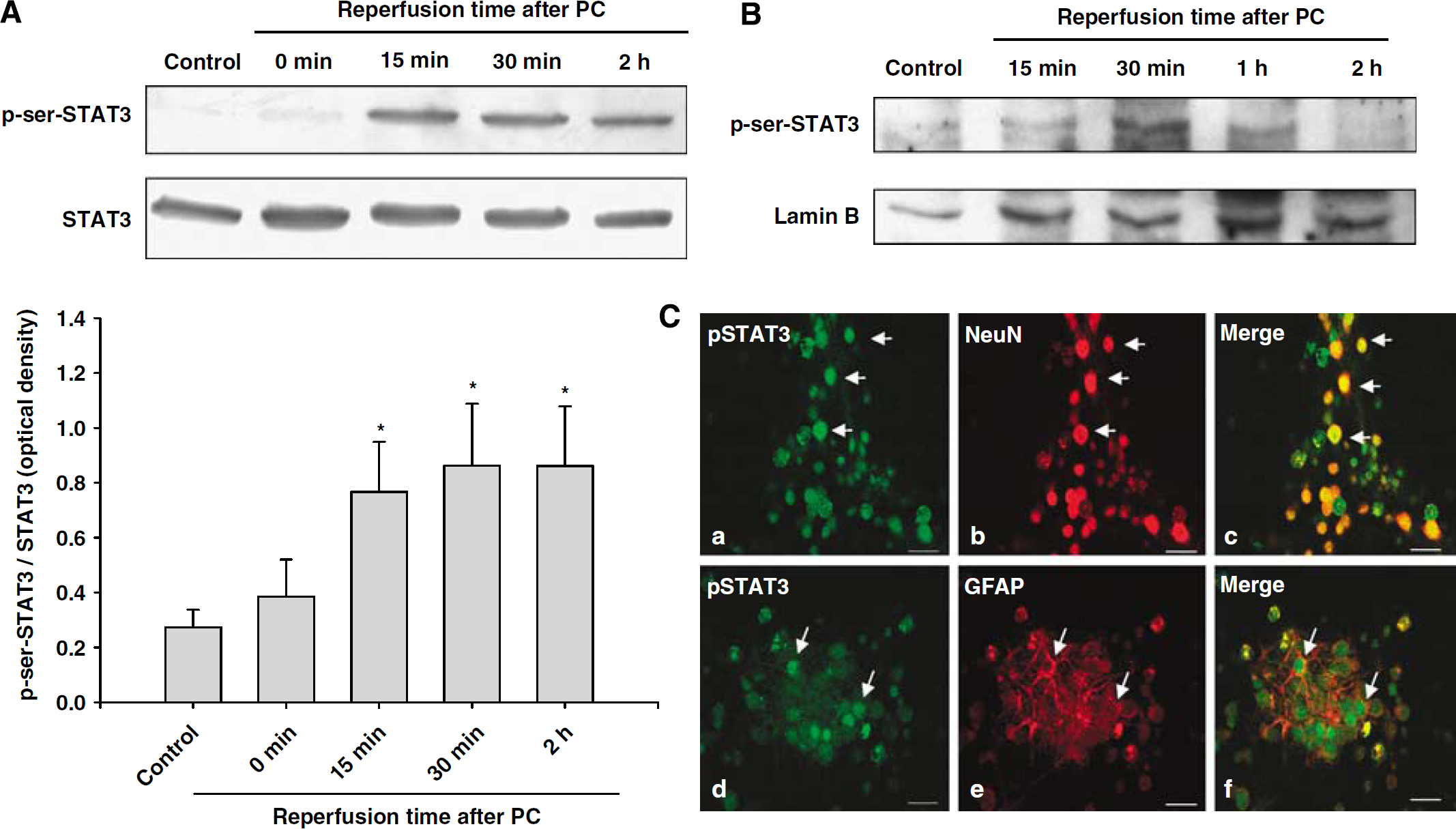
Preconditioning induced phosphorylation of STAT3 at the serine residue. (
As a transcription factor, p-STAT3 needs to be translocated into the nucleus to bind a specific DNA sequence. To determine whether the serine phosphorylated form of STAT3 translocated to the nucleus, we prepared the nuclear fraction of cultured neuronal cells and analyzed it for p-serine 727-STAT3 by western blots. Western blot analysis revealed that PC induced serine phosphorylation of STAT3 at 15 and 30 mins of reperfusion after PC in this fraction, indicating that the serine phosphorylated STAT3 translocated to the nucleus (Figure 2B).
To identify the cell type(s) in which STAT3 is being phosphorylated after PC, we performed immunocytochemistry for p-serine-STAT3 in mixed cortical neuron/astrocyte cell cultures. The results showed that the p-serine-STAT3 immunoreactivity was expressed in both neurons and astrocytes at 30 min of reperfusion after PC (Figure 2C).
In the heart, STAT3 activation via Janus kinase-dependent tyrosine phosphorylation pathways has been shown to be essential for PC-induced cardioprotection (Bolli et al, 2003). However, it is not known whether ischemic PC in neurons induces phosphorylation of STAT3 at the tyrosine residue. Therefore, we determined whether PC induces tyrosine phosphorylation of STAT3 similar to the serine phosphorylation of STAT3. Our results showed that PC induced phosphorylation of STAT3 at tyrosine 705 residue at 15 and 30 mins of reperfusion after PC (Figure 3A). These results suggested that PC induced phosphorylation of STAT3 at both serine and tyrosine residues.
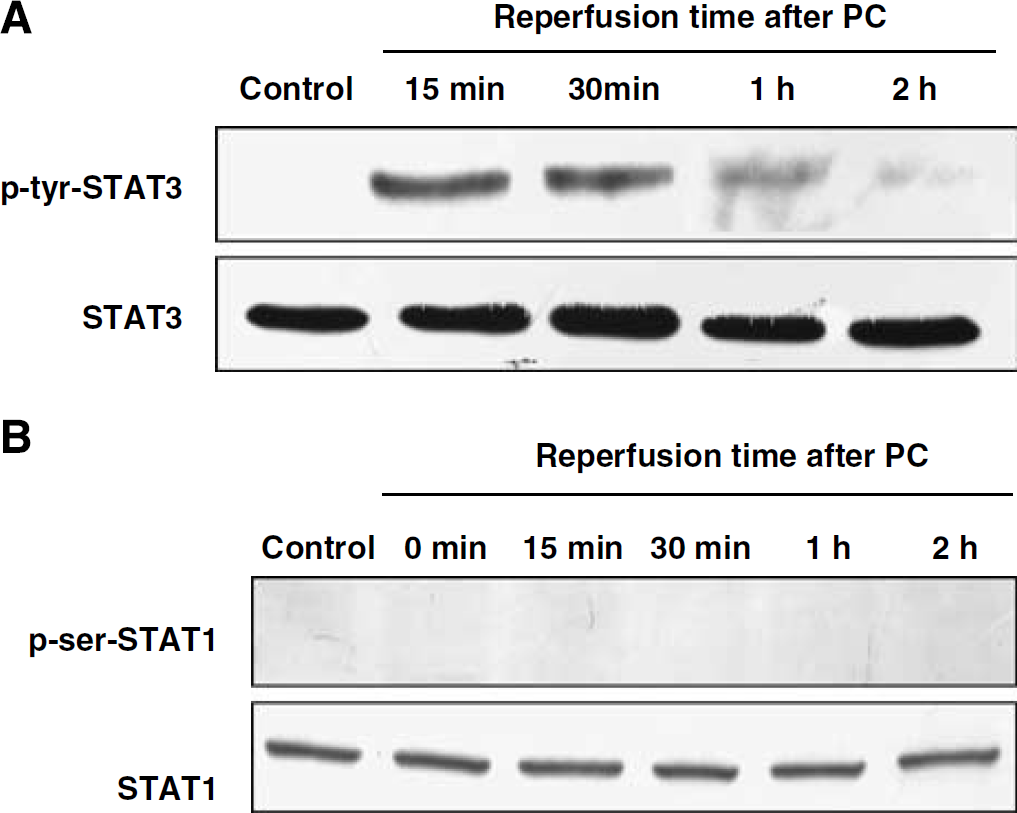
Preconditioning induced phosphorylation of STAT3 at the tyrosine residue, but not STAT1 at the serine residue. Cells were lysed immediately after 1 h of PC and at 15 mins, 30 mins, 1 h, and 2 h of reperfusion after 1 h of PC. Western blotting for p-tyrosine 705-STAT3 (
Next, we determined whether PC induced phosphorylation of STAT1 as well as STAT3. The analysis of western blots revealed no serine phosphorylation of STAT1 after PC (Figure 3B). To further validate whether the p-serine-STAT1 antibody we used (Cell Signaling Technology) was working, we confirmed phosphorylation of STAT1 by exposing cultured astrocytes to hydrogen peroxide (300 μmol/L) for 1 h as in a previous study (Gorina et al, 2005) (data not shown). The results suggest that the PC-induced signal transduction pathway requires the phosphorylation of STAT3 but not STAT1 at a serine residue.
εPKC Activation is Involved in the Phosphorylation of STAT3 at Serine 727 Residue After Preconditioning
Previously, we have reported that activation of εPKC plays a key role in the PC signaling cascade in the organotypic hippocampal slice and cell cultures by activating the ERK1/2 pathway (Kim et al, 2007; Lange-Asschenfeldt et al, 2004; Raval et al, 2003). Thus, we tested the hypothesis that serine/threonine kinase εPKC is an upstream regulator of STAT3 phosphorylation at the serine residue after PC. To characterize this signaling pathway, we inhibited εPKC activation with an εPKC-specific inhibitory peptide (εV1–2, 100 nmol/L) during PC and reperfusion in cell cultures. Inhibition of εPKC activation reduced PC-induced phosphorylation of STAT3 at serine 727 residue, 15 and 30 mins after PC (
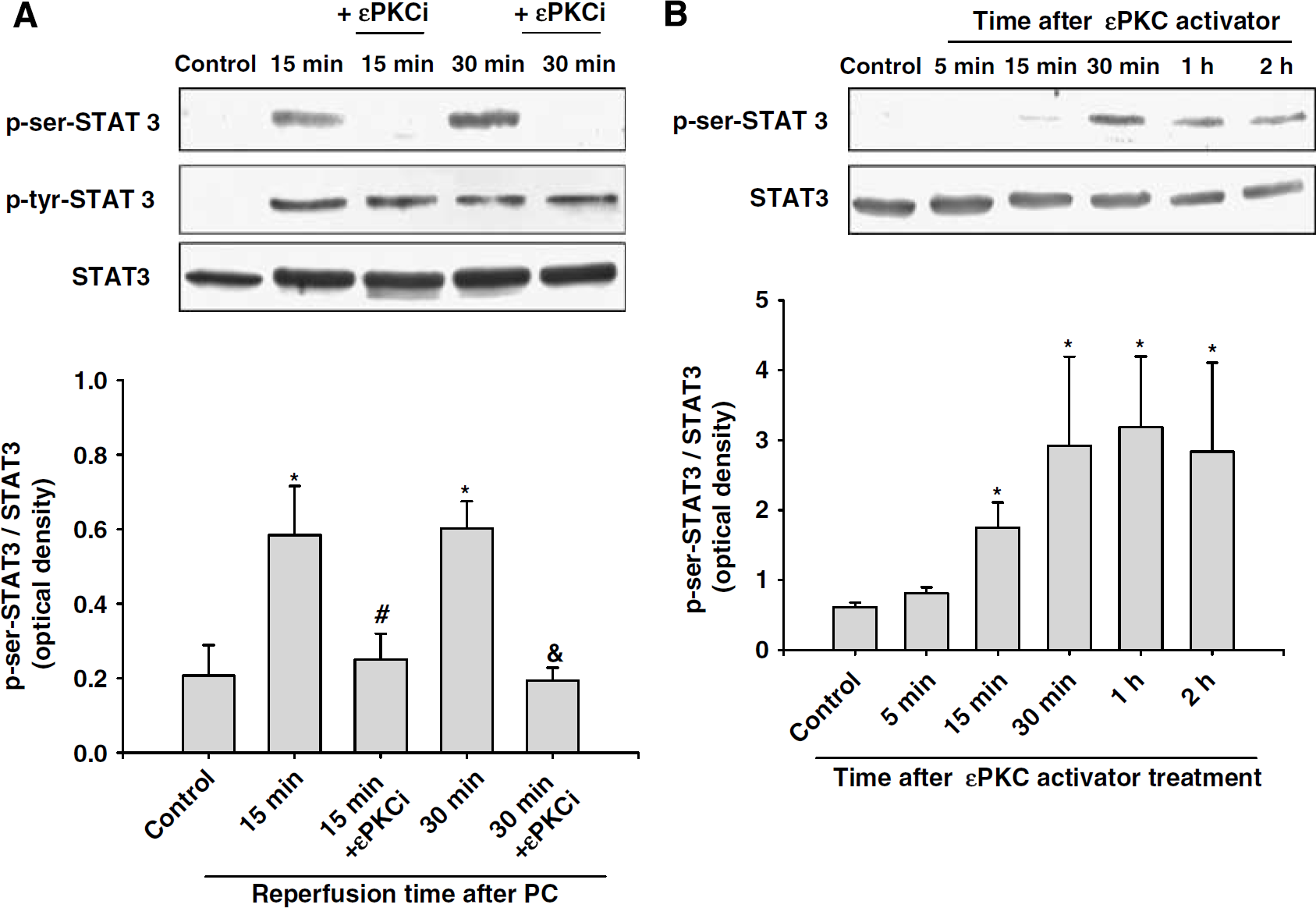
Activation of εPKC is involved in the phosphorylation of STAT3 at the serine residue after PC. (
ERK1/2 Activation is Involved in the Phosphorylation of STAT3 at Serine 727 Residue After Preconditioning
In a previous study, we showed that εPKC activation led to phosphorylation of ERK1/2 in neuronal cell cultures (Kim et al, 2007). In the next series of experiments, we tested whether PC induced phosphorylation of ERK1/2 and whether phosphorylated ERK1/2 in turn phosphorylated STAT3 at serine 727 residue. Preconditioning induced phosphorylation of ERK1/2 at 15 mins, which peaked at 30 mins and persisted for 1 h (Figure 5A). Next, we investigated whether blockade of ERK1/2 phosphorylation reduced PC-induced phosphorylation of STAT3. For this purpose, ERK1/2 activation was inhibited in cell cultures by administration of the MAPK-K inhibitor PD98059 (10 μmol/L) during PC and reperfusion. Inhibition of ERK1/2 activation completely inhibited PC-induced serine phosphorylation of STAT3 at all the time points under investigation (Figure 5B). However, inhibition of ERK1/2 during and after PC showed no effect on the tyrosine phosphorylation of STAT3 (Figure 5B).
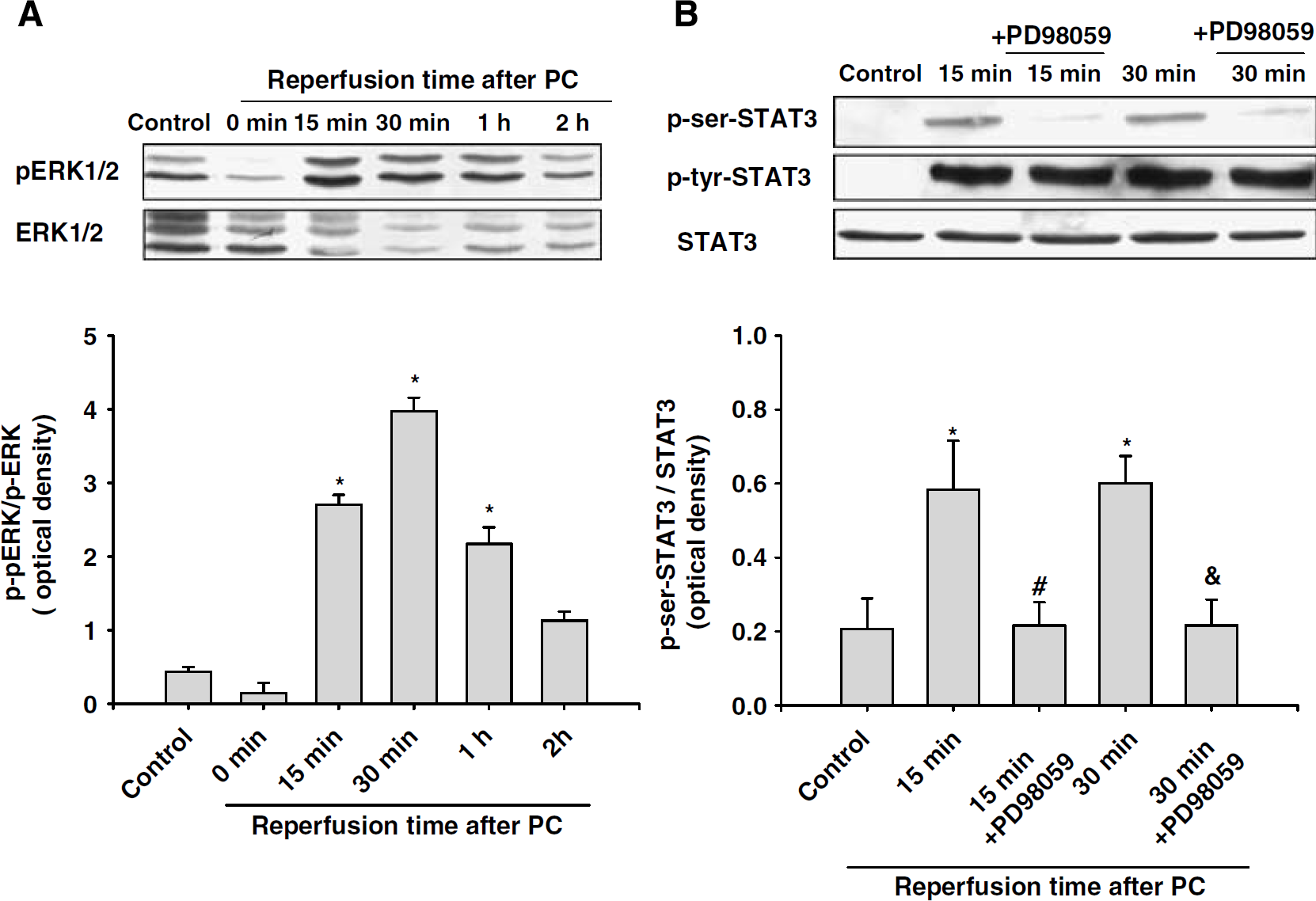
Preconditioning induced ERK1/2 phosphorylation and inhibition of ERK1/2 activation reduced PC-induced phosphorylation of STAT3 at the serine residue. (
Neither ERK1/2 nor εPKC Directly Interacts with STAT3 After Preconditioning
Our results suggested that PC induced phosphorylation of STAT3 through εPKC and ERK1/2 activation. However, the direct interaction of either ERK1/2 or εPKC with STAT3 still remained undefined. To determine whether there is a direct interaction between ERK1/2 and STAT3, we performed immunoprecipitation experiments followed by western blotting. In this experiment, cell culture protein extracts were isolated at 5, 15, and 30 mins after 1 h of PC. The samples were immunoprecipitated with pERK1/2 antibody, and for western blotting p-serine 727-STAT3 antibody was used. The co-immunoprecipitation experiments showed that phosphorylated ERK1/2 did not directly interact with phosphorylation of STAT3 (Figure 6A). Next, we determined whether there was a direct interaction between εPKC and STAT3 using immunoprecipitation with εPKC antibody and western blotting analysis with p-serine 727-STAT3 antibody. This experiment also failed to show a direct link between εPKC and STAT3 phosphorylation (Figure 6B). To further confirm our results, we repeated the immunoprecipitation experiments using both polyclonal and monoclonal antibodies for p-serine 727-STAT3. Regardless of the source of antibody, the results were identical to those for co-immunoprecipitation. These results indicated that neither ERK1/2 nor εPKC directly interacted with STAT3, suggesting the role of another serine kinase in phosphorylating serine 727 of STAT3, which in turn should be activated by ERK1/2 or εPKC.
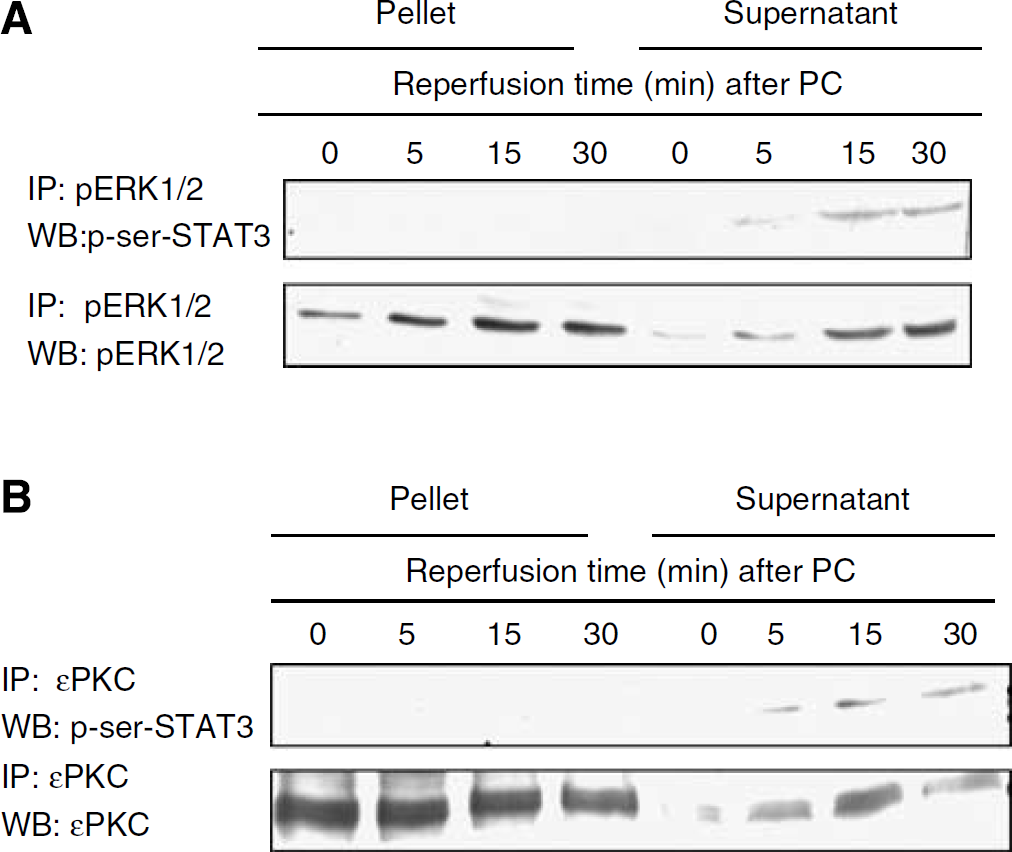
Neither ERK1/2 nor εPKC directly interacted with STAT3. Cells were lysed immediately at 5, 15, and 30 mins of reperfusion after 1 h of PC. (
STAT3 Activation Mediates COX-2 Induction and Confers Neuroprotection
Recently, we showed that ischemic PC induced COX-2 via the εPKC → ERK1/2 pathway in an
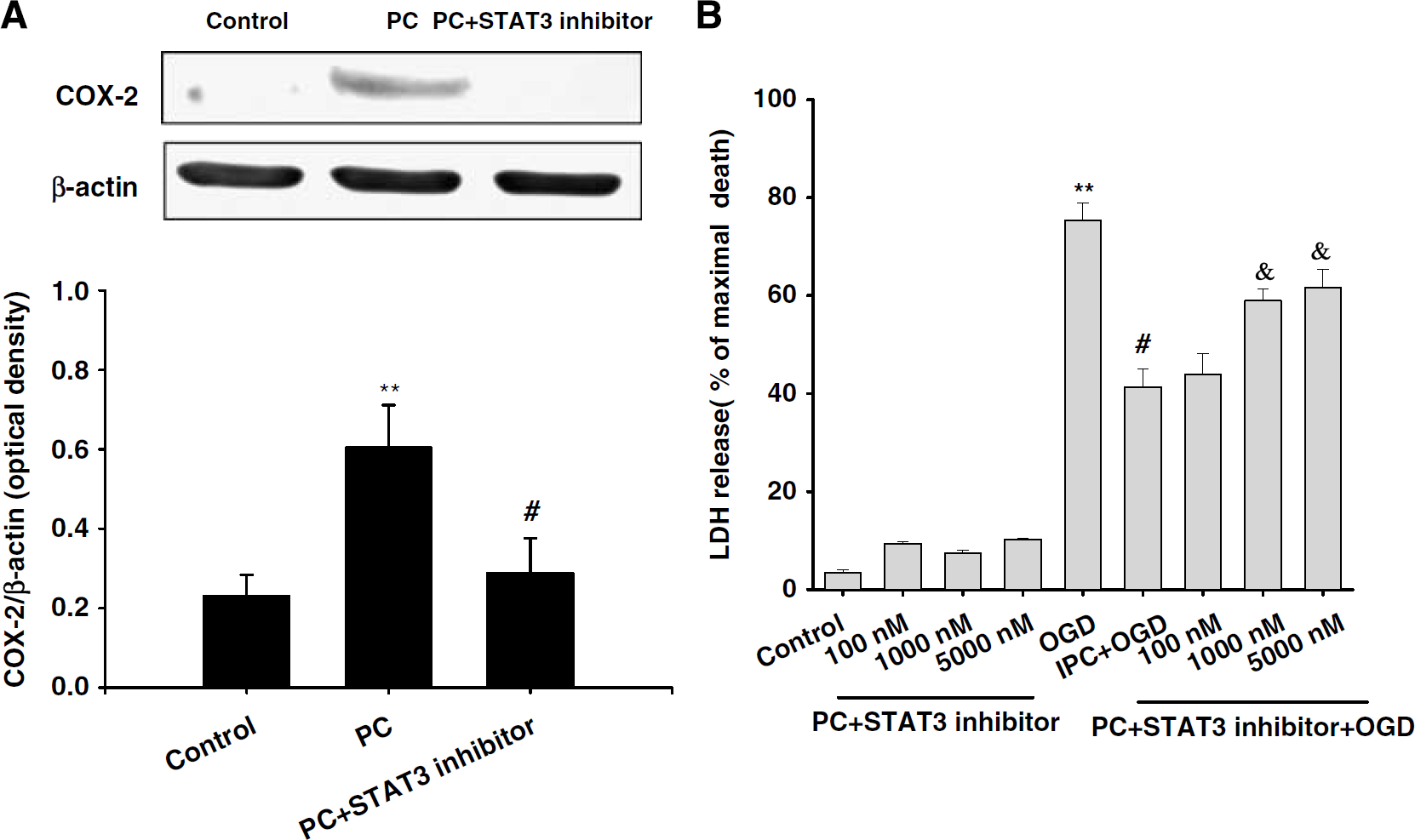
Inhibition of STAT3 activation reduced PC-induced COX-2 expression and neuroprotection. (
Discussion
This is the first study, to the best of our knowledge, to show that STAT3 activation mediated COX-2 expression after sublethal OGD (PC paradigm) and played a critical role in the protection against OGD in neurons. The main findings from our study include (1) the signal transduction pathway initiated by PC requires STAT3 phosphorylation in cortical neuron/astrocyte cell cultures, (2) εPKC activation and ERK1/2 indirectly mediated STAT3 phosphorylation at a serine residue after PC, and (3) STAT3 activation was crucial for COX-2 expression induced by PC and conferred neuroprotection.
In general, the transcriptional activity of the STATs involves their dimerization, nuclear translocation, DNA binding, and recruitment of transcriptional coactivators (Klampfer, 2006; Vinkemeier, 2004). The process of dimerization requires phosphorylation of STAT at tyrosine 705 residue by the Janus kinases and an additional phosphorylation at serine 727 residue by members of the MAP kinase family for complete activation (Aziz et al, 2007; Darnell, 1997; Li and Shaw, 2004). The dimerized STAT then undergoes nuclear translocation (Klampfer, 2006; Vinkemeier, 2004). The tyrosine phosphorylation of STAT mediated by Janus kinase signaling pathways has been shown to be essential for late ischemic PC in the heart (Bolli et al, 2003). However, a study has shown that activation of STAT1 and STAT3 requires two phosphorylations at both tyrosine and serine residues after ischemic PC in the heart. Cyclooxygenase-2 was upregulated by STAT1 and STAT3 phosphorylation at both tyrosine and serine residues (Xuan et al, 2005). In agreement with this study, our study showed that PC induced phosphorylation of STAT3 at both serine and tyrosine residues and inhibition of STAT3 activation reduced COX-2 upregulation, which mediated PC neuroprotection. The STAT3-specific inhibitor used in this study was the SH2 domain-binding peptide, which interferes with phosphorylation of the tyrosine residue, and disrupts STAT3 dimerization and STAT3 activity
The role of STAT activation in ischemic PC protection and apoptosis has been well documented in the heart (Butler et al, 2006; Hilfiker-Kleiner et al, 2004; Jacoby et al, 2003; Yamaura et al, 2003); however, it remains to be explored in neurons. In isolated perfused hearts of rats, ischemic PC induced STAT3, STAT5, and STAT6 activation (Yamaura et al, 2003). Several studies using knockout mice for STAT3 and STAT5A showed an increased susceptibility to myocardial ischemic reperfusion injury (Hilfiker-Kleiner et al, 2004; Jacoby et al, 2003; Yamaura et al, 2003). Thus, it is agreed that STAT3 and STAT5A activation resulted in cardioprotective effects of myocardial ischemic PC. In contrast to STAT3 or STAT5A, STAT1 plays a role in apoptotic cell death in cardiomyocytes (Stephanou et al, 2001). STAT1 has been shown to enhance proapoptotic genes encoding caspase-1, Fas, and Fas ligand and also inhibit antiapoptotic genes encoding Bcl-2 and Bcl-X proteins and to promote cardiac cell death (Stephanou et al, 2000, 2001). Our findings showing phosphorylation of STAT3 but not STAT1 after PC indicated that STAT3 phosphorylation plays an important role in the neuroprotective effects of ischemic PC in the brain. In contrast, a recent study showed STAT3 activation after ischemia alone in the brain (Dziennis et al, 2007). In this context, a number of other neuroprotective signaling pathways activated by ischemic PC appear to be activated by ischemia as well (e.g., HSP70, BDNF, etc.; Dirnagl et al, 2003). Although not completely defined, the activation of these neuroprotective signaling pathways did not appear to be sufficient to overcome cell death pathways, suggesting that the differences in the role of STAT3 after PC and ischemia are worth exploring.
In our previous study, we showed that εPKC mediated ischemic PC neuroprotection
Recently, several studies have reported that genes involved in cell protection against apoptosis are regulated by STAT signaling (Bhattacharya et al, 2005; Harada et al, 2005; Zhang et al, 2007). Harada et al (2005) reported that in transgenic mice expressing dominant-inhibitory STAT3, Bcl-2 and Bcl-xL upregulation mediated by granulocyte colony-stimulating factor was diminished in cardiomyocytes (Harada et al, 2005). This suggested that STAT3 could regulate antiapoptotic genes Bcl-2 and Bcl-xL. In hippocampal neurons, Zhang et al (2007) showed that STAT5 phosphorylation occurred after erythropoietin stimulation, which upregulated antiapoptotic genes such as Bcl-xL and X-linked inhibitor of apoptosis (Zhang et al, 2007).
One of the protective pathways downstream of STAT activation involves COX-2. STAT3 has been shown to bind the interferon gamma-activated sequence motifs on COX-2 promoter and be essential for COX-2 transcriptional activity in human colonic epithelial cells (Koon et al, 2006). Xuan et al (2007) reported that COX-2 and endothelial nitric oxide expression through STAT1 and STAT3 activation after ischemic PC resulted in cardioprotection against ischemic injury. Furthermore, Wang et al (2007b) reported that upregulation of the inflammatory cytokine tumor necrosis factor-α mediated late PC cardioprotective effects, showing COX-2 and inducible nitric oxide synthase expression by STAT3 in unstable angina patients (Wang et al, 2007b). In neurons, cortical spreading depression induces COX-2 expression and confers neuroprotection against ischemia (Horiguchi et al, 2006). In our previous study using cortical neuron/astrocyte cultures, we showed that PC induced COX-2 expression in neurons but not in astrocytes and that COX-2 activation is crucial for induction of ischemic tolerance (Kim et al, 2007). In this study, we characterized that STAT3 phosphorylation was expressed in both neurons and astrocytes after PC. These results suggest a possible interaction between neurons and astrocytes, which resulted in STAT3 activation and COX-2 expression in neurons and development of ischemic tolerance.
Finally, the role of COX-2 activation and COX-2-derived prostaglandins in neuroprotection or neurotoxicity is still controversial. It is likely to be dependent on different cell types and on different intensity of injury. Exogenous prostaglandin E2 administration leads to neuroprotection in cerebral ischemia (McCullough et al, 2004). In cortical neurons, COX-2-induced prostaglandin E2 release promoted neuroprotection against OGD in a PC model (Gendron et al, 2005). Thus, COX-2 could be one of the protective target genes regulated by STAT3 after PC even though the downstream effectors of COX-2 expression, prostaglandins, after ischemic PC need to be defined.
In conclusion, PC induced by sublethal OGD increased the phosphorylation of STAT3, which is mediated by εPKC and ERK1/2 activation in cortical neuron/astrocyte cell cultures. This study also showed that neither εPKC nor ERK1/2 directly phosphorylated STAT3 after PC, suggesting the requirement of a yet undefined intermediate kinase. Finally, our findings showed that STAT3 activation induced COX-2 expression after PC and conferred neuroprotection against OGD.
Footnotes
Acknowledgements
The authors thank Dr Beata Frydel (Miami Project Core Facilities) for her encouraging guidance on use of confocal microscope.