
Abstract
Lipopolysaccharide (LPS) preconditioning provides neuroprotection against subsequent cerebral ischemic injury. Tumor necrosis factor-α (TNFα) is protective in LPS-induced preconditioning yet exacerbates neuronal injury in ischemia. Here, we define dual roles of TNFα in LPS-induced ischemic tolerance in a murine model of stroke and in primary neuronal cultures in vitro, and show that the cytotoxic effects of TNFα are attenuated by LPS preconditioning. We show that LPS preconditioning significantly increases circulating levels of TNFα before middle cerebral artery occlusion in mice and show that TNFα is required to establish subsequent neuroprotection against ischemia, as mice lacking TNFα are not protected from ischemic injury by LPS preconditioning. After stroke, LPS preconditioned mice have a significant reduction in the levels of TNFα (~ threefold) and the proximal TNFα signaling molecules, neuronal TNF-receptor 1 (TNFR1), and TNFR-associated death domain (TRADD). Soluble TNFR1 (s-TNFR1) levels were significantly increased after stroke in LPS-preconditioned mice (~ 2.5-fold), which may neutralize the effect of TNFα and reduce TNFα-mediated injury in ischemia. Importantly, LPS-preconditioned mice show marked resistance to brain injury caused by intracerebral administration of exogenous TNFα after stroke. We establish an in vitro model of LPS preconditioning in primary cortical neuronal cultures and show that LPS preconditioning causes significant protection against injurious TNFα in the setting of ischemia. Our studies suggest that TNFα is a twin-edged sword in the setting of stroke: TNFα upregulation is needed to establish LPS-induced tolerance before ischemia, whereas suppression of TNFα signaling during ischemia confers neuroprotection after LPS preconditioning.
Introduction
Endotoxin (lipopolysaccharide (LPS)), a surface component of gram-negative bacteria, modulates the immune system through activation of Toll-like receptor 4. Administration of high doses of LPS induces a robust inflammatory response that can result in lethal septic shock. In contrast, administration of low doses of LPS induces a protective state of tolerance to subsequent exposure to LPS at doses that would ordinarily cause serious injury (Fan and Cook, 2004). Low-dose exposure to LPS also induces cross-tolerance wherein protection occurs against heterologous injury unrelated to LPS, such as ischemia. This protective state known as ‘LPS preconditioning or tolerance’ is not well understood, although emerging evidence suggests that modulation of inflammatory responses and release of cytokines, particularly tumor necrosis factor-α (TNFα), play an important role in elicitation of the tolerant state (Bordet et al, 2000; Rosenzweig et al, 2004; Tasaki et al, 1997; Toyoda et al, 2000).
Tumor necrosis factor-α is a particularly intriguing effector molecule because it is protective in the setting of preconditioning (Ginis et al, 1999; Nawashiro et al, 1997c), yet deleterious in ischemic brain injury after stroke (Barone et al, 1997). Support for a beneficial effect of TNFα in preconditioning is documented by the findings that neutralization of TNFα in the systemic circulation blocks LPS preconditioning (Tasaki et al, 1997), whereas pretreatment with either TNFα or its downstream-signaling mediator ceramide induces neuroprotection against ischemic injury in vivo (Furuya et al, 2001; Nawashiro et al, 1997c) and in vitro (Ginis et al, 1999). In contrast to the beneficial effects of TNFα administered before stroke injury, there is substantial evidence that TNFα is induced by stroke and that this induction worsens ischemic damage. Tumor necrosis factor-α is increased very early after stroke in mice (Gong et al, 1998; Uno et al, 1997) and affects numerous inflammatory responses, including microglial and vascular endothelial activation, coagulation cascades, and upregulation of enzymes such as COX-2, all of which contribute to the pathogenesis of brain damage (Hallenbeck, 2002). In addition, TNFα causes cell death directly by activating apoptotic signaling pathways mediated by the Fas-associated death domain and caspase-8 (Muppidi et al, 2004). A cytotoxic role for TNFα in ischemic brain is evinced by a reduction of infarct size in rodent models of stroke after systemic or central nervous system blockade of TNFα at the time of cerebral ischemia (Barone et al, 1997; Dawson et al, 1996; Nawashiro et al, 1997a; Yang et al, 1998).
The fact that TNFα plays a protective role in preconditioning by LPS and a damaging role during ischemic injury led us to speculate that LPS primes the neuroprotective process via TNFα production, and that this effect ultimately suppresses TNFα pathway activation after an ischemic event. We reasoned that the deleterious effects of TNFα during ischemia may be reduced in LPS preconditioning by dampened TNFα production and/or by impaired signal transduction after stroke. In a series of in vitro and in vivo experiments, we show for the first time that the effects of TNFα may be mitigated by altered ligand production and suppressed signaling via the TNFα pathway.
We examined the effects of LPS preconditioning on systemic TNFα production over time and using a mouse model of middle cerebral artery occlusion (MCAO) assessed the influence of LPS preconditioning on the following proximal mediators of TNFα signaling: TNF-receptor type 1 (TNFR1) and its soluble form s-TNFR1 and the intracellular adaptor TNFR-associated death domain (TRADD) molecule. We established a novel in vitro model system of LPS-ischemic tolerance to test directly the capacity of LPS preconditioning to alter neuronal responsiveness to cytotoxic TNFα in setting of ischemia. We show that LPS preconditioning protects primary neuronal cultures from the deleterious effects of TNFα during ischemia. Importantly, we also show that LPS preconditioning protects ischemic brain from centrally administered TNFα. Our findings help explain the beneficial role of TNFα induced by LPS preconditioning and also its deleterious role during ischemia as we show that LPS preconditioning changes the neuronal response to TNFα after ischemia in vivo and in vitro, and attenuates ischemic brain damage through suppressed ligand production and decreased expression of proximal signaling molecules. Thus, these experiments offer new insights into the mechanisms by which a single mediator (in this case TNFα) may play both a protective and a damaging role in the setting of brain ischemia.
Materials and methods
Mice
Age-matched male (8 to 10 weeks) C57Bl/6 mice, TNFα knockout mice (B6129SF-tnf) and its control strain (B6129F2/J) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. Procedures were conducted according to National Institute of Health guidelines and Oregon Health and Science University Institutional Animal Care and Use Committee. In all experiments, at the time of killing, mice were transcardiacally perfused to remove blood from the brain vasculature before harvesting the brain.
Lipopolysaccharide Preconditioning and Ischemia in Mice
Mice were preconditioned with phenol-extracted LPS from Escherichia coli 055:B5 (L-2880, L-2630; Sigma, St Louis, MO, USA) by an intraperitoneal injection of 200 μL volume 3 days before challenge with MCAO. Control mice received an intraperitoneal injection of sterile saline. Because of the differences in LPS purity and EU activity that exist between lots of LPS, the optimal preconditioning dose was determined for each lot of LPS. Mice were treated with doses of LPS that ranged between 7500 and 25,000 EU. For surgery, mice were anesthetized with 4% halothane and subjected to MCAO using the monofilament suture method described previously (Rosenzweig et al, 2004). Briefly, a silicone-coated 8 to 0 monofilament nylon surgical suture was threaded through the external carotid artery into the internal carotid artery to block the middle cerebral artery, and maintained intraluminally for 33 or 60 mins. The suture was then removed to restore blood flow. Regional cerebral blood flow was monitored by laser Doppler flowmetry throughout surgery. Body temperature was maintained at 35°C with a thermostat-controlled heating pad.
Intracerebral Ventricular Injection of Tumor Necrosis Factor-α after Middle Cerebral Artery Occlusion
The effect of LPS preconditioning on ischemic injury induced by central administration of exogenous TNFα was studied in mice after 33 mins MCAO. At 25 min after termination of MCAO, recombinant mouse TNFα (Chemicon, Temecula, CA, USA; 30 ng/1.5 μL volume) was injected into the right lateral ventricle, according to previously described techniques (Meller et al, 2005). A control group of animals received an injection of the same volume of sterile, artificial cerebral spinal fluid. In all animals, infarct volume was then measured 24 h after stroke.
Infarct Measurement
Infarct measurements were assessed, as described previously (Meller et al, 2005). Briefly, dissected mouse brains (minus the olfactory bulb and cerebellum) were sliced into seven, 1 mm coronal sections from the rostral end (using a Stoelting tissue slicer, Wood Dale, IL, USA). Sections were incubated with the vital dye, 2% 2,3,5-triphenyltetrazolium chloride (Sigma) for 15 mins at 37°C and fixed in 10% formalin. This technique provides quantification of infarction (Tureyen et al, 2004). A masked observer to treatment groups measured the area of infarction and the area of the ischemic hemisphere of each individual brain section using NIH Image 1.62 (Bethesda, MD, USA). Infarct volume was then calculated by the infarct area of each section multiplied by the section thickness (1 mm) and summed over the entire brain. Percent infarct volume = infarct volume/ischemic hemisphere volume × 100. The % volume was calculated for all experiments except for Figure 1B, where % area, rather than volume was calculated using the fifth brain section. This was done so that the remaining brain tissue sections could be used for the immunofluorescent studies performed in Figure 2. We have documented previously that % area infarct of the fifth coronal brain section correlates highly (r2 = 0.96) with infarct volume in our MCAO model (Hill et al, 1999).
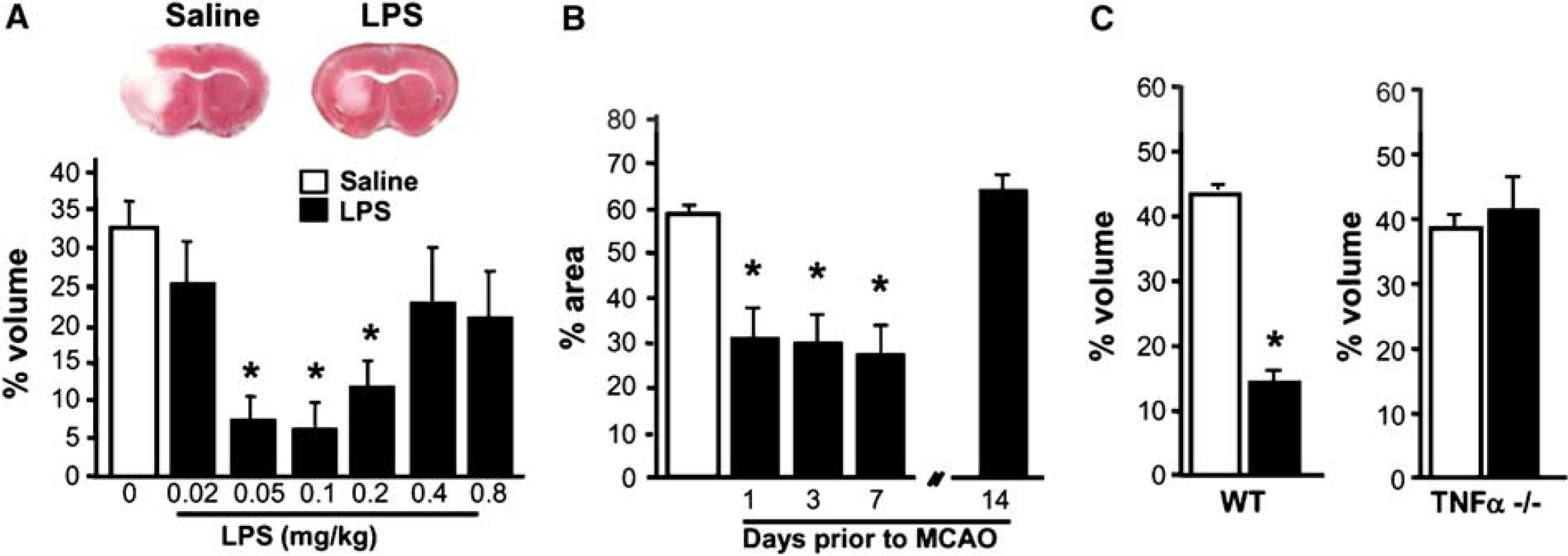
Tumor necrosis factor-α plays a necessary role in LPS preconditioning in mice. (
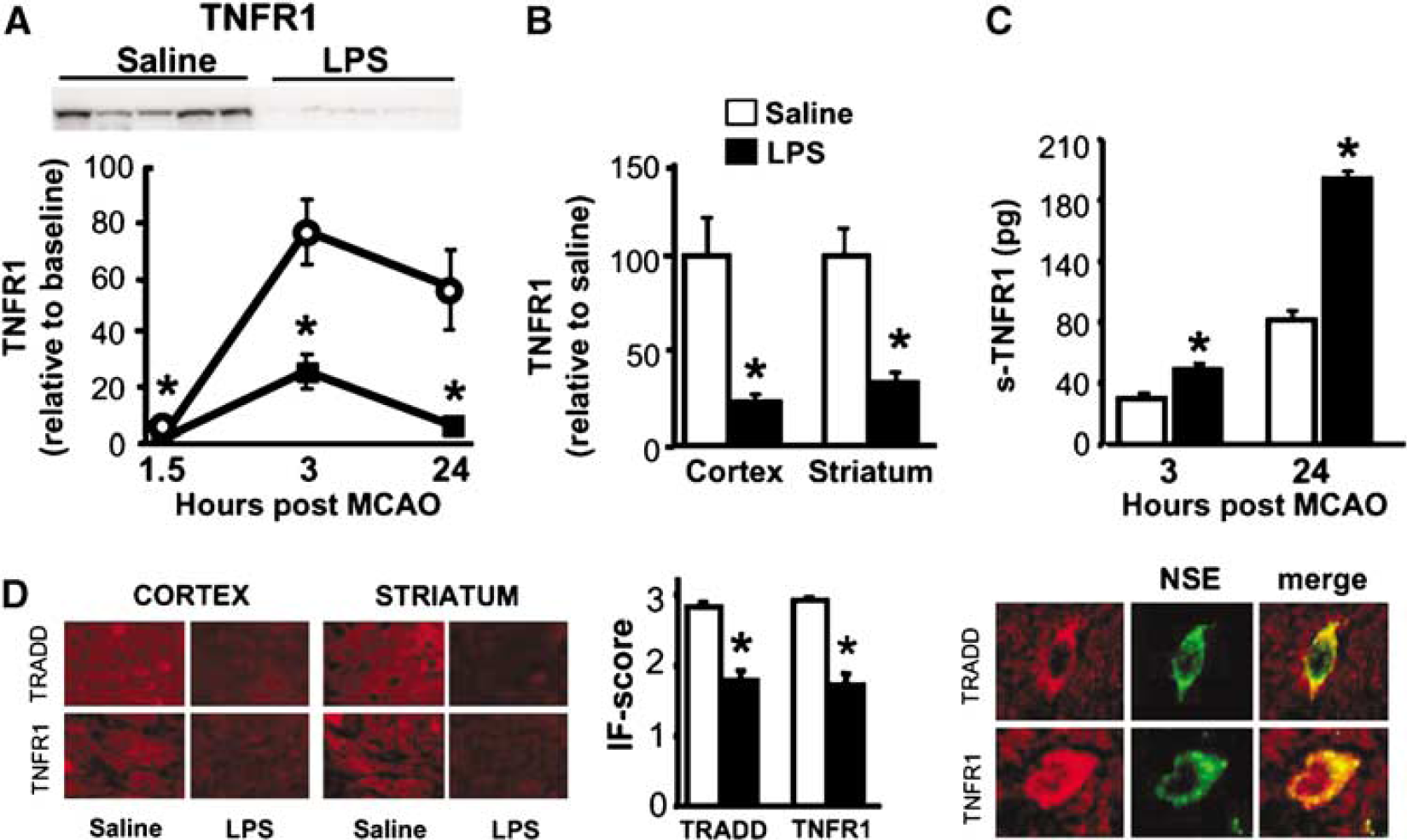
Lipopolysaccharide preconditioning modulates proximal mediators of TNFα signaling pathway after MCAO. C57Bl/6 mice were treated with 0.2 mg/kg LPS at 72 h before60 mins MCAO. (
Physiological Measurements
In a parallel series of experiments, various physiological parameters such as mean arterial blood pressure, blood gases, and body temperature were assessed at 72 h after LPS administration (at the time of surgery). Mean arterial blood pressure and arterial blood gases were measured via a femoral artery catheter in mice anesthetized with 1.5% halothane. Blood pressure values were collected using a Statham P23ID pressure transducer (Gould Inc., Oxnard, CA, USA) in line with a Grass Model 7 polygraph (Grass Instruments, Quincy, MA, USA) and expressed as an average across 30 mins of sampling (sampling rate of 100 Hz). Blood gases were measured using an Instrument Laboratory Synthesis 10 (Barcelona, Spain). Body temperature was measured by a rectal probe.
Lipopolysaccharide Preconditioning and Ischemia In Vitro
Preparation of primary rat cortical neuronal cultures and oxygen—glucose deprivation (OGD) was performed according to our previously published methods (Meller et al, 2005; Stenzel-Poore et al, 2003). Cultures were prepared from 1-to 2-day-old Sprague—Dawley rat pups (Harlan, Indianapolis, IN, USA). Cortices were dissected and dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA) and plated at a density of 1 × 106 cells/mL onto coverslips coated with poly-
Acidosis Exposure
Acidosis was induced according to a previously published method (Xiong et al, 2004). Cortical neuronal cultures were exposed to extracellular pH 6.0 for 1.5 h during a 2-h exposure of OGD. Exposure to acidosis and OGD was terminated by replacement of the medium with Neurobasal-A medium, pH 7.2 (supplemented with Glutamax), and return of the cells to a normoxic incubator.
Cell Death Evaluation In Vitro
Cell death in vitro was examined 24 h after OGD by means of fluorescent, cell permeable, DNA-binding dyes: propidium iodide (PI), as an indicator of cell death, and 4′,6-diamidino-2-phenylindole (DAPI), as an indicator of the total number of cells. Coverslips were incubated with PI (1.5 μg/mL; Sigma) for 2 mins, washed with phosphate-buffered saline, and fixed with Vectashield-mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Labs, Burlingame, CA, USA). Stained cells were visualized with a fluorescent microscope (Leica GMBH, Bannockburn, IL, USA) and analyzed using Bioquant software. The number of PI- and 4′,6-diamidino-2-phenylindole-stained cells were counted in two random fields of view on each coverslip, and percent death was calculated as mean (PI)/(DAPI) × 100 per field of view. Each treatment was performed on duplicate coverslips within an experiment and the entire experiment was repeated three or more times.
Western Blotting
Protein extraction was performed as described previously (Meller et al, 2005) with some modifications. Briefly, tissue samples were dissected from the cortex or striatum of each hemisphere and lysed in a buffer containing a protease inhibitor cocktail (Roche, Mannheim, Germany). Protein concentrations were determined using a BCA kit (Pierce-Endogen, Rockford, IL, USA). Protein samples (50 μg) were denatured in a gel-loading buffer (Bio-Rad Labs, Hercules, CA, USA) at 100°C for 5 mins and then loaded onto 12% Bis—Tris polyacrylamide gels (Bio-Rad Labs). After electrophoresis, proteins were transferred to polyvinylodene difluoride membranes (Bio-Rad Labs) and incubated with anti-TNFR1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. Membranes were then incubated with an anti-mouse immunoglobulin G antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) and detected by chemiluminescence (NEN Life Science Products, Boston, MA, USA) and exposure to Kodak film (Biomax, Kodak Company, New Haven, CT, USA). Images were captured using an Epson scanner and, the densitometry of the gel bands, including α-tubulin as a loading control, was analyzed using scanning-integrated optical density software (Image J, Bethesda, MD, USA).
Immunofluorescence
Brain tissue was prepared for immunofluorescence as described previously (Rosenzweig et al, 2004). Brain sections were treated with anti-TNFR1 or anti-TRADD antibodies (Santa Cruz Biotechnology), which were detected with a Cy3-conjugated antibody (Jackson Immuno Research, PA, West Grove, USA). Tumor necrosis factor-receptor type 1 and TRADD immunofluorescence was quantified from 10 randomly selected fields of view at × 20 located on the brain section that was taken from within the cortex or striatum of each individual mouse. The tissue section was scored by a masked observer on a scale of 0 to 3 (0 = no staining (i.e., equivalent to background of negative control), 1 = light staining, 2 = moderate staining, and 3 = heavy staining). Cell phenotype was determined by counterstaining sections with a neuronal specific antibody (antineuron-specific enolase (NSE) antibody, Chemicon) and detected by an anti-fluorescein isothiocyanate-conjugated antibody (Jackson ImmunoResearch). Colocalization of TNFR1 and TRADD with NSE was quantified from five different fields of view at × 40 and the mean count obtained. Images were collected using a Leica microscope with an Optonics DEI-750 3-chip camera equipped with a BQ 8000sVGA frame grabber and analyzed using BioQuant (Nashville, TN, USA).
Soluble Tumor Necrosis Factor Receptor 1 Measurement
Protein extraction and quantification was performed as described above on tissue dissected from the cortex of each mouse brain hemisphere. Soluble TNFR1 was measured with a commercial mouse s-TNFR1 ELISA kit (R&D Systems, Minneapolis, MN, USA). Equal amounts of protein (267 μg) for each sample were added in duplicate wells, and measured according to a standard curve.
Tumor Necrosis Factor-α Measurements
Plasma TNFα levels in mice were measured by a commercial mouse TNFα ELISA kit (BD Pharmingen) from blood samples obtained via the retro-orbital plexus. Tumor necrosis factor-α bioactivity in the supernatants of cortical neuronal cultures in vitro was determined using a cytotoxic bioassay with the TNFα-sensitive indicator cell line WEHI-164/clone-20 (Gold et al, 2002) derived from WEHI 164 (CRL-1751, American Type Culture Collection, Manassas, VA, USA). WEHI-164 cells were cultured in RPMI-1640 medium (supplemented with 10% heat-inactivated fetal bovine serum, 50 μmol/L 2-mecaptoethanol, 2% penicillin/streptomycin/glutamine). For assessment of TNFα activity, WEHI cells were plated at a density of 40,000 cells/well in 96-well plates and then sensitized with LiCl2 and actinomycin D (25 mmol/L and 2 μg/mL, respectively; Sigma) before adding samples. WEHI cells were then incubated overnight at 37°C and cell death was assessed by reduction of Alamar Blue dye (BioSource, Carlsbad, CA, USA) based on the absorbance at 570 nm (for reduced) and 600 nm (for oxidized). Tumor necrosis factor-α levels were determined in duplicate compared with a standard curve of known amounts of recombinant rat TNFα (Chemicon).
Reagents
Recombinant mouse or rat TNFα was purchased from Chemicon. Rabbit anti-TNFα neutralizing antibody (3 μg/mL) was purchased from Pierce-Endogen; TAPI-1 (8 μmol/L) was purchased from Calbiochem (Darmstadt, Germany).
Statistical Analysis
Mean differences were analyzed using two-way and oneway analysis of variance with Bonferroni's post hoc test. Data are represented as mean ± s.e.m. and differences were considered statistically significant when P < 0.05.
Results
TNFα Plays a Necessary Role in Lipopolysaccharide Preconditioning in Mice
We investigated whether TNFα played an essential role in LPS preconditioning in a mouse model of stroke. Previous work in a rat model showed that neutralization of TNFα at the time of LPS administration blocked neuroprotection against subsequent stroke (Tasaki et al, 1997). Here we examined whether TNFα knockout mice could be administered a low dose of LPS to induce neuroprotection against subsequent stroke injury. Because all previous studies involving Lipopolysaccheride preconditioning had been performed in rats, we first established the optimal neuroprotective conditions (dose and time) of LPS preconditioning in a mouse model of MCAO. This is particularly important as mice and rats differ in their sensitivity and response to LPS. Mice were administered increasing doses of LPS systemically 72 h before MCAO and stroke outcome was assessed 24 h later (Figure 1A). We found that mice treated with doses of LPS between 0.05 and 0.2 mg/kg showed significant protection compared with saline-treated controls. To determine the duration of neuroprotection induced by LPS treatment, mice were preconditioned with LPS for different time intervals before MCAO (Figure 1B) and stroke outcome was assessed. We found that LPS-induced neuroprotection developed within 1 day after administration and extended through day 7. Protection was no longer evident 14 days after treatment with LPS. Interestingly, the time window of LPS-induced neuroprotection in mice is similar to that reported for ischemic-tolerance rodents (Chen and Simon, 1997). Importantly, such neuroprotection by LPS preconditioning was not associated with physiological differences between treatment groups in mean arterial blood pressure, arterial blood gases, cerebral blood flow, or body temperature at the time of MCAO (72 h after LPS treatment, data not shown). These data define the specific dose range and time frame of LPS preconditioning in mice. We then asked whether TNFα knockout mice could be preconditioned against stroke injury by LPS. We administered LPS to TNFα knockout (TNFα−/–) and wild-type control mice 72 h before MCAO and assessed stroke outcome. As expected, wild-type mice pretreated with LPS showed a significant reduction in ischemic injury; however, TNFα−/–mice did not exhibit protection by LPS preconditioning (Figure 1C). Thus, there is a critical role for TNFα in mediating the neuroprotective effects of LPS preconditioning against ischemic injury.
Tumor Necrosis Factor-α Production is Suppressed in Lipopolysaccharide-Preconditioned Mice after Middle Cerebral Artery Occlusion
We next tested whether production of TNFα in the plasma in response to stroke was altered in LPS-preconditioned mice. Lipopolysaccharide-preconditioned mice showed increased TNFα levels in the plasma within 1 h after LPS administration, which returned to baseline within 24 h. In response to MCAO, plasma TNFα levels increased in LPS-treated and untreated mice similar levels at 1.5 and 3 h. However, by 24 h after MCAO, plasma TNFα levels were threefold higher in mice treated with saline compared with those preconditioned with LPS before MCAO, which did not increase (Table 1).
Effects of LPS preconditioning on production of TNFα in the plasma over time
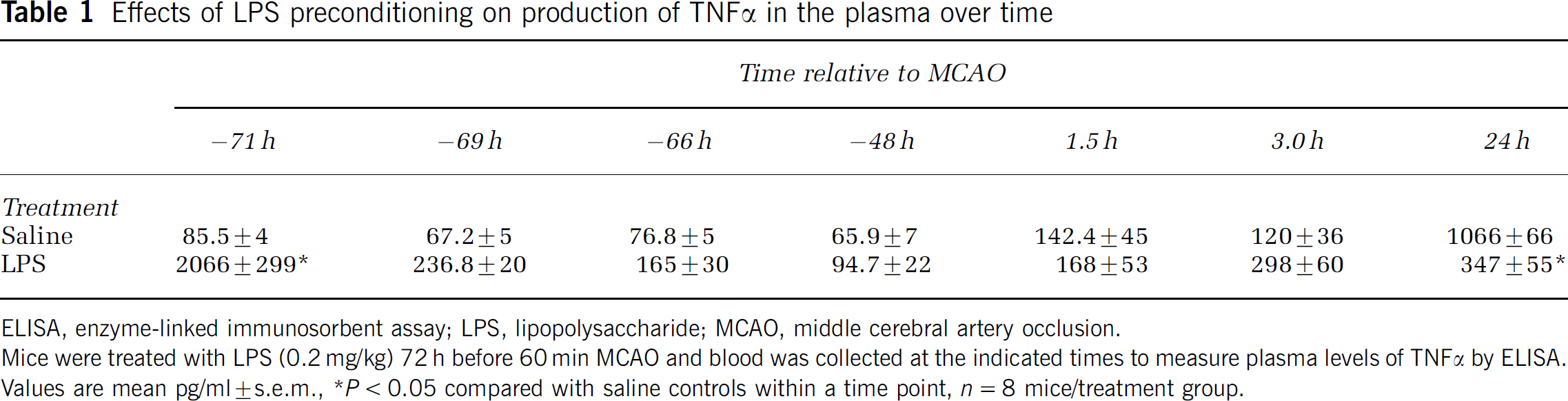
ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion.
Mice were treated with LPS (0.2 mg/kg) 72 h before 60 min MCAO and blood was collected at the indicated times to measure plasma levels of TNFα by ELISA. Values are mean pg/ml±s.e.m.
P<0.05 compared with saline controls within a time point, n = 8 mice/treatment group.
Lipopolysaccharide Preconditioning Modulates Proximal Mediators of the Tumor Necrosis Factor-α Signaling Pathway after Middle Cerebral Artery Occlusion
We went on to assess whether LPS preconditioning alters proximal mediators of TNFα signaling in addition to TNFα (Figure 2). We first tested whether TNFR1 expression in the brain was altered by MCAO in mice and found a marked increase in TNFR1 as early as 1.5 h after MCAO, which remained elevated 24 h after MCAO in the ischemic hemisphere of saline-treated control mice (Figure 2A). In contrast, mice preconditioned with LPS showed very low induction of TNFR1 after MCAO. Since no differences in TNFR1 expression between saline and LPS-pretreated mice before stroke were observed (data not shown), LPS preconditioning does not alter TNFR1 expression before stroke but appears to preclude its induction in response to stroke.
The LPS-induced suppression of TNFR1 occurred in uninfarcted cortex (region or protection) as well as infracted striatum (Figure 2B), which indicates that diminished TNFR1 expression was not simply a result of less injury in the cortex, but is specifically associated with LPS pretreatment. Next we examined the effect of LPS preconditioning on the s-TNFR1, which binds and inhibits the actions of TNFα. After MCAO, LPS preconditioned mice showed significantly greater levels of s-TNFR1 in ischemic brain hemispheres compared with saline-treated control mice (Figure 2C). This difference was evident at 3 h and was sustained out to 24 h after MCAO. It should be noted that, in contrast to brain, s-TNFR1 levels in plasma were not different in LPS-preconditioned or saline-treated mice 5 h after MCAO (data not shown).
We also used immunofluorescence to examine TNFR1 and its adaptor molecule, TRADD. This approach allowed us to determine the cellular localization of the TNFR1 complex (Figure 2D). We found that expression of TNFR1 and TRADD was increased after MCAO in the ischemic hemisphere; LPS preconditioning suppressed expression of both molecules equally in the cortex and the striatum, consistent with our finding above. Costaining for neuronal cells with NSE revealed that 95% ± 1% of the time, TNFR1 and TRADD expression colocalized with neurons, as shown in a representative picture in Figure 2D (far right). The neuronal phenotype indicated by NSE staining was also consistent with neuronal morphology.
Blockade of Tumor Necrosis Factor-α Abrogates Lipopolysaccharide Preconditioning in Modeled Ischemia In Vitro
Our results indicate that LPS preconditioning may attenuate TNFα signaling in ischemia. We postulated that LPS preconditioning reduces neuronal sensitivity to the injurious effects of TNFα in the setting of ischemia. To test this directly, at the cellular level in the absence of confounding systemic changes associated with TNFα, we developed an in vitro model of LPS preconditioning in cortical neuronal cultures. We show that LPS treatment of cortical neurons for 24 h confers protection against injury induced by exposure to OGD, as shown in Figure 3A. The neuroprotective effect of LPS was dependent on de novo protein synthesis, as the addition of cyclohexamide, an inhibitor of protein translation, reversed neuroprotection against OGD (data not shown). This is consistent with previous reports regarding the effect of LPS preconditioning in vivo (Bordet et al, 2000). To assess the involvement of TNFα in our in vitro system of LPS preconditioning, we measured TNFα levels after LPS treatment of neuronal cultures and found a marked increase in TNFα levels before OGD (Figure 3B). To test whether TNFα activity was important in the neuroprotective effect of LPS, neuronal cultures were treated with a TNFα-neutralizing antibody to block the effect of TNFα at the time of LPS preconditioning. Neutralization of TNFα reversed the neuroprotective effects of LPS preconditioning (Figure 3C). Treatment with either anti-TNFα antibody or control immunoglobulin G alone for 24 h before OGD had no effect on cell viability or OGD-induced cell death (data not shown).
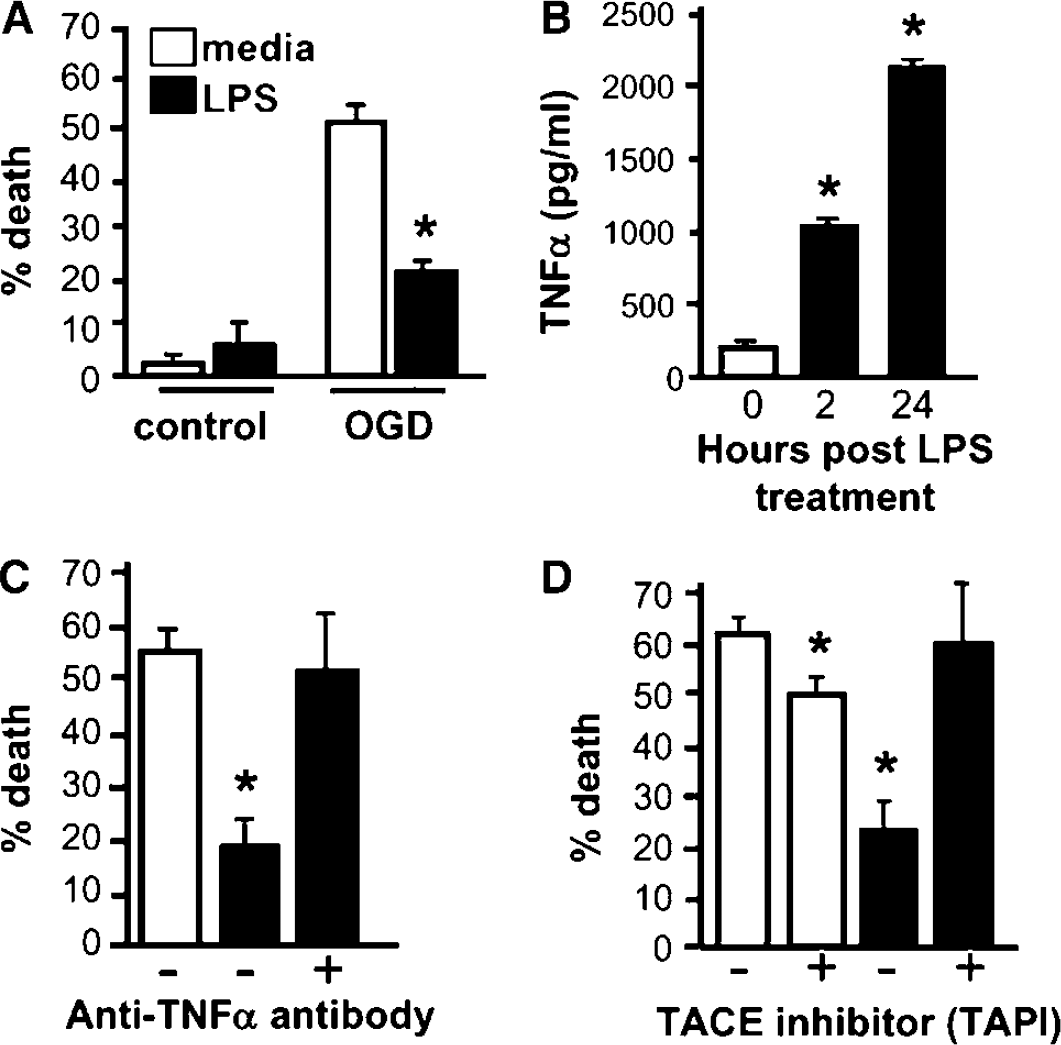
Soluble TNFα is essential for LPS preconditioning in an in vitro model of ischemia. (
TNFα is a type II transmembrane protein (mTNFα) that can bind directly to its receptors through cell-to-cell contact. mTNFα can also undergo cleavage (via the protease TNFα-converting enzyme (TACE)) and subsequently bind its receptors as the soluble protein, TNFα (Eissner et al, 2004). To establish whether the neuroprotective effect of TNFα was mediated through a soluble form of the molecule, we inhibited cleavage of mTNFα by treatment with TNFα protease inhibitor (TAPI), which inhibits TACE, at the time of LPS preconditioning and found that LPS-induced neuroprotection against OGD-induced injury was lost completely (Figure 3D). There was a modest reduction in cell death in control TAPI-treated cells after OGD, which may result from residual TAPI that remained after washing prior to OGD. This is consistent with the fact that TACE is upregulated after OGD and contributes to ischemic injury (Hurtado et al, 2002; Wang et al, 2004). Treatment with TAPI alone in control cultures not exposed to OGD had no effect on cell viability (data not shown). This is the first report of the role of TACE in LPS preconditioning. Overall, these data reveal that the soluble form of TNFα mediates the protective actions of TNFα during LPS preconditioning.
Lipopolysaccharide Preconditioning Ameliorated TNFα-Exacerbated Neuronal Injury after Ischemia
We next assessed whether TNFα exacerbated ischemic injury to neurons in the setting of prior LPS preconditioning. Not unexpectedly, we found that endogenous release of TNFα during OGD is cytotoxic as evinced by the fact that treatment with TNFα-neutralizing antibody after OGD limits cell death (Figure 4A). To test whether LPS preconditioning reduces the susceptibility of ischemia-exposed neuronal cells to TNFα-induced injury, TNFα was added to LPS-preconditioned cortical neuronal cultures after exposure to OGD (Figure 4B). We found that exogenous TNFα enhanced OGD-induced cell death in control, nonpreconditioned cortical neuronal cultures. However, LPS-preconditioned cortical neuronal cultures were protected against TNFα-induced injury after OGD. In control cultures not exposed to OGD, TNFα treatment alone did not affect cell viability (data not shown), which supports the deleterious role of TNFα in the setting of ischemia. These data show that LPS preconditioning changes the neuronal response to the cytotoxic actions of TNFα in the setting of ischemia—an effect that may contribute to the neuroprotective process of LPS preconditioning. To assess whether LPS preconditioning changed the neuronal response to injurious stimuli other than TNFα in the setting of ischemia, we examined the effect of acidosis on OGD-induced cell injury. Acidosis occurs after ischemia, which in turn causes neuronal damage via membrane acid-sensing ion channels (Xiong et al, 2004). After titrating the duration of acidosis to induce moderate injury, we tested whether LPS protected against acidosis-induced injury (exposure to extracellular pH 6.0) in the setting of OGD. Figure 4C shows that lowering the extracellular pH to 6.0 for 1.5 h during exposure to OGD induces marked cell death in the presence or absence of LPS treatment. Thus, LPS preconditioning protects against TNFα but not acidosis-induced injury in the setting of ischemia, which suggests that acidosis-mediated damage is independent of TNFα.
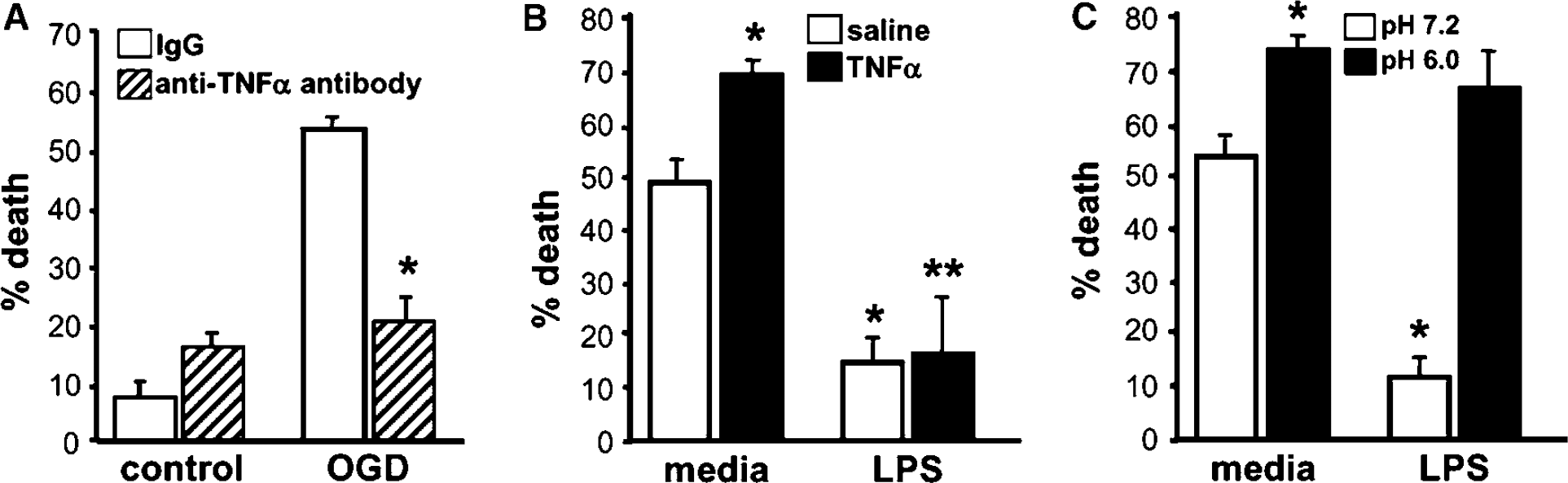
Lipopolysaccharide preconditioning protects neurons against TNFα-mediated injury in the setting of ischemia in vitro. (
We went on to assess whether LPS preconditioning protects against TNFα cytotoxicity during ischemia in vivo. We reasoned that in the absence of LPS preconditioning, the addition of TNFα would exacerbate stroke injury. The duration of MCAO was reduced to 33 mins (from 60 mins), to induce less damage and thereby allow detection of increased damage by exogenous TNFα. We injected TNFα (30 ng) or artificial cerebral spinal fluid into the right lateral ventricle 25 mins after termination of MCAO in LPS preconditioned mice or saline controls and infarct size was evaluated 24 h later (Figure 5). Tumor necrosis factor-a administration failed to worsen stroke damage in mice preconditioned with LPS, whereas mice not preconditioned suffered significantly larger stroke injury with the administration of TNFα. We did not observe any brain injury due to TNFα treatment after sham surgery (data not shown). This is the first direct evidence that TNFα administration to rodents at the time of stroke exacerbates brain injury. Moreover, our findings that LPS preconditioning blocked the TNFα-induced exacerbation of ischemic injury suggest that neuroprotective effects of LPS preconditioning are mediated partially through diminished sensitivity of the brain to the injurious effects of TNFα at the time of stroke.
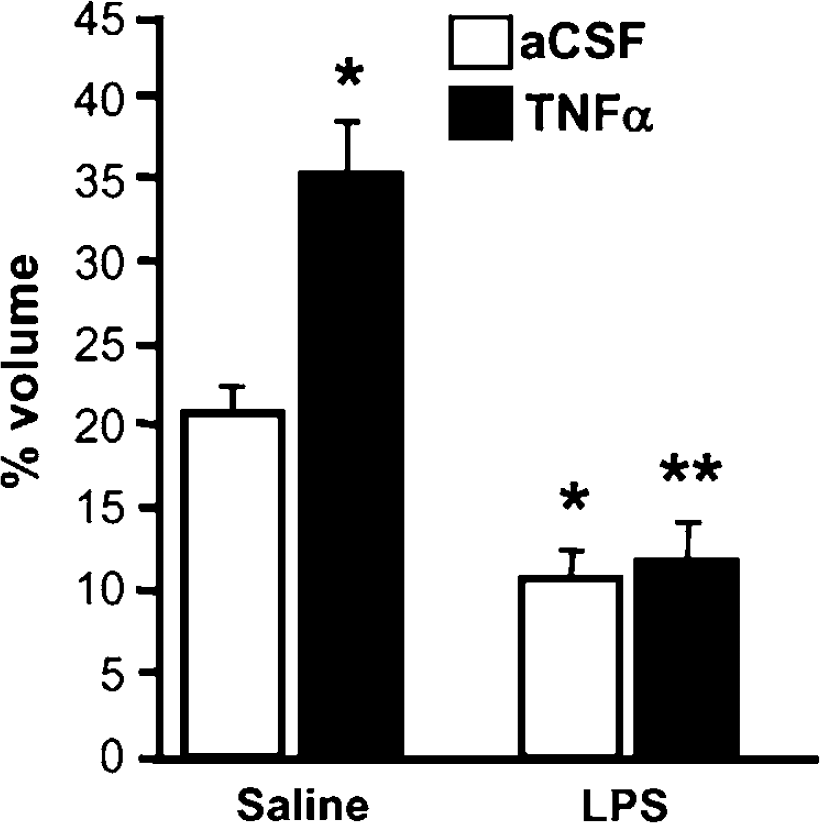
Lipopolysaccharide preconditioning protects against TNFα-induced ischemic brain damage after cerebral ischemia in vivo. Mice that were preconditioned with LPS 72 h before MCAO (33 mins) were then administered an intracerebroventricular injection of TNFα (30 ng) or artificial cerebral spinal fluid after stroke. Infarct volume was measured 24 h after MCAO by 2,3,5-triphenyltetrazolium chloride staining. Values are mean ± s.e.m., *P < 0.05 versus saline controls treated with intracerebroventricular injection of artificial cerebral spinal fluid, **P < 0.05 versus saline controls treated with i.c.v. injection of TNFα, n =8 mice/treatment group.
Discussion
Here we report the novel finding that LPS preconditioning suppresses the TNFα response to cerebral ischemic injury. We show that LPS preconditioning alters proximal mediators of the TNFα signaling pathway after stroke, as TNFα, TNFR1, and TRADD were reduced and s-TNFR1 was increased. We showed that cortical neuronal cultures preconditioned by LPS were less susceptible to TNFα-induced injury after ischemia in vitro. Moreover, the capacity of LPS preconditioning to protect against TNFα was also evident in vivo, as TNFα treatment failed to exacerbate stroke injury in LPS preconditioned mice. These findings suggest that LPS preconditioning may provide neuroprotection against ischemic injury by diminishing the deleterious actions of TNFα induced after stroke.
Somewhat paradoxically, we also show that TNFα plays an essential and beneficial role as an initiator of LPS preconditioning against ischemic injury in mice. This is indicated by the fact that LPS preconditioning in TNFα knockout mice does not protect against injurious MCAO. It should be noted that when briefer periods of MCAO were used in the absence of LPS preconditioning, TNFα knockout mice showed reduced stroke size (data not shown), consistent with the fact that TNFα is known to be deleterious in the setting of ischemia. In addition, our studies suggest that the soluble form of TNFα mediates the neuroprotective effects of LPS preconditioning in vitro because cortical cultures treated with a TACE inhibitor that blocks the release of soluble TNFα are not protected by LPS preconditioning. This latter finding implicates TNFR1 as the mediator of protective signaling because the soluble form of TNFα primarily signals through this receptor subtype rather than TNFR2 (Grell et al, 1995).
Preconditioning by administration of LPS increased TNFα levels in the circulation before ischemia, but levels returned to baseline by the time of stroke (72 h). Early induction of TNFα may be essential in the emergence of neuroprotection, as inhibition of TNFα at the time of LPS preconditioning by systemic administration of TNFα-binding protein reversed neuroprotection against MCAO in rats (Tasaki et al, 1997). These data suggest that TNFα may be an early signal that primes the brain against subsequent ischemic injury. The mechanisms by which TNFα mediates LPS-ischemic tolerance are not known, although studies in vitro have shown that TNFα pretreatment alone is protective against ischemic injury and that activation of the transcription factor nuclear factor (NF)-κB plays an essential role in the induction of tolerance by TNFα. Nuclear factor-κB activation by TNFα has been shown to increase expression of cell survival and neuroprotective proteins such as bcl-2 and MnSOD (Tamatani et al, 1999; Wilde et al, 2000), which could protect against the damaging effects of cerebral ischemia. Strong evidence also points to a protective role for TNFα-induced signaling events and activation of NF-κB in the induction of ischemic tolerance by other preconditioning stimuli, such as subinjurious ischemia (Pradillo et al, 2005). Thus, it is reasonable that similar TNFα-induced signaling events may be involved in LPS-ischemic tolerance. This would also be consistent with the reported beneficial role of TNFα in neuroprotection against metabolic poisoning in vitro induced by LPS pretreatment (Lastres-Becker et al, 2006).
TNFα may mediate LPS-induced ischemic tolerance by suppression of subsequent TNFα signaling in the setting of ischemia. Studies show that TNFα pretreatment in cortical brain cells suppresses subsequent TNFα-induced signaling events, as NF-κB activity was reduced and ICAM-1 expression was inhibited on re-exposure to TNFα (Ginis et al, 2002; Ginis et al, 1999). The negative autocrine regulation induced by prior TNFα treatment is thought to occur through increased expression of negative feedback inhibitors such as MnSOD, A20, c-IAP, and c-FLIP that inhibit TNFα-signaling events (Muppidi et al, 2004). Such features of TNFα tolerance could be protective against the cytotoxic effects of TNFα during ischemia. Indeed, it has been shown that preconditioning with LPS or the diphosphoryl lipid A component of LPS increased superoxide dismutase (SOD) activity during MCAO in rats (Bordet et al, 2000; Toyoda et al, 2000). This finding is consistent with TNFα tolerance where MnSOD expression is increased during re-exposure to TNFα (Ginis et al, 2002) and that MnSOD inhibits TNFα-signaling responses and apoptosis (Lin et al, 2005; Manna et al, 1998).
Our data suggest the possibility that neuroprotection induced by LPS preconditioning depends on TNFα production before stroke, which ultimately causes suppression of proximal mediators of the TNFα-signaling pathway after stroke. Lipopolysaccharide-induced suppression of effectors of the TNFα-signaling pathway after MCAO is evinced by our data that show reductions in systemic TNFα production and neuronal TNFR1 and TRADD expression. Tumor necrosis factor-receptor type 1 expression was suppressed at early times after MCAO and sustained out to 24 h—a critical time window in the development of brain injury. Suppression of TNFR1 was coincident with enhanced s-TNFR1 and together these changes would be expected to decrease the effect of soluble TNFα. This observation further underscores the importance of modulating TNFR1 for neuroprotection by LPS preconditioning. In addition, it has been shown previously that improved stroke outcome results from exogenous treatment with s-TNFR1 at the time of ischemia (Nawashiro et al, 1997b). Such improvement occurs presumably by neutralization of the actions of TNFα. Our data support a similar protective role for enhanced levels of endogenous s-TNFR1 after stroke and suggest one way that TNFR1-mediated signaling may be dampened during the acute response to stroke injury. We found that TNFR2 expression was not induced substantially until later times after stroke (24 h after MCAO)—an upregulation that was suppressed by prior LPS preconditioning (data not shown). Although others have suggested that TNFR2 may play a beneficial role in neuronal survival in the setting of ischemia (Marchetti et al, 2004), our data suggest that TNFR2 is not a major mediator of acute neuroprotective responses in the unique setting of LPS preconditioning. Collectively, our findings showing increased levels of s-TNFR1 in association with decreased expression of neuronal TNFR1 and TRADD indicate that LPS preconditioning may limit TNFα signaling and thereby enhance neuronal survival in the setting of ischemia.
Lipopolysaccharide preconditioning may alter the neuronal responses to the injurious effects of TNFα is bolstered further by our finding that LPS preconditioning decreases the vulnerability of neurons to TNFα-mediated injury after ischemia in vitro. Mitigation of the effect of TNFα would be expected to be beneficial in ischemia as indicated by our studies in vitro showing that inhibition of TNFα at the time of OGD is protective. This is consistent with a deleterious role for endogenous TNFα in neuronal survival in this model system as suggested by studies done previously showing that the absence of TNFα at the time of OGD is neuroprotective in cortical cultures (Martin-Villalba et al, 2001). Robust protection against TNFα-mediated injury may be a specific response to LPS preconditioning, as there was no protection against the deleterious effects of acidosis during ischemia in LPS preconditioned cortical cultures. That protection is not present in the setting of acidosis may suggest that LPS preconditioning specifically alters TNFα-associated effects but does not alter more general responses to injury such as acid-sensing ion channel-induced changes in intracellular calcium flux that lead to neuronal damage (Xiong et al, 2004). Thus, LPS preconditioning may protect against TNFα-induced injury by inhibition of TNFα-induced cell death directly, as TNFα triggers apoptosis through Fas-associated death domain and the caspase-8 signaling pathway, or by diminishing neuronal sensitivity to glutamate toxicity, oxidative and/or mitochondrial stress (Cheng et al, 1994; Lastres-Becker et al, 2006; Manna et al, 1998). Furthermore, our findings in vivo show that TNFα administered into the brain at the time of stroke does not exacerbate ischemic brain damage in mice that have been preconditioned, which suggests that neuroprotection by LPS may also involve diminished inflammatory responses. In the absence of LPS preconditioning, TNFα exacerbates reperfusion injury by initiating inflammatory responses such as microglial and endothelial activation, increasing vascular permeability and peripheral cellular infiltration. In support of attenuated inflammation as an effect of LPS-induced neuroprotection, we have shown previously that LPS preconditioning reduces neutrophil infiltration and activation of microglia and monocytes after MCAO (Rosenzweig et al, 2004). In addition, others have shown that LPS preconditioning results in preservation of microvascular perfusion and endothelial cell function after MCAO in rats (Bastide et al, 2003; Dawson et al, 1999).
Our finding that LPS preconditioning decreases the ratio of TNFα to s-TNFR1 resembles effects seen in endotoxin tolerance, wherein a low dose of LPS is protective against a greater, lethal dose of LPS. Pretreatment with low-dose LPS suppresses production of proinflammatory mediators such as TNFα, IL-1β, and IL-6 after subsequent challenge with a large dose of endotoxin. Furthermore, LPS preconditioning leads to protection against subsequent LPS-induced cell death via a process referred to as ‘reprogramming,‘ wherein priming by exposure to low-dose LPS alters the response to subsequent LPS. Such reprogramming leads to sustained or enhanced production of anti-inflammatory mediators such as s-TNFR1 and IL-10 (Fan and Cook 2004; Shnyra et al, 1998). It is possible that LPS preconditioning also reprograms the response to ischemic injury leading to increased cell survival. Genomic expression patterns observed in LPS-preconditioned animals provide supportive evidence of such reprogramming (Stenzel-Poore and Simon, 2004) and indicate that protection may result, in part, from marked suppression of deleterious proinflammatory pathways such as those mediated by TNFα and also by induction of beneficial anti-inflammatory and neuroprotective pathways that enhance cell survival. Such mechanisms may define targets for further drug discovery. These findings also have important implications for therapeutic treatment of patients at risk of stroke, as LPS preconditioning offers the potential to minimize the deleterious effects of TNFα while enhancing beneficial neuroprotective mediators after stroke.
Footnotes
Acknowledgements
We acknowledge Sonya King and Emily Dykhuizen for their outstanding technical support, and Dr Dan Hatton for expert assistance in physiological measurements.