
Abstract
ORP150—150-kd oxygen-regulated protein—is a novel stress protein localized in the endoplasmic reticulum (ER). To investigate the role of ORP150 in delayed neuronal cell death, the authors examined its expression in the gerbil brain after an ischemic insult. The expression of ORP150 antigen, as well as its transcripts, was observed in the CA1 region after the occlusion of the common carotid artery, and the preconditioning enhanced this expression. In cultured neurons, exposure either to hypoxia or to glutamate induced the expression of ORP150, and this effect was also observed by treating the culture with breferdin A or thapsigargin, indicating that both glutamate and hypoxia can cause stress in the ER (ER stress). Neurons became more vulnerable to these stresses following treatment with cycloheximide or after infection with an adenovirus carrying the ORP150-antisense structure. In contrast, the overexpression of ORP150 by an adenovirus suppressed neuronal cell death, and this was accompanied by the suppression of Ca2+ elevation and proteolytic activity induced by glutamate. Further, overexpression of ORP150 in CA1 neurons by an adenovirus carrying the ORP150-sense structure suppressed delayed neuronal cell death after ischemia. These data suggest a possible function of ORP150 as an intracellular apparatus that participates in a protective response in ischemic tolerance.
As a common denominator, oxygen deprivation (hypoxia) plays a central role in the pathogenesis of cerebral ischemia (Siesjoe, 1988). In cultured cells, hypoxia induces a set of stress proteins, termed oxygen-regulated proteins (ORPs) (Heacock and Sutherland, 1986), as well as a shift in the energy metabolism to anoxic process (Niitsu et al., 1999). The ORPs overlap with proteins induced by glucose deprivation (GRPs), and most ORPs/GRPs are located in the endoplasmic reticulum (Lee, 1992), suggesting that stress caused by an environmental decline either in oxygen tension or in the glucose level specifically targets the intracellular organelle, the ER.
Among the constituents of the central nervous system, neurons demonstrate a marked vulnerability in altered environments (Siesjoe, 1988). Some of their subtypes show a more enhanced vulnerability to environmental stresses that do not necessarily exert any toxic effect on other cell types. Furthermore, some neuron subtypes show an enhanced vulnerability to other subtypes of neurons. The hippocampal pyramidal cells, for instance, demonstrate a phenotype that is more sensitive to both ischemia and excitotoxicity (Monaghan et al., 1983; Wieloch, 1985). In these cells, the access binding of glutamate to its receptors, especially under ischemic conditions (Felipo et al., 1998; Mitani and Kataoka, 1991), leads to an impairment of intracellular Ca2+ homeostasis, which finally triggers the activation of several cell death pathways (Coyle and Puttfarchen, 1993; Kitao et al., 2001; Lee et al., 2000; Mody and Macdonald, 1995). Against this glutamate excitotoxicity, the stress response is initiated in the ER, which is represented by the expression of ER-located stress protein (Yu et al., 1999). Exposure of cultured neurons to glutamate induces expression of the major stress protein in the ER, the 78-kd glucose-regulated protein GRP78, suggesting that neurons respond to excitotoxicity as a stress response to protect the ER. Since glutamate-calcium axis plays a central role in neuronal cell death in neurodegenerative diseases and in brain ischemia, the response to the stress in the ER (ER stress) could be a novel target for the regulation of ischemia-induced neuronal cell death.
The delayed neuronal cell death (DND) observed in the gerbil hippocampus after a short and deep ischemia is characterized by a slow progressive cell death, which can be rescued by a prior nonlethal ischemic insult (Kirino and Sano, 1984; Kirino et al., 1992; Kitagawa et al., 1990). These observations suggest that DND is the final outcome in a series of stress responses that could have saved the neurons. Furthermore, the importance of the glutamate-calcium axis on DND substantiates the role of ER-stress proteins as a deference mechanism in neurons. These considerations led us to hypothesize that any stress protein whose expression is enhanced by the preconditioned state might suppress DND, a more chronically progressive death. ORP150 (150-kd ORP) was identified from cultured astrocytes based upon a phenotype showing marked resistance to ischemic stress (Kuwabara et al., 1996). Expression of ORP150 is essential to maintain cellular viability under hypoxia (Ozawa et al., 1999), and neurons that overexpress ORP150 show resistance to acute ischemic damage (Tamatani et al., 2001).
In this report, we investigated whether the stress response in the ER occupies a central part in the defense mechanisms of neurons against DND. Such a finding raises the possibility that this intracellular organelle can regulate neuronal cell death in ischemic brain.
MATERIALS AND METHODS
Introduction of delayed neuronal cell death in gerbil brain
Delayed neuronal cell death was introduced in Mongolian gerbils (20–24 weeks old) as described previously (Matsuyama et al., 1993). Briefly, after the animals were lightly anesthetized with inhaled ether, a bilateral cerebral ischemia was produced by bilateral occlusion of the common carotid arteries. Body temperature was maintained at 36.5°C to 37°C during the ischemic insult, and for at least 1 hour following the ischemic period. Animals (n = 5 per group) were divided into a sham-operated group (control), a group that received a single ischemic insult for either 2 or 5 minutes, and a group that received a 2-minute ischemic insult followed by 5-minute ischemia, which was performed 24 hours after the first insult.
Histochemistry
Twenty-four hours after the final ischemic insult, the animals were perfused with a 4% paraformaldehyde solution under deep anesthesia, and 20-mm-thick coronal brain sections were cut using a vibratome. Sections were processed for immunohistochemistry using either monoclonal mouse anti—microtubule-associated protein 2 (MAP2; 1:1,000 dilution; Sigma, St. Louis, MO, U.S.A.) or rabbit anti-ORP150 antibody (5 μg/mL) (Matsushita et al., 1998). For detection of the administered adenovirus containing LacZ, immunocytochemistry for β-galactosidase was performed using a polyclonal rabbit antibody (1:1,000 dilution; ICN-Cappel, Aurora, OH, U.S.A.). The binding site of the primary antibody was visualized by avidin-biotin-peroxidase complex method, followed by the semiquantitative analysis (Matsuyama et al., 1993). In brief, the number of ORP150 immunoreactive neurons was counted in the CA1 pyramidal layer by a blind observer (12 slices per animal, n = 5 per group). Results were presented as percentage of total pyramidal neurons.
In situ hybridization
Animals were divided into control, 2-minute ischemia, and 5-minute ischemia groups (n = 5 per group). In situ hybridization analysis was performed on the ischemic gerbil brain during the 24-hour reperfusion period as described previously (Matsushita et al., 1998). In brief, a rat ORP150 cDNA fragment (151–381) was subcloned into the EcoRV site of the pBluescript KS vector (Stratagene Inc., La Jolla, CA, U.S.A.). After linearization of the plasmid, digoxigenin-labeled single-stranded RNA was synthesized by T7 and T3 RNA polymerase with digoxigenin-UTP as well as unlabeled ATP, GTP, and CTP. Hybridization of ORP150 mRNA was performed at 50°C for 16 hours, and signals were localized using the Nucleic Acid Detection Kit (Roche Molecular Biochemicals, Mannheim, Germany).
Cell culture of hippocampal neurons
Cultured hippocampal neurons were prepared from newborn C57BL/6 mice (Japan SLC, Inc.) within 1 day of birth, as described previously (Kitao et al., 2001). In brief, the hippocampus was separated and incubated in calcium- and magnesium-free Hank's balanced salt solution containing papain (0.2%). Cells were plated in plastic wells precoated with poly-L-lysine (10 μg/mL), and the cultures were then maintained for 3 days in minimum essential medium supplemented with heat-inactivated fetal calf serum (10%), glucose (30 mmol/L), L-glutamine (2 mmol/L), pyruvate (1 mmol/L), KCl (20 mmol/L), sodium bicarbonate (10 mmol/L), and HEPES (1 mmol/L, pH = 7.2).
Construction of adenovirus and their applications
A replication-deficient recombinant adenovirus containing the cDNA for ORP150 was constructed according to the COS/TPC method as described elsewhere (Miyake et al., 1996). Control adenovirus carrying β-galactosidase (Ad/LacZ) and GFP (Ad/GFP) were obtained from The Institute of Physical and Chemical Research (Tokyo, Japan).
In primary hippocampal neuron culture, infection was carried out by adding recombinant Ad(s) to serum-containing medium. The cells were incubated at 37°C for 60 minutes with constant agitation. The medium was changed, and the cells were further incubated at 37°C for 48 hours before exposure to hypoxia or treatment with chemical materials (Tamatani et al., 1999).
In the gerbil ischemic model, a mixture of Ad/ORP150S and Ad/LacZ (5 × 107 pfu each) was directly administered into the gerbil CA1 region as described previously (Yagi et al., 2000) 5 days prior to the ischemic insult. As the control experiment, Ad/LacZ alone (108 pfu) was administered in the CA1 of the other hemisphere in the same animal.
Assessment of cell death in vitro
Cell viability after hypoxic or chemical challenge was assessed with morphologic criteria (Yu et al., 1999). In brief, a neuron with intact neurites and cell body that was smooth and round-to-oval was considered viable. A neuron with fragmented neurites and a cell body that was shrunken and rough in appearance was considered nonviable.
Assessment of cell death in vivo
Neuronal cell viability was assessed in five adenovirus-treated animals according to a previously reported method, with some modification (Hoehn et al., 2001). Seven days after the ischemic onset, gerbil brain was perfusion-fixed with 4% PFA and postfixed with 4% PFA for 1 to 2 days. Twenty-micrometer frozen sections in the coronal plane were taken at 100-μm increments 1 mm anterior and posterior to the needle track. Slices were first subjected to immunohistochemistry for β-galactosidase to select the slice that showed the maximal immunoreactivity in CA1 pyramidal layer, and the region of interest (ROI)—a 1-mm span in the pyramidal layer of the slice where β-galactosidase showed the maximal spread—was determined. The adjacent section was stained with MAP2 antibody, and MAP2-positive neurons were counted by a blinded observer in 100-μm spans from the center to the lateral or central direction in the ROI. Data are shown as both peak and mean number of MAP2-positive neurons in the ROIs.
Western blot
Levels of ORP150 antigen in brain tissue or primary neuron culture were determined by immunoblotting as described previously (Kuwabara et al., 1996; Matsushita et al., 1998). Hippocampal tissue obtained from the four groups of animals (three independent experiments in duplicate) was separated 24 hours after the last ischemic insult, homogenated in 1 mL phosphate-buffered saline (PBS) containing NP-40 (1%), and subjected to Western blot analysis. Where indicated, Western blotting with anti-KDEL (StressGen biotechnologies) or anti–β-actin (Sigma, St. Louis) monoclonal antibody was used to assess the levels of GRP78, GRP94, or β-actin, as described previously (Ozawa et al., 2001; Tamatani et al., 1999). The ORP150 level in brain tissue was semiquantitatively determined by densitometry analysis. In each case, total protein content in the mixture was determined by the method of Lowry et al. (1951).
Measurement of intracellular calcium level
Measurement of the intracellular calcium in cultured neurons was performed as described previously (Wahl et al., 1989). After infection with the adenovirus, cultured hippocampal neurons in 96-well plates (≈3 × 105 cells) were washed three times in Locke's buffer and then incubated at 20°C for 15 minutes with 25 μL Fluo-3/AM (final concentration, 20 μmol/L) dissolved in HEPES-buffered saline (20 μmol/L, pH = 7.4). Cells were then washed with HEPES-buffered saline and the fluorescence was recorded at 37°C with a Fluoroscan II (Labsystems) microtiter fluorometer using fluoroscan filter pairs (excitation wavelength, 485 nm; emission wavelength, 533 nm).
The calibration was performed on cells incubated with the ionophore A-23187 (10 μmol/L) to obtain the Fmax and with CaCl2 (2 mmol/L) and A-23187 (10 μmol/L) dissolved in NaCl (0.9%) to obtain the Fmin. Cytosolic-free Ca2+ was calculated by the following formula:
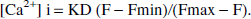
Activation of cathepsin B activity
Activation of Ca2+-dependent proteinase was assessed as described elsewhere (Tsuchiya et al., 1999). To evaluate cathepsin B activity, the culture homogenate in PBS (10 μL) was incubated in sodium acetate buffer (2.5 μL, 400 mmol/L, pH = 5.5) containing cysteine (8 mmol/L) at 37°C for 5 minutes. Synthetic peptide substrate, Z-Arg-Arg-AMC (1 mmol/L; Peptide Research Institute, Japan), was added and the mixture was incubated for a further 5 minutes at 37°C. The reaction was terminated by the addition of SDS (5% final concentration) to the sample and dilution in Tris-HCl (2 mL, 100 mmol/L). Release of 7-amino-4-methylcoumarin (AMC) was measured by fluorescence spectrophotometry using the wavelengths of 370 nm (excitation) and 460 nm (emission). Where indicated, an inhibitor of μ-calpain (ZLLLal, 10 μmol/L; Tsubuki et al., 1996) was added to the mixture prior to the assay.
Data analysis
Statistical analysis was performed using the nonpaired t-test or ANOVA followed by multiple comparison analysis using the Newman-Keuls equation. Where indicated, the data were analyzed by a two-way ANOVA followed by the multiple contrast analysis. For nonparametric data, Kruskal-Wallis analysis or χ2 analysis was applied.
RESULTS
Expression of ORP150 in gerbil hippocampus
The basal expression of ORP150 in hippocampus of control mice was low, and its expression in animals subjected to a 2-minute temporal occlusion became evident (an approximate fivefold increase compared with controls). In animals treated with a 5-minute ischemia, the expression was decreased compared with 2-minute ischemia, whereas pretreatment of the animals with a nonlethal ischemia (2 minutes, in this case) enhanced the expression of ORP150 antigen after a 5-minute occlusion (an approximate eightfold increase compared with controls) (Figs. 1A and 1B). Immunohistochemical analysis using an anti-ORP150 antibody revealed the enhanced expression of ORP150 antigen in pyramidal cells (Fig. 1C– Fig. 1G), and this was in parallel with the observations by Western blot analysis. In situ hybridization analysis using a cRNA probe demonstrated the expression of ORP150 transcripts following the ischemic insult. At the basal level, the ORP150 transcript was only slightly expressed in pyramidal cells of the CA1 region (Figs. 1H and 1K). The expression of ORP150 message was more clearly detected after a 2-minute occlusion of the carotid artery (Figs. 1I and 1L), and it was most remarkable after a 5-minute occlusion (Figs. 1J and 1M).
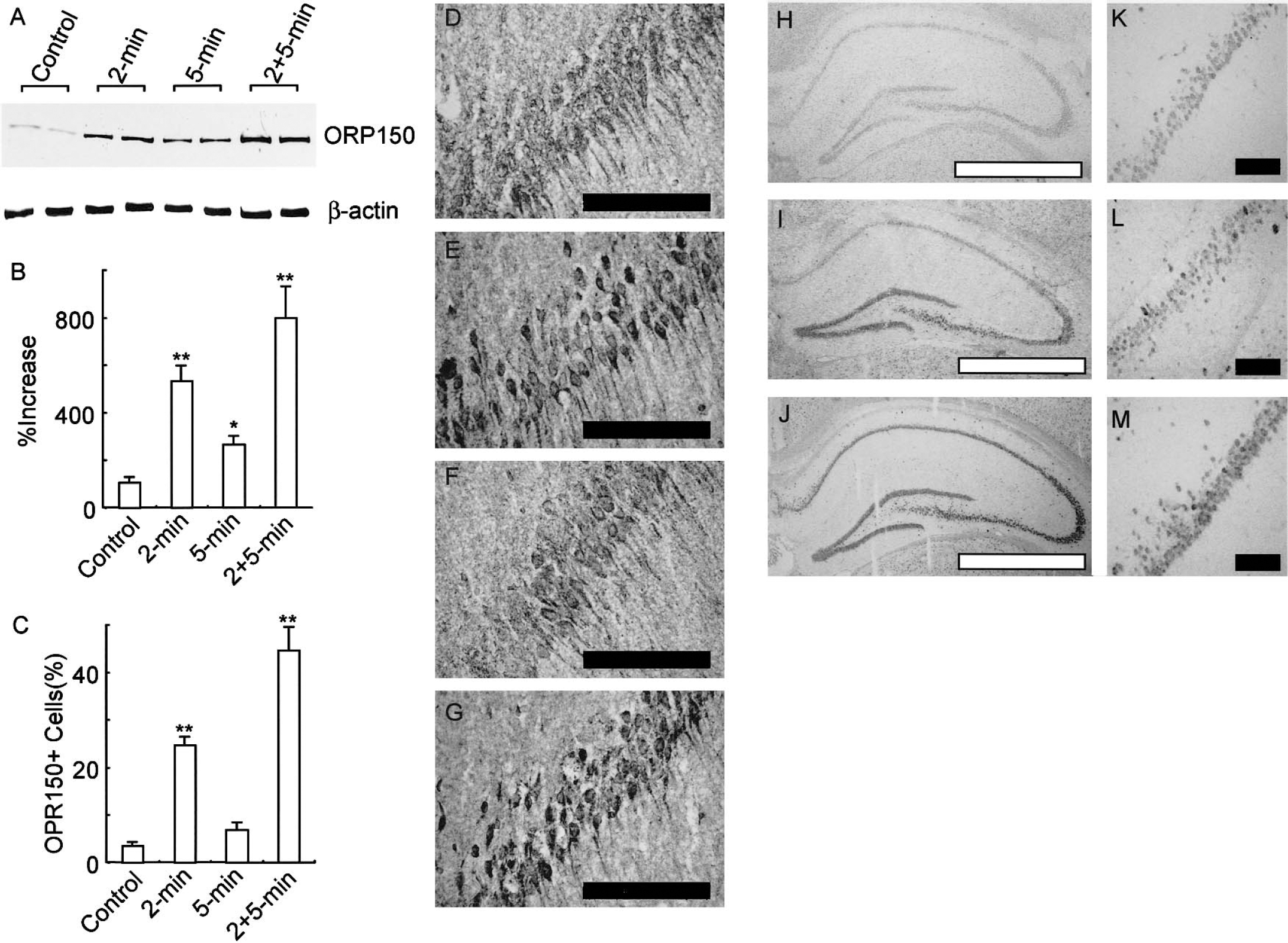
Expression of ORP150 in the gerbil hippocampus after an ischemic insult.
Expression of ORP150 in cultured hippocampal neurons
These in vivo observations suggest a possible relationship between ORP150 expression and the tolerance of neurons in DND. In cultured neurons, both exposure to hypoxia and treatment with glutamate induced the expression of ORP150 (Fig. 2A). In each case, pretreatment with cycloheximide (0.1 μg/mL), which suppresses protein synthesis as assessed by the incorporation of 3H-leuscine (by ≈30% compared with control cultures), inhibited the expression of ORP150 (Fig. 2A, lanes Cx + Hypoxia and Cx + Glutamate). At the same time, pretreatment of cultures with cycloheximide accelerated the cell death under both glutamate and hypoxia (Figs. 2B and 2C), suggesting a central role for protein synthesis in the stress response of neurons to hypoxia and glutamate.
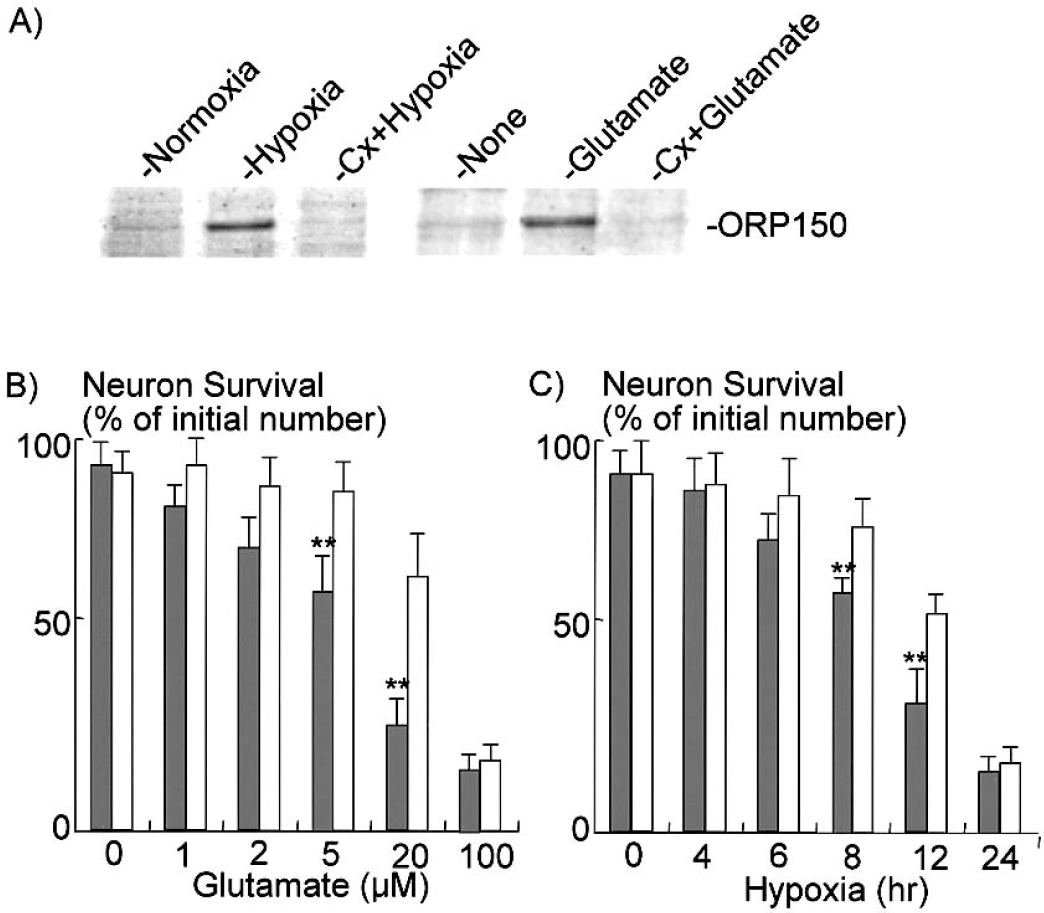
Inhibition of protein synthesis and neuronal cell death in vitro. Hippocampal neurons were prepared from newborn mice and cultured on six-well plates (≈104 cells).
To focus on the mechanism of ORP150 induction in cultured neurons, the expression of ORP150 was assessed after the sublethal chemical stimuli. Exposure of cultured hippocampal neurons to both hypoxia and glutamate induced the expression of ORP150, which reached a maximum 8 to 12 hours after the onset of stimuli (Fig. 3A to Fig. 3I, III). Exposure of cultures to other chemical stimuli including breferdin A (5 ng/mL; Fig. 3A to Figs. 3I, 3III) and thapsigargin (0.3 μg/mL; Fig. 3A—V), which cause ER-stress (Low et al., 1991; Yu et al., 1999), also induced the expression of ORP150. In contrast, other chemical stressors including NaAs (100 μmol/L; Uto et al., 1995), staurosporine (1 μmol/L; Deshmukh and Johnson, 2000), and hydrogen peroxide (10 μmol/L; See and Loeffler, 2001), failed to induce ORP150 in cultured neurons (Figs. 3A—V, 3VI, 3VII). Several doses of chemical materials were added to hippocampal neurons and the similar results were obtained (Fig. 3B). These data suggest that expression of ORP150 in cultured neurons represents their response to maintain the function of the ER. The expression of ORP150 in the ischemic hippocampus, therefore, represents the existence of environmental alterations in ischemic hippocampus, which, at least in part, cause ER stress.
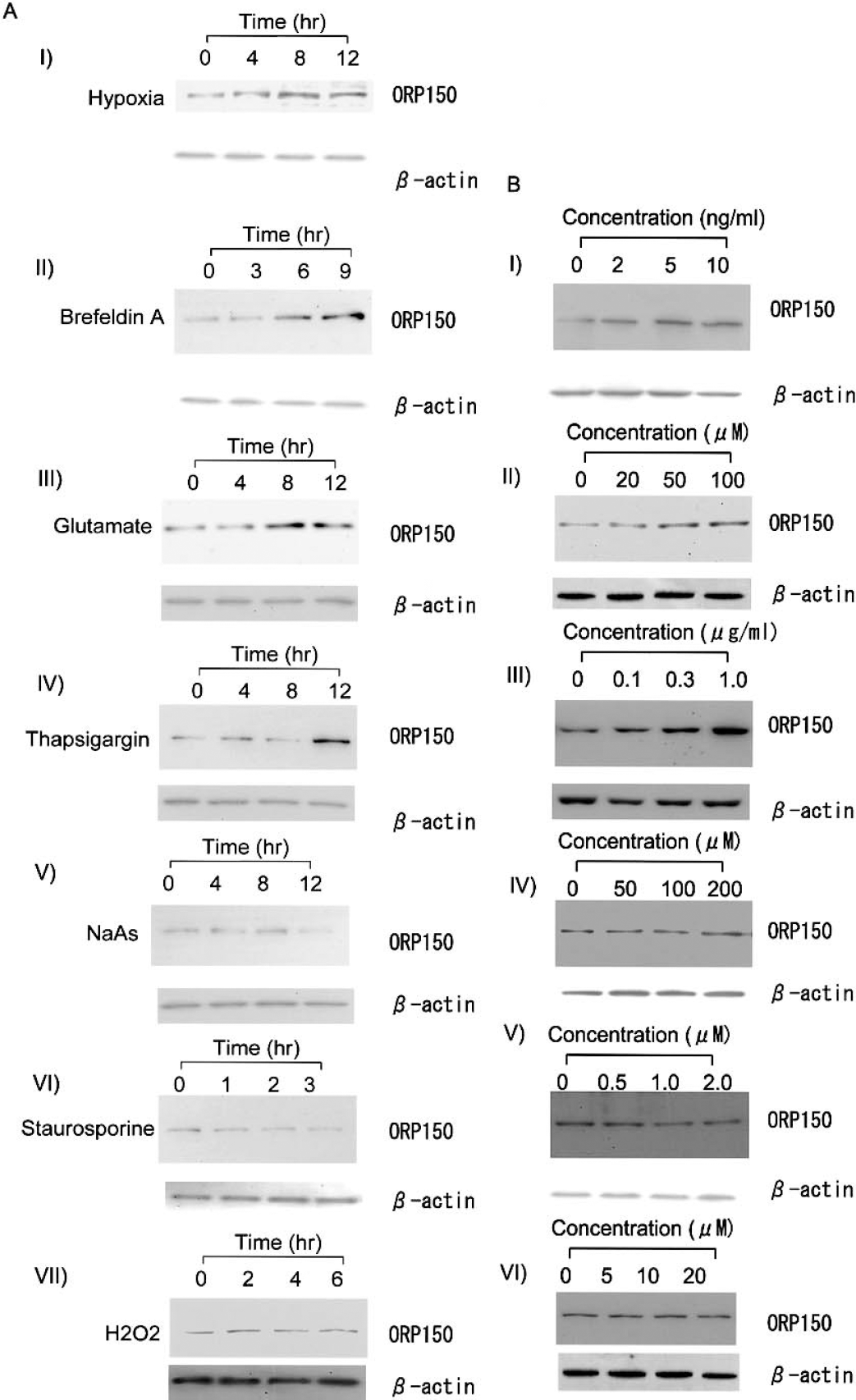
Expression of ORP150 in cultures neurons.
Role of ORP150 in cultured neurons under endoplasmic reticulum stress
To further identify the role of ORP150 in ER stress of cultured neurons, cultures were infected with adenovirus to overexpress sense ORP150 (Ad/ORP150S), antisense ORP150 (Ad/ORP150AS) (Tamatani et al., 2001), β-galactosidase (Ad/LacZ), or GFP (Ad/GFP). Immunostaining of MAP2 in infected cultures showed that more than 90% of neuronal cells were GFP positive (data not shown) at the concentration of 50 multiplicities of infection (moi). Infection of neurons with Ad/ORP150S increased the ORP150 antigen in cultured neurons, while treatment of cultures with Ad/ORP150AS decreased the ORP150 antigen level compared with control (Fig. 4A, upper panel). In each case, the levels of other molecular chaperons in the ER (78-kd and 94-kd GRP78 and GRP94, respectively) remained unchanged (Fig. 4A, lower panel). Further, treatment of the culture with either LacZ or GFP adenovirus did not alter the levels of ORP150, GRP94, or GRP78 (Fig. 4A, right panel), suggesting that the ORP150 sense/antisense structure can specifically alter the level of ORP150 antigen in cultured neurons.
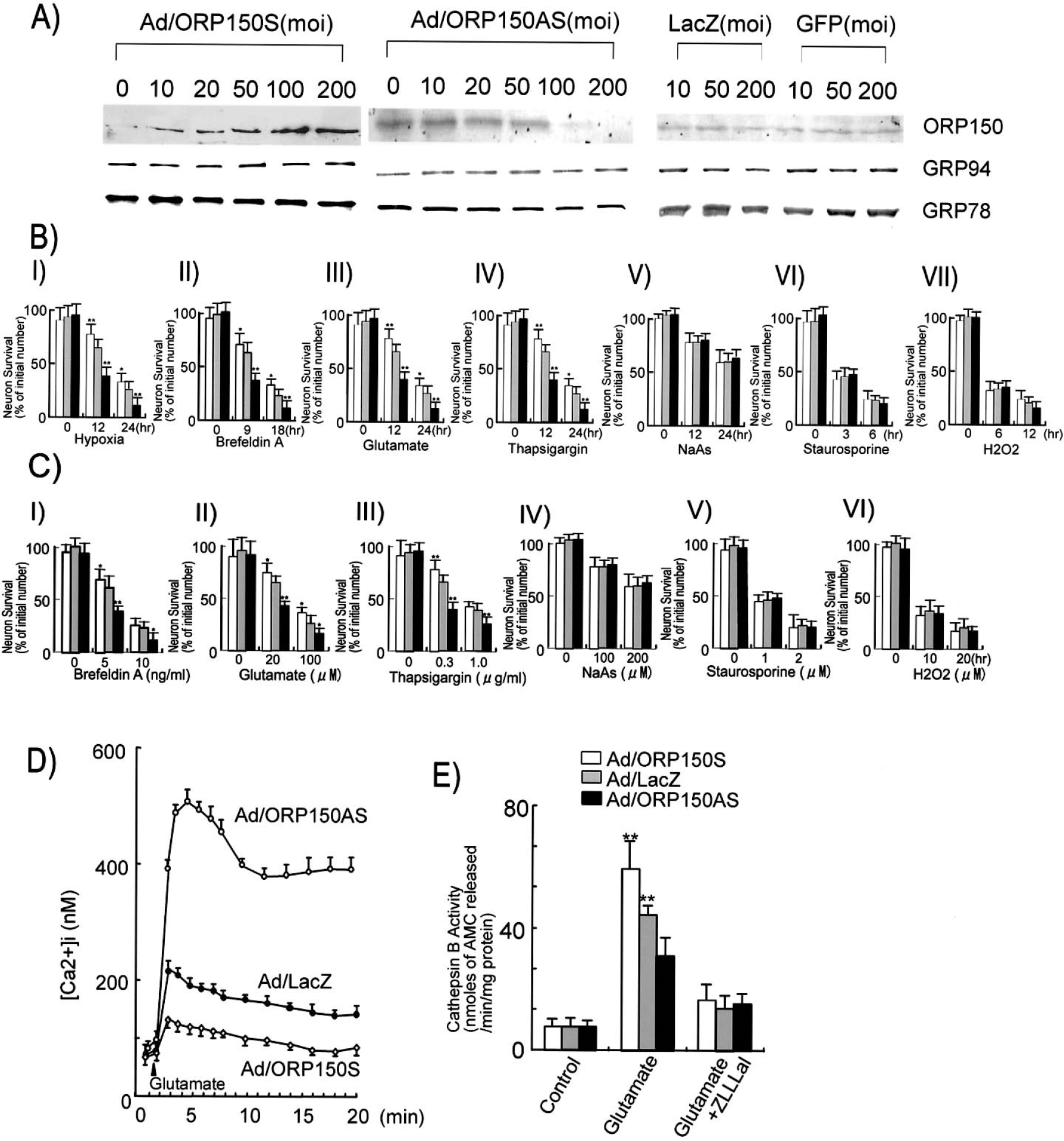
Overexpression of ORP150 rescues neurons from ER stress.
Exposure of cultured neurons to hypoxia, breferdin A, glutamate, or thapsigargin, all of which induced ORP150, caused neuronal cell death. Overexpression of ORP150 with Ad/ORP150S suppressed the neuronal cell death, whereas suppression of ORP150 with Ad/ORP150AS accelerated neuronal cell death in these paradigms (Figs. 4B I—IV, 4C I—III). In contrast, these adenoviruses had no effect on the neuronal cell death induced by other chemical stimuli (NaAs, staurosporine, and hydrogen peroxide) (Figs. 4B V—VII, 4C IV—VI), suggesting that cell death caused by other chemical stresses occurs independent of ORP150. On the other hand, overexpression of ORP150 suppressed the glutamate-mediated increase of intracellular Ca2+ in a dose-dependent manner, whereas the antisense adenovirus treatment caused a reverse effect (Fig. 4D). Furthermore, treatment with glutamate resulted in a rise in cathepsin B activity most prominently in neurons infected with Ad/ORP150AS, whereas neurons infected with Ad/LacZ showed a diminished response, and neurons infected with Ad/ORP150S showed only an approximate threefold increase in cathepsin B activity (Fig. 4E). These data suggest a link between the protective role of ORP150 under stress and the maintenance of ER function.
Overexpression of ORP150 rescues neurons from delayed neuronal cell death
To further investigate the role of ORP150, neural viability in the hippocampus after infection of the ORP150 adenovirus (Ad/ORP150S) was assessed in delayed neuronal death. Immunostaining with anti–β-galactosidase revealed expression of infected gene in hippocampus CA1 region in both cases (i.e., treatment with Ad/LacZ alone; Figs. 5A and 5B) and with a combination of Ad/LacZ and Ad/ORP150S (Figs. 5F and 5G), suggesting the successful infection with adenovirus vector in both cases. Immunostaining of adjacent section using an anti-MAP2 antibody revealed the almost complete loss of MAP2 signals of hippocampus pretreated with Ad/LacZ (Fig. 5C– Fig. 5E), whereas treatment of Ad/ORP150S resulted in survival of pyramidal neurons after the ischemic insult (Fig. 5H– Fig. 5J). Quantitative analysis of repeated experiments revealed that this improvement of neuronal cell viability by the overexpression of ORP150 was statistically significant (Fig. 5K). Taken together, these data demonstrate that the overexpression of ORP150 in hippocampal neurons by local administration of adenovirus can rescue neurons from DND.
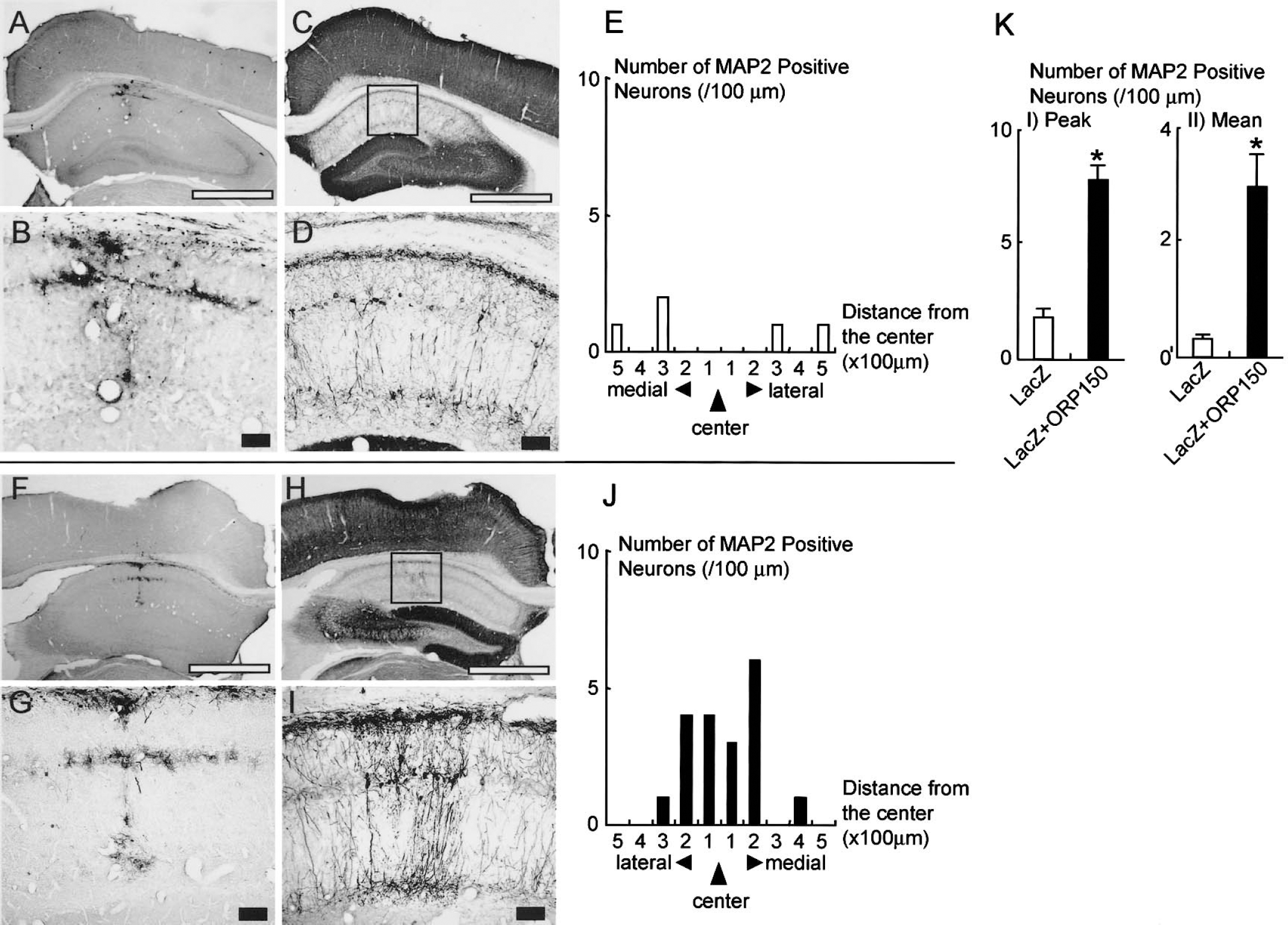
Overexpression of ORP150 rescues neuronal cell death in the hippocampus. Ad/LacZ alone (108 pfu in 2 μL phosphate-buffered saline (PBS;
DISCUSSION
Delayed neuronal cell death and ischemic tolerance in hippocampus CA1 of gerbil brain are well-known phenomena; however, the molecular mechanisms involved are not yet fully delineated. To determine the function of ORP150 in DND of gerbil brain, we performed a line of experiments. We showed that ORP150 is induced by transient ischemia in CA1 pyramidal neurons. However, the expression of ORP150 antigen was less induced by 5-minute ischemic insult than by 2-minute ischemia, whereas ORP150 message is strongly induced. This finding is consistent with those of previous reports that an overall suppression of protein synthesis occurs in DND (Hossmann, 1993) and that some stress proteins, including ubiquitin (Nowak, 1991), HSP27, HSP70, HSP90 (Wagstaff et al., 1996), and HSP40 (Tanaka et al., 2002), are upregulated only at the message level. Protein synthesis of ORP150 was restored in CA1 by 2-minute ischemia followed by 5-minute occlusion (Fig. 1) in a manner similar to that shown in the cases of other stress responders. ORP150 may rescue neurons from DND by relieving ER stress, which plays a pivotal role in the suppression of protein synthesis (DeGracia et al., 2002). These data suggest the possibility that ORP150 is one of the factors responsible for the acquisition of ischemic tolerance.
We demonstrated that the exposure of hippocampal neurons to hypoxia, glutamate, and other reagents that cause ER stress resulted in the expression of ORP150, whereas no such response was observed with other stressors. Further, the overexpression of ORP150 by adenovirus improved the cellular viability in neurons to glutamate and other ER stressors, whereas its suppression accelerated neuronal cell death, suggesting the protective role of ORP150 in neurons following glutamate exposure. These results are consistent with those of our previous study (Kitao et al., 2001; Tamatani et al., 2001), and indicate that the neuroprotective effect of ORP150 is not directly involved in reactive oxygen species or caspase activation. Moreover, we demonstrated that overexpression of ORP150 by adenovirus injection to hippocampus also suppressed the DND of CA1 pyramidal neuron. Taking these findings into consideration, it is possible that transient ischemia might damage the ER and that the ER dysfunction may, at least in part, lead to DND.
We have already demonstrated that the chaperone function of ORP150 can rescue neurons from hypoxia-mediated cell death, by an eventual suppression of caspase-3–like activity (Tamatani et al., 2001). On the other hand, our present data indicate the possible ability of ORP150 to maintain Ca2+ metabolism in the ER under glutamate excitotoxicity, which may be more prolonged in ischemic conditions (Mitani and Kataoka, 1991), by suppressing Ca2+ dependent proteolysis activity (Figs. 4C and 4D). ORP150 shares several components similar to GRP78, the 78-kd GRP, which is a molecular chaperon located in the ER. GRP78 is also induced by glutamate stress in neurons, and suppression of GRP78 increased the vulnerability of neurons to glutamate (Yu et al., 1999). GRP78 protects neurons through its ability to maintain Ca2+ homeostasis, which is consistent with the finding that GRP78 can bind to Ca2+, although the mechanism has not yet been clearly demonstrated (Lievremont et al., 1997). Furthermore, ORP150 is a homologue of CPB140, a 140-kd calcium-binding protein (Naved et al., 1995), which was identified as an ER protein with the capacity to bind Ca2+. These reports are consistent with our finding that ORP150 is involved in calcium homeostasis in neurons exposed to glutamate.
Although increasing evidence suggests that an active process similar to programmed cell death contributes to DND in gerbil, the mechanism of neuronal death still remains controversial. It has been suggested, mainly based on biochemical criteria, that postischemic hippocampal CA1 damage is classified as apoptosis (Honkaniemi et al., 1996), though the typical patterns of apoptotic morphologic change often do not appear in DND (Colbourne et al., 1999). Even in the cultured neurons, cell death caused by hypoxia or chemical stresses may include both apoptotic and necrotic pathways. In this report, therefore, viability of hippocampal neuron culture was assessed by morphologic criteria, and apoptotic cell death was not evaluated.
Although there still exists a gap between the function of ORP150 in cultured neurons and the role of this stress protein in the brain ischemia, our data suggest that the maintenance of ER function may be a novel target for the strategies of intervention medicine against ischemic neuronal cell death.