
Abstract
Neuroprotection against cerebral ischemia can be realized if the brain is preconditioned by previous exposure to a brief period of sublethal ischemia. The present study was undertaken to test the hypothesis that nitric oxide (NO) produced from the neuronal isoform of NO synthase (NOS) serves as a necessary signal for establishing an ischemia-tolerant state in brain. A newborn rat model of hypoxic preconditioning was used, wherein exposure to sublethal hypoxia (8% oxygen) for 3 hours renders postnatal day (PND) 6 animals completely resistant to a cerebral hypoxic-ischemic insult imposed 24 hours later. Postnatal day 6 animals were treated 0.5 hour before preconditioning hypoxia with the nonselective NOS inhibitor L-nitroarginine (2 mg/kg intraperitoneally). This treatment, which resulted in a 67 to 81% inhibition of calcium-dependent constitutive NOS activity 0.5 to 3.5 hours after its administration, completely blocked preconditioning-induced protection. However, administration of the neuronal NOS inhibitor 7-nitroindazole (40 mg/kg intraperitoneally) before preconditioning hypoxia, which decreased constitutive brain NOS activity by 58 to 81%, was without effect on preconditioning-induced cerebroprotection, as was pretreatment with the inducible NOS inhibitor aminoguanidine (400 mg/kg intraperitoneally). The protective effects of preconditioning were also not blocked by treating animals with competitive [3-(2-carboxypiperazin-4-yl)propyl-1-phosphonate; 5 mg/kg intraperitoneally] or noncompetitive (MK-801; 1 mg/kg intraperitoneally) N-methyl-D-aspartate receptor antagonists prior to preconditioning hypoxia. These findings indicate that NO production and activity are critical to the induction of ischemic tolerance in this model. However, the results argue against the involvement of the neuronal NOS isoform, activated secondary to a hypoxia-induced stimulation of N-methyl-D-aspartate receptors, and against the involvement of the inducible NOS isoform, but rather suggest that NO produced by the endothelial NOS isoform is required to mediate this profound protective effect.
Neuroprotection from ischemic brain injury can be realized in experimental models of stroke if the tissue is previously “preconditioned” with a sublethal hypoxic or ischemic challenge (for recent review see Chen and Simon, 1997). Because preconditioning harnesses the often robust endogenous protective potential of the tissue, elucidation of the mechanisms responsible for the induction of ischemic tolerance may have practical value in the search for effective means of therapeutic intervention in stroke. In animal models, hours to days must ensue between the preconditioning stimulus and the establishment of a state of ischemic tolerance; thus, the consensus from many studies is that the preconditioning stimulus induces changes in gene expression that require a finite time period to become manifest (Chen and Simon, 1997). Although this phenomenon has been reproducibly documented in a number of small animal models of cerebral ischemia, little is known of the mechanistic pathways operative in its induction, the genes so activated by the preconditioning stimuli, and the changes that characterize its final expression.
The present study was undertaken to examine the early induction phase of preconditioning to identify principal molecular players required to signal the subsequent adaptive genomic changes that follow the preconditioning stimulus. We hypothesized that nitric oxide (NO) might be an important proximal component of this transduction pathway, given the ongoing identification of its many diverse paracrine actions in the central nervous system, including the modulation of cerebral blood flow, neurotransmitter release and electrocortical activity, neuroendocrine balance, nociception, cardiomedullary reflex activity, sexual and other behaviors, and synaptic plasticity (Garthwaite and Boulton, 1995; Zhang and Snyder, 1995). NO is formed when arginine is oxidized to NO by the action of one of three different NO synthase (NOS) isoforms, either constitutive or inducible, which are uniquely distributed among particular cell types. In brain, constitutive, calcium-dependent neuronal NOS (nNOS) is found in discrete populations of neurons with highly branched processes, allowing for widespread influence of NO after its production (Dawson and Snyder, 1994). Vascular endothelial cells, also ubiquitously distributed in brain, contain endothelial NOS (eNOS), a constitutive NOS isoform distinctly separate from nNOS. Calcium-independent inducible NOS (iNOS) is generally expressed by glia, resident macrophages, and circulating neutrophils under specific conditions such as after ischemia; interestingly, in the perinatal period, cerebral blood vessels stain positively for a constitutive form of iNOS (Galea et al., 1995).
Neuronal NOS is activated by calcium influx through voltage-gated N-methyl-D-aspartate (NMDA) receptor channels following glutamate binding to these receptors (Zhang and Snyder, 1995); this pathway is now implicated in the physiologic coupling of neuronal metabolism to cerebral blood flow and in neuronal toxicity following stroke (Iadecola, 1997). We hypothesized that this same pathway is also activated in response to sublethal preconditioning ischemia and that the NO so formed by this isoform is critical to the induction of an ischemia-tolerant state. Using a reproducible model of ischemic tolerance in the immature rat (Gidday et al., 1994), nonselective and isoform-selective NOS inhibitors, and competitive and noncompetitive NMDA receptor antagonists, we identified an obligatory role for NO in preconditioning-induced protection, but, unexpectedly, our results suggest that eNOS, not nNOS or iNOS, is the isoform responsible for producing the NO required for the development of ischemic tolerance.
METHODS
Animal model for hypoxic-ischemic brain injury and ischemic tolerance
Fifty-seven litters of newborn Sprague-Dawley rats were used for these studies. All protocols met with institutional approval and were consistent with NIH policy on the use of animals in experimental research. The hypoxia-ischemia model utilized involves left unilateral carotid ligation in halothaneanesthetized postnatal day (PND) 7 animals followed 90 minutes later by a 3-hour exposure to 8% oxygen (Rice et al., 1981; Hagberg et al., 1997), which causes a marked hypoxia (Welsh et al., 1982) and a fall in cerebral blood flow (Silverstein et al., 1984). The animals are exposed to the hypoxic gas mixture in small chambers immersed in 36.5°C water, which we previously verified maintains the animal's core temperature normothermic. The hypoxia-ischemia intervention results in unilateral brain injury in the majority of littermates, characterized by selective neuronal necrosis to infarction in the cortex, hippocampus, thalamus, striatum, and white matter (Rice et al., 1981); this injury can be quantified by several highly correlative criteria (Gidday et al., 1995).
The protocol for inducing ischemic tolerance in these animals has been detailed previously (Gidday et al., 1994). In brief, PND 6 animals are exposed to 8% oxygen for 3 hours in humidified, normothermic chambers; when subjected to the standard hypoxic-ischemic insult 24 hours later (unilateral carotid ligation followed by 3 hours of hypoxia), the animals exhibit complete cerebroprotection, without evidence of selective neuronal loss in the vast majority of animals (Gidday et al., 1994). We and others have shown previously that exposure to hypoxia alone is not associated with any morphological indexes of cellular injury (Rice et al., 1981; Gidday et al., 1994).
Experimental protocols and drug administration
Hypoxic preconditioning was always performed on PND 6 animals, and all animals were rendered hypoxic-ischemic 24 hours later on PND 7. Except where indicated, all drug-treated litters were divided into the following four subgroups: nonpreconditioned animals with and without drug treatments and preconditioned animals with and without drug treatments. In this way, each litter contained two subgroups of animals that served as intralitter controls to ensure (a) that the litter was vulnerable to hypoxic-ischemic injury (nonpreconditioned, vehicle treated) and (b) that drug given on PND 6 with the express intention of affecting the preconditioning mechanism did not change outcome from hypoxia-ischemia induced 24 hours later (nonpreconditioned, drug treated). Failure to meet this second criteria in the MK-801-treated animals led us to undertake studies in animals pretreated with MK-801 on PND 5 and 4 as described in detail below.
To identify which, if any, NOS isoform might be important in initiating the requisite mechanisms responsible for establishing an ischemia-tolerant state in response to the hypoxic preconditioning stimulus, the nonselective constitutive NOS inhibitor L-nitroarginine (L-NA; 2 mg/kg), the nNOS inhibitor 7-nitroindazole (7-NI; 40 mg/kg), or the iNOS inhibitor aminoguanidine (AG; 400 mg/kg) was administered intraperitoneally to PND 6 animals 30 minutes before the 3-hour period of preconditioning hypoxia was initiated. All drugs except the nNOS inhibitor 7-NI were dissolved in saline and administered intraperitoneally at a volume of 10 mL/kg body wt. 7-Nitroindazole was dissolved in 100% corn oil maintained at 80°C for 1 hour followed by sonication for 5 minutes. We also sought independent verification of the effectiveness of the intraperitoneal doses of NOS inhibitors used in this model. It was shown previously (Hamada et al., 1994) that pretreating PND 7 rat pups with 2 mg/kg L-NA attenuated the magnitude of hypoxicischemic brain injury, so these studies were not repeated. We used separate PND 7 litters (n = 3 for 7-NI and n = 4 for AG) to assess the efficacy of 40 mg/kg 7-NI and 400 mg/kg AG in protecting against hypoxic-ischemic brain injury in the absence of preconditioning. Litters were randomly divided into two groups: Half the pups in each litter were treated with 7-NI or AG 30 minutes before hypoxia (1 hour after unilateral carotid ligation), and the other half received appropriate vehicle.
To assess the involvement of NMDA receptor activation in preconditioning-induced protection, animals were pretreated with either a competitive or a noncompetitive NMDA receptor antagonist. 3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonate (CPP), a competitive antagonist for the NMDA receptor, was injected at 5 mg/kg intraperitoneally to PND 6 animals 30 minutes before preconditioning hypoxia. In pilot studies, we observed unacceptably high mortality when CPP was administered to PND 6 animals at a dose of 10 mg/kg. Because of long-lasting protective effects of the noncompetitive NMDA receptor antagonist MK-801, this drug was administered (1 mg/kg) at three separate time points in three different groups of litters: In one group (n = 6 litters), PND 6 animals received MK-801 30 minutes before hypoxic preconditioning. In a second group (n = 5 litters), MK-801 was administered to PND 5 animals 24 hours before preconditioning on PND 6. In the third group (n = 6 litters), MK-801 was administered to PND 4 animals 48 hours prior to preconditioning hypoxia on PND 6. As with the other drug-treated groups, each of these MK-801-treated litters was divided into four subgroups of animals: One subgroup was preconditioned at 0.5, 24, or 48 hours after MK-801 as described above, the second and third received MK-801 or vehicle, respectively, at the same time points but were not preconditioned, and the fourth served as vehicle-treated, nonpreconditioned controls.
Injury quantification and NOS measurements
Following hypoxia-ischemia on PND 7, all pups were returned to their dams, and acceptance of the pups by the mother, as indicated by nursing and nesting behavior, was verified to ensure the maintenance of postischemic normothermia for the pups. Within 1 to 2 hours, animals were returned to our institutional vivarium until PND 14, when unilateral brain injury was assessed by comparison of ligated and unligated hemispheric weight differences as described previously (Gidday et al., 1994, 1995). We and others have shown that results from this method of injury analysis are highly correlated to changes in infarct area measured in coronal sections, biochemical indexes of ischemic injury, and electrophysiologic criteria (Gidday et al., 1995).
Constitutive, calcium-dependent NOS activity was measured in brains of PND 6 animals exposed to either 45 minutes or 3 hours of preconditioning hypoxia (three litters) and also in brains of PND 6 animals 0.5, 2.0, 3.5, 7.0, and 24 hours after L-NA or 7-NI administration (five litters). Roughly half the drug-treated animals in the latter study were exposed to 3 hours of preconditioning hypoxia 30 minutes after drug administration. At the defined times, animals were completely immersed in liquid nitrogen, their brains were removed on dry ice, and the cerebellum and olfactory lobes were removed. Calcium-dependent NOS was measured in the remaining cerebral hemispheres using the [14C]arginine to [14C]citrulline method (Wang et al., 1995), and values were normalized within each litter to nonhypoxic vehicle-treated littermates.
Statistical analyses
All values are shown as means ± SD. Because hypoxicischemic brain injury in this model is often not distributed normally, within-litter differences in the extent of brain injury between treatment groups were assessed by Mann-Whitney nonparametric tests. Drug-induced differences in cerebral NOS activity were assessed by one-way analysis of variance. P values of <0.05 were accepted as significant.
RESULTS
Protection by hypoxic preconditioning
Since the publication of our first report describing our model of ischemic tolerance (Gidday et al., 1994), we have obtained data from an additional 53 litters that continue to confirm the robust nature of cerebroprotection in this model (Fig. 1). In this large sample, subjecting nonpreconditioned animals to hypoxia-ischemia resulted in a 40 ± 16% reduction in hemispheric weight measured 7 days after the insult (n = 153), whereas littermates (n = 149) preconditioned with hypoxia on the previous day exhibited only a 2 ± 8% reduction in hemispheric weight 7 days after hypoxia-ischemia, which was not different from matched nonischemic controls (3 ± 5%; n = 73). About 10% of nonpreconditioned animals subjected to hypoxia-ischemia were not injured, which is a common finding in the standard PND 7 hypoxia-ischemia model. The success rate of hypoxic preconditioning in protecting against hypoxic-ischemic injury in this neonatal rat model approaches 96%.
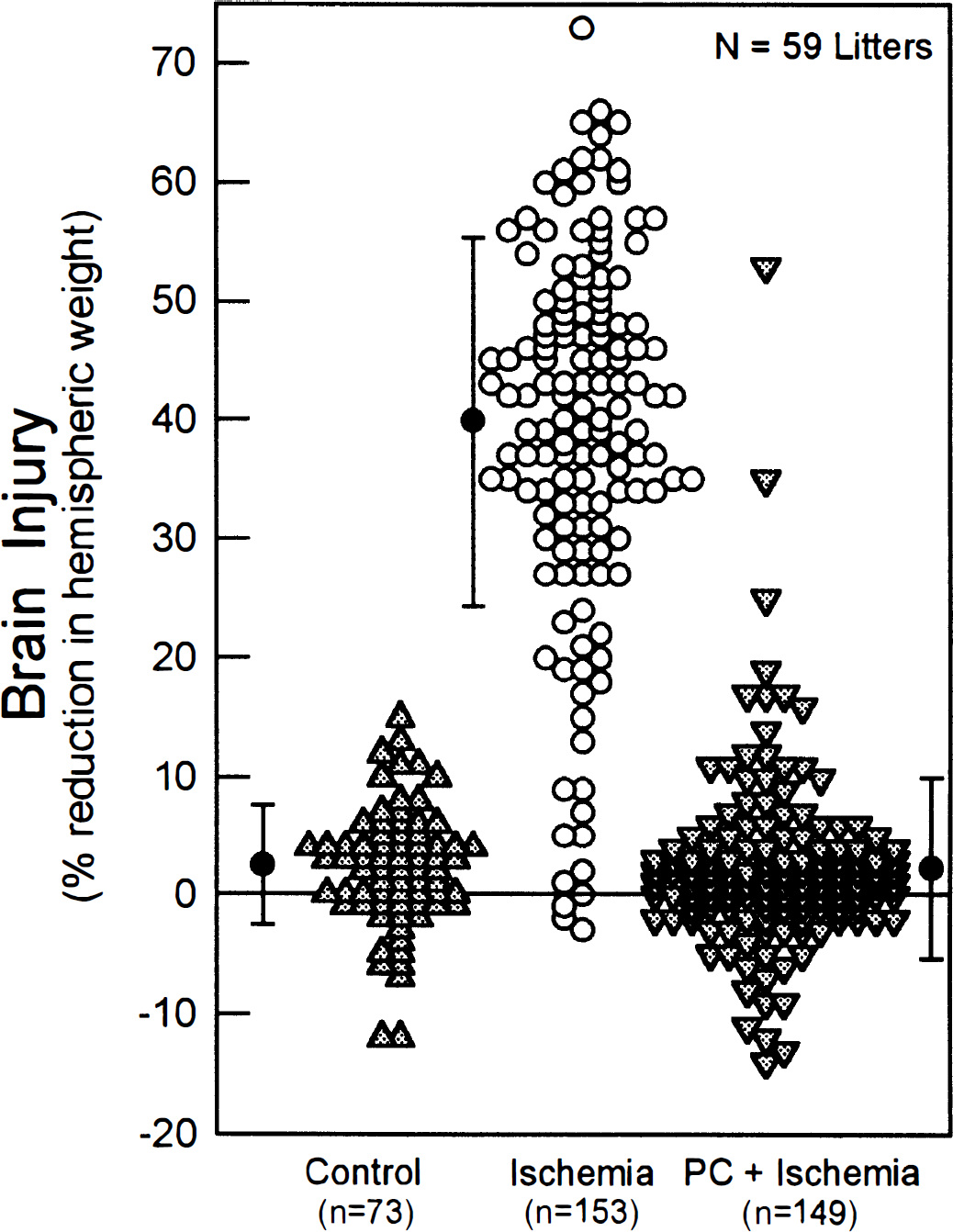
Preconditioning (PC)-induced protection from hypoxic-ischemic brain injury in 59 litters of PND 7 rat pups. Brain injury in preconditioned animals (3-hour hypoxic preconditioning on PND 6; n = 149) was significantly less than that measured in littermates that were not preconditioned (n = 153) and did not differ significantly from that measured in nonischemic controls (n = 73). Each point shown represents one animal. Means and standard deviations for each of the three animal groups are shown as filled symbols to the side of each distribution. Brain injury was quantified as the percent reduction in hemispheric weight, as previously described (Gidday et al., 1995).
Effect of L-NA on brain NOS and preconditioning protection
Our serial brain NOS measurements documented that 2 mg/kg L-NA significantly inhibited brain NOS. The extent of NOS inhibition by L-NA was not significantly affected by exposing the animals to hypoxia (data not shown), so the results from both hypoxia-exposed and normoxic subgroups were combined for display in Fig. 2. Note that NOS was inhibited 67 to 81% in L-NA-treated animals (n = 15) relative to vehicle-treated littermates (n = 12) during the initial 3.5 hours after intraperitoneal delivery, coinciding with the period during which the animals were exposed to preconditioning hypoxia. It was shown previously that this same dose of L-NA also reduced the extent of hypoxic-ischemic brain injury when given to PND 7 animals immediately before hypoxia-ischemia (Hamada et al., 1994); however, our data indicate that this protective effect is lost when L-NA is administered 24 hours before hypoxia-ischemia to nonpreconditioned PND 6 animals (Fig. 3), even though brain NOS activity remains inhibited.
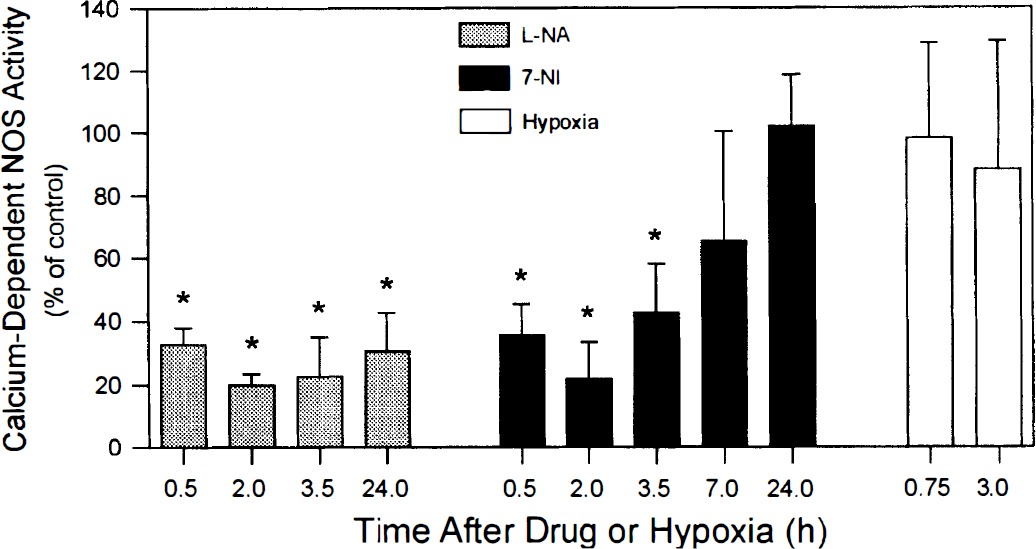
Changes in calcium-dependent, constitutive NOS activity at indicated times following L-NA (2 mg/kg intraperitoneally; shaded bars), 7-NI (40 mg/kg intraperitoneally; filled bars), and preconditioning hypoxia (0.75 or 3.0 hours). Values shown (means and standard deviations) were normalized to NOS levels measured in untreated nonischemic littermate controls. Each data point shown represents values from 4 to 12 brains. *P < 0.05 versus untreated controls.
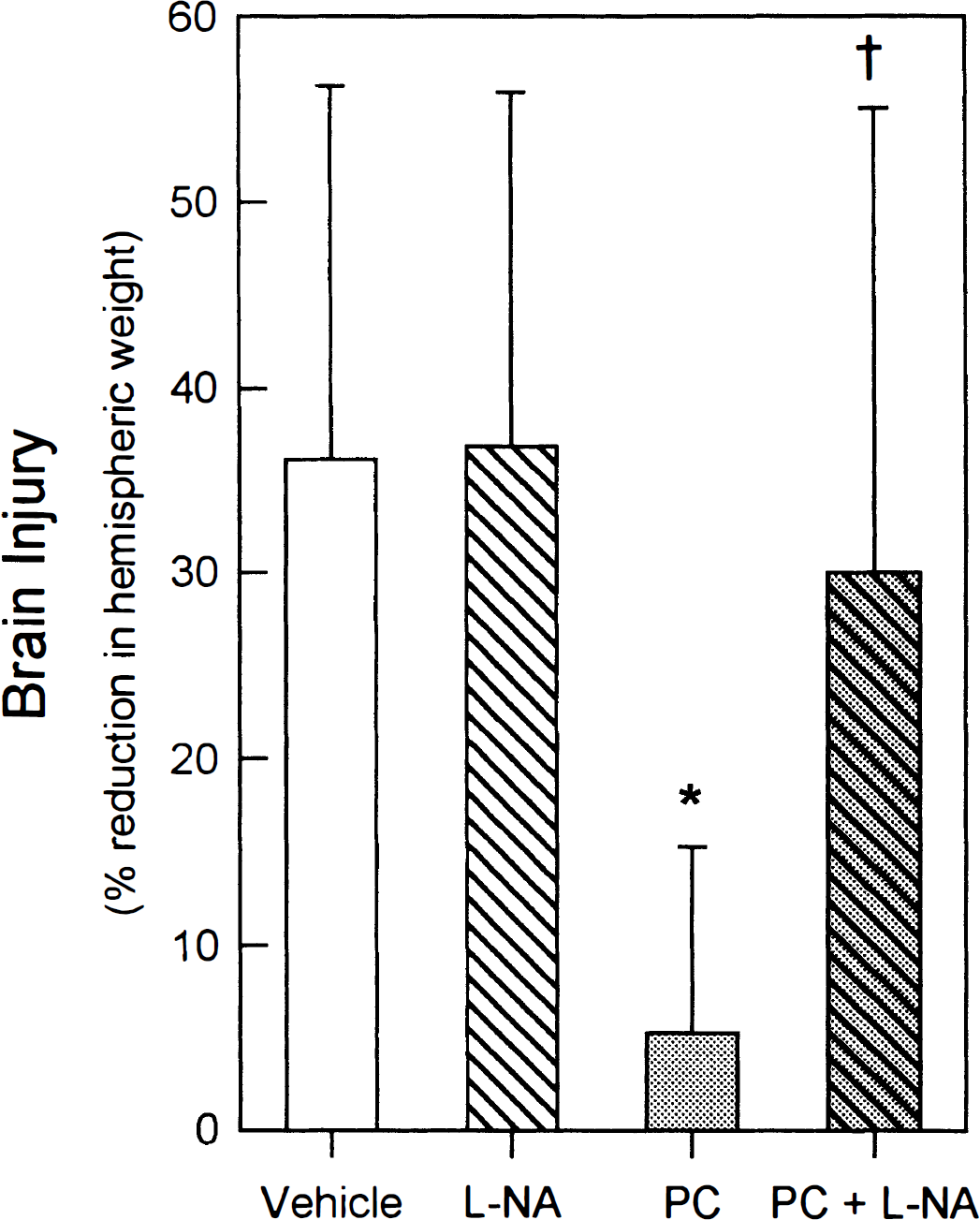
Pretreatment with the constitutive NOS inhibitor L-NA (2 mg/kg intraperitoneally) blocks preconditioning (PC)-induced protection against hypoxic-ischemic brain injury. Resultant brain injury (means and standard deviations) in the four intralitter subgroups is shown: Vehicle, vehicle-treated animals; L-NA, nonpreconditioned animals receiving L-NA on PND 6; PC, preconditioned animals without L-NA pretreatment; and PC + L-NA, animals pretreated with L-NA 30 minutes before preconditioning. *P < 0.05 versus vehicle group; †P < 0.05 versus PC group.
Fig. 3 shows that pretreating animals (n = 21) with L-NA 30 minutes before preconditioning hypoxia resulted in a near complete abolition of preconditioning-induced protection, indicative of an important role for NO in the induction of an ischemia-tolerant state. The extent of brain injury in these animals, manifested as a 30 ± 25% reduction in hemispheric weight, was not significantly different from that measured in nonpreconditioned vehicle-treated animals (36 ± 20%; n = 17). Conversely, the subgroup of preconditioned littermates not receiving L-NA (n = 17) exhibited only a 5 ± 10% reduction in weight, not significantly different from nonischemic controls.
Effect of 7-NI on brain NOS, hypoxic-ischemic injury, and preconditioning protection
To ensure that the 40-mg/kg 7-NI dose we used significantly inhibited nNOS, we serially measured brain NOS levels after drug administration and we verified that 40 mg/kg 7-NI would protect against hypoxic-ischemic brain injury when given to nonpreconditioned PND 7 animals subjected to hypoxia-ischemia. As shown in Fig. 2, within 30 minutes of 7-NI administration, NOS was inhibited 65%; the maximum extent of inhibition attained at 2 hours (75 to 81%); by 3.5 hours, the extent of inhibition was reduced slightly to 58%. As with L-NA, the magnitude of NOS inhibition during the initial 3.5 hours was not affected by exposing the animals to hypoxia, indicating that this nNOS inhibitor blocked a significant portion of nNOS activity during the period of preconditioning. Unlike the L-NA-treated animals, however, the inhibitory effect of 7-NI was transient, with NOS levels returning to baseline by 24 hours.
When nonpreconditioned PND 7 animals were pretreated with 40 mg/kg 7-NI 30 minutes before hypoxiaischemia, brain injury was reduced by 56%. In these litters, vehicle-treated animals (n = 15) exhibited a reduction in hemispheric weight of 35 ± 20%, whereas littermates (n = 14) pretreated with 7-NI showed only a 16 ± 20% reduction in hemispheric weight.
Fig. 4 shows that, in contrast to the inhibitory effects on preconditioning that we observed with L-NA, no attenuation of preconditioning-induced protection was noted in animals that were pretreated with the nNOS inhibitor 7-NI (40 mg/kg) 30 minutes before preconditioning hypoxia. In particular, the robust protection seen in preconditioned animals (9 ± 15%; n = 18) was not affected by pretreating animals with 7-NI immediately before preconditioning (a 5 ± 12% reduction in hemispheric weight; n = 21). With respect to the remaining littermate control groups, the nonpreconditioned vehicle-treated littermates (n = 21) exhibited a 43 ± 16% reduction in ipsilateral hemispheric weight, similar to the hemispheric weight loss observed in littermates treated on PND 6 with 7-NI but not subsequently preconditioned (38 ± 15%; n = 19).
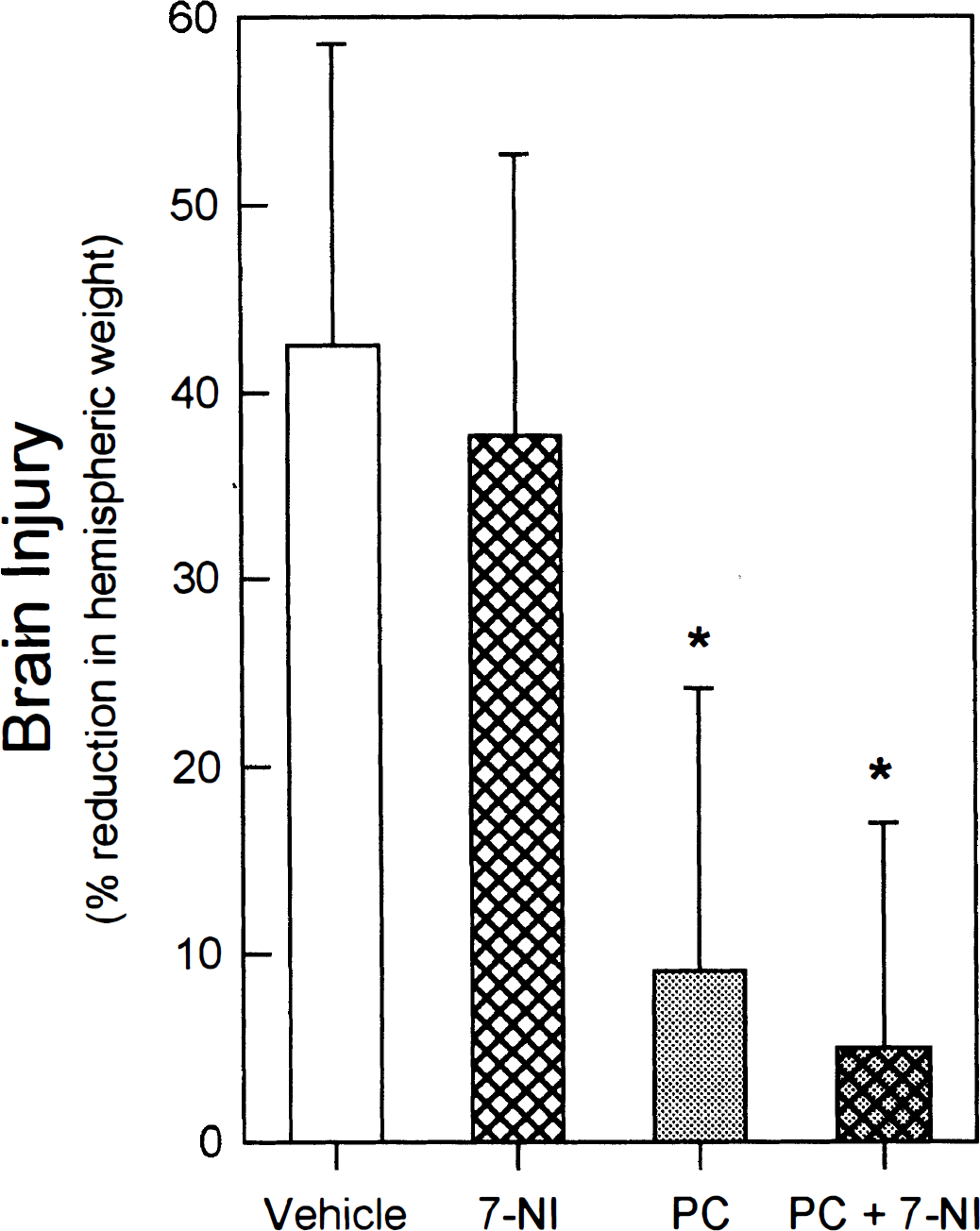
Pretreatment with the nNOS inhibitor 7-NI (40 mg/kg intraperitoneally) has no effect on preconditioning (PC)-induced protection against hypoxic-ischemic brain injury. Resultant brain injury (means and standard deviations) in the four intralitter subgroups is shown: Vehicle, vehicle-treated animals; 7-NI, nonpreconditioned animals receiving 7-NI on PND 6; PC, preconditioned animals without 7-NI pretreatment; and PC + 7-NI, animals pretreated with 7-NI 30 minutes before preconditioning. *P < 0.05 versus vehicle group.
Effect of AG on preconditioning protection and hypoxic-ischemic injury
Fig. 5 shows that the development of tolerance was also unaffected by AG pretreatment. The extent of preconditioning-induced protection in untreated animals (a 4 ± 13% reduction in hemispheric weight; n = 12) was not different from that measured in preconditioned animals pretreated with AG (an 8 ± 17% reduction in hemispheric weight; n = 20). In these litters, a 35 ± 17% reduction in ipsilateral hemispheric weight was found in the nonpreconditioned vehicle-treated animals (n = 9), which was similar to that found in littermates treated on PND 6 with AG but not subsequently preconditioned (32 ± 31%; n = 10).
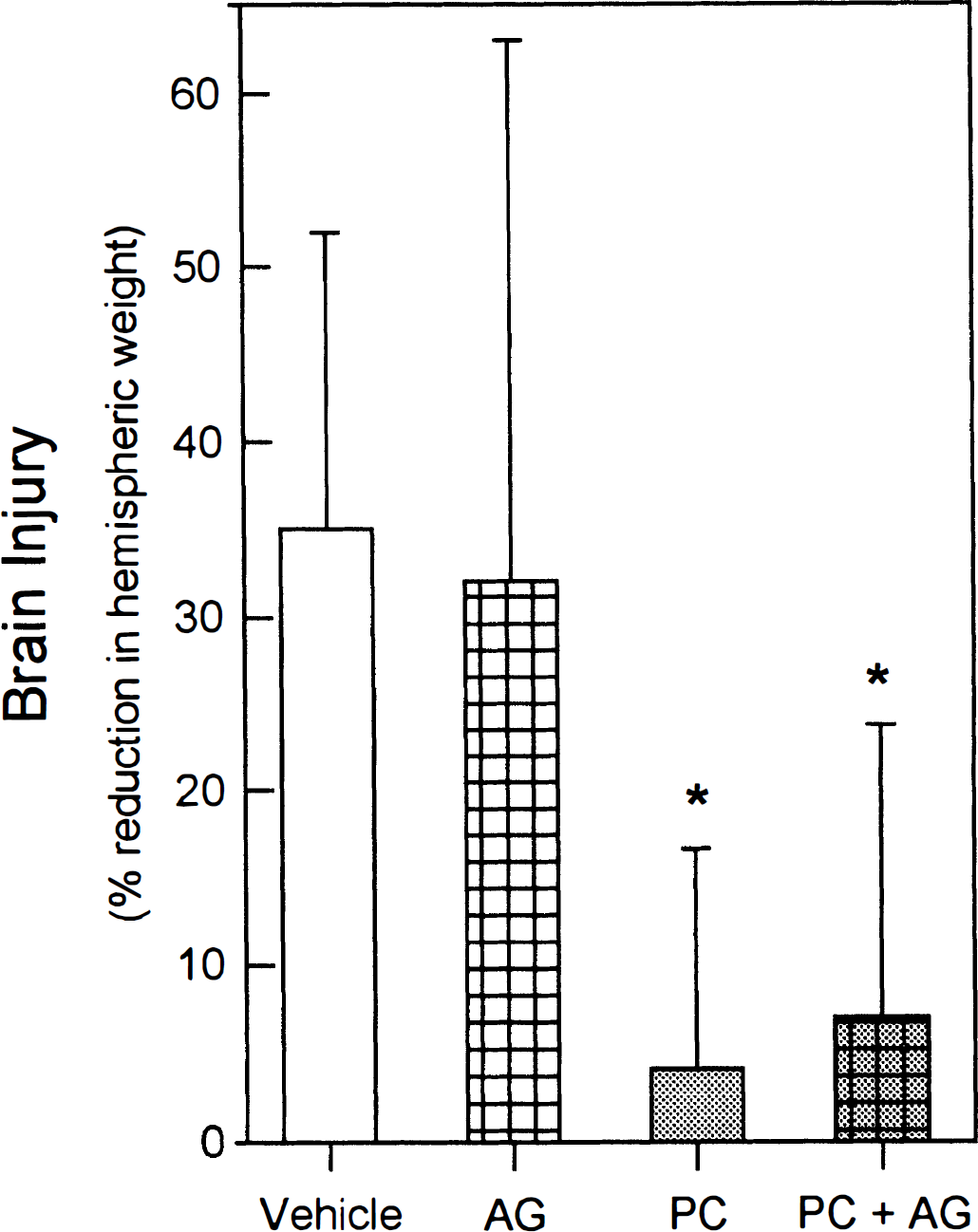
Pretreatment with the iNOS inhibitor AG (400 mg/kg intraperitoneally) has no effect on preconditioning (PC)-induced protection against hypoxic-ischemic brain injury. Resultant brain injury (means and standard deviations) in the four intralitter subgroups is shown: Vehicle, vehicle-treated animals; AG, nonpreconditioned animals receiving AG on PND 6; PC, preconditioned animals without AG pretreatment; and PC + AG, animals pretreated with AG 30 minutes before preconditioning. *P < 0.05 versus vehicle group.
We also verified that the 400-mg/kg dose of AG we used was sufficient to affect outcome from cerebral ischemia. Treating nonpreconditioned PND 7 animals with 400 mg/kg AG 30 minutes before hypoxia-ischemia resulted in a 41% reduction in brain injury, measured 1 week later. Specifically, vehicle-treated animals in these litters (n = 22) exhibited a reduction in hemispheric weight of 43 ± 16%, whereas AG-treated littermates (n = 32) showed only a 26 ± 20% reduction in hemispheric weight.
Effect of hypoxia on brain NOS
Calcium-dependent NOS activity in brains of animals subjected to 45 minutes or 3 hours of preconditioning hypoxia (no drug treatments) was not elevated relative to NOS activity levels measured in littermate controls (Fig. 2).
Effect of NMDA receptor antagonism on preconditioning protection
We used both CPP and MK-801, competitive and non-competitive antagonists at the NMDA receptor, respectively, to test the hypothesis that NMDA receptor activation was a necessary step in the induction of a state of ischemic tolerance. Neither pretreatment blocked preconditioning-induced protection. In the five litters used for the CPP studies, injury in the CPP-pretreated preconditioned animals (a 3 ± 5% reduction in hemispheric weight; n = 12) did not differ from values in untreated preconditioned littermates (1 ± 12%; n = 10). There was a trend for CPP to have a lasting protective effect against hypoxic-ischemic injury induced 24 hours after a single dose, but the magnitude of injury in this subgroup (29 ± 24% reduction in hemispheric weight; n = 13) was not significantly different from that measured in vehicle-treated littermates (42 ± 12%; n = 13) (Fig. 6).
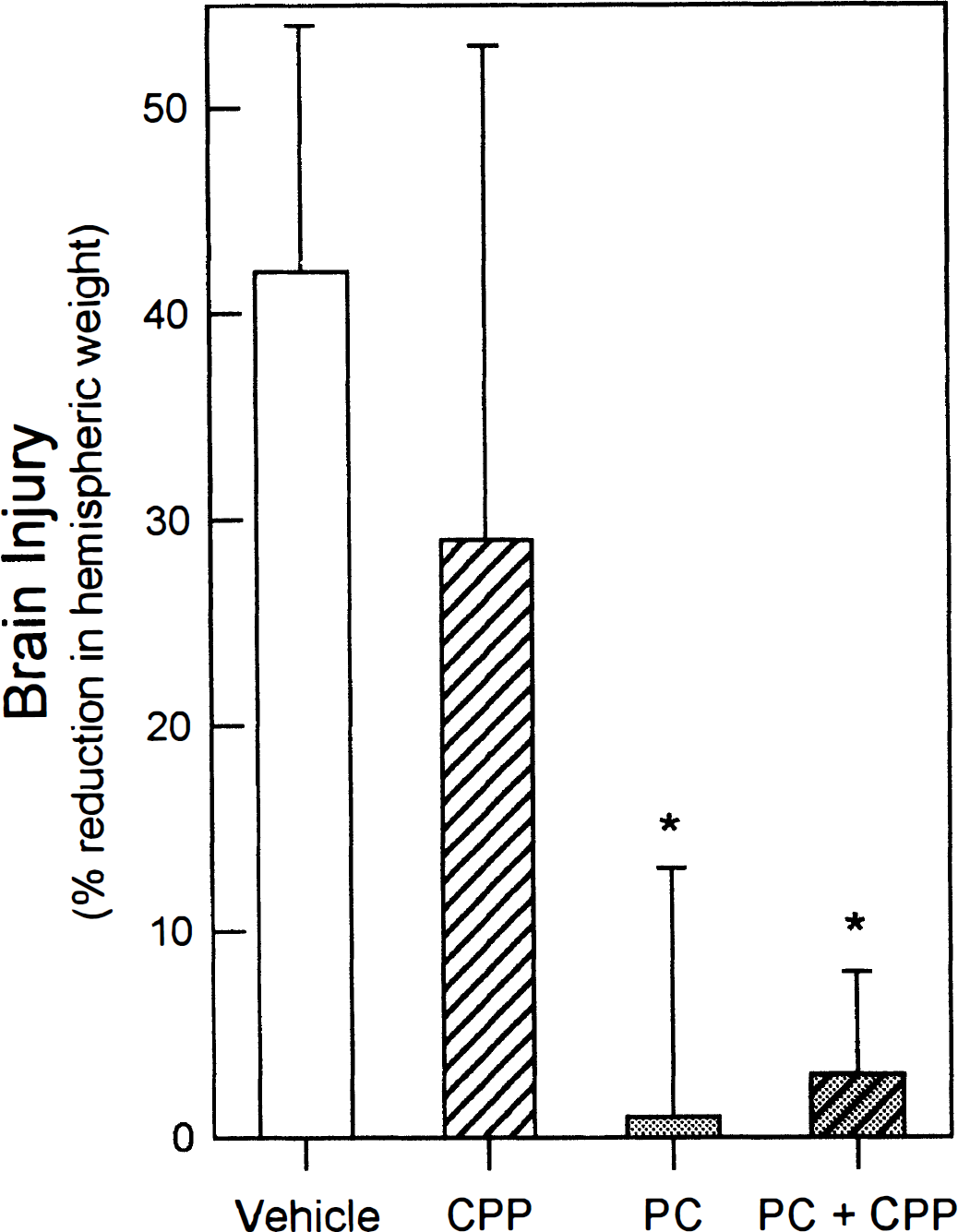
Pretreatment with the competitive NMDA receptor antagonist CPP (5 mg/kg intraperitoneally) is without effect on preconditioning (PC)-induced protection against hypoxic-ischemic brain injury. Resultant brain injury (means and standard deviations) in four intralitter subgroups is shown: Vehicle, vehicle-treated animals; CPP, nonpreconditioned animals receiving CPP on PND 6; PC, preconditioned animals without CPP pretreatment; and PC + CPP, animals pretreated with CPP 30 minutes before preconditioning. *P < 0.05 versus vehicle group.
With respect to the MK-801 studies, data from the one subgroup of nonpreconditioned MK-801-treated animals revealed that MK-801 significantly protected the PND 7 brain from hypoxic-ischemic injury when administered 24 hours (6 ± 7% reduction in hemispheric weight; n = 11) or even 48 hours (12 ± 19% reduction in hemispheric weight; n = 14) earlier, but this protection was lost if the pretreatment time was extended to 72 hours (36 ± 16% reduction in hemispheric weight; n = 14). Thus, the effects of MK-801 on preconditioning could be assessed only with a 48-hour pretreatment time; the resulting protection at shorter pretreatment times precluded us from separately identifying an effect of this drug on preconditioning. In light of this pharmacokinetic profile, we treated PND 4 animals with MK-801 so that the potential NMDA receptor stimulation resulting from hypoxic preconditioning 48 hours later on PND 6 would be blocked, but not the potential NMDA receptor activation resulting from hypoxia-ischemia 72 hours later on PND 7. We found that MK-801 pretreatment in these animals (n = 21) did not affect the extent of protection produced by subsequent hypoxic preconditioning (4 ± 10%). The extent of protection in vehicle-treated preconditioned animals did not differ between the three remaining subgroups of animals, nor did the extent of injury in vehicle-treated, nonpreconditioned animals, so these data were averaged, resulting in a 3 ± 7% (n = 47) and a 37 ± 16% (n = 46) reduction in hemispheric weight, respectively (Fig. 7).
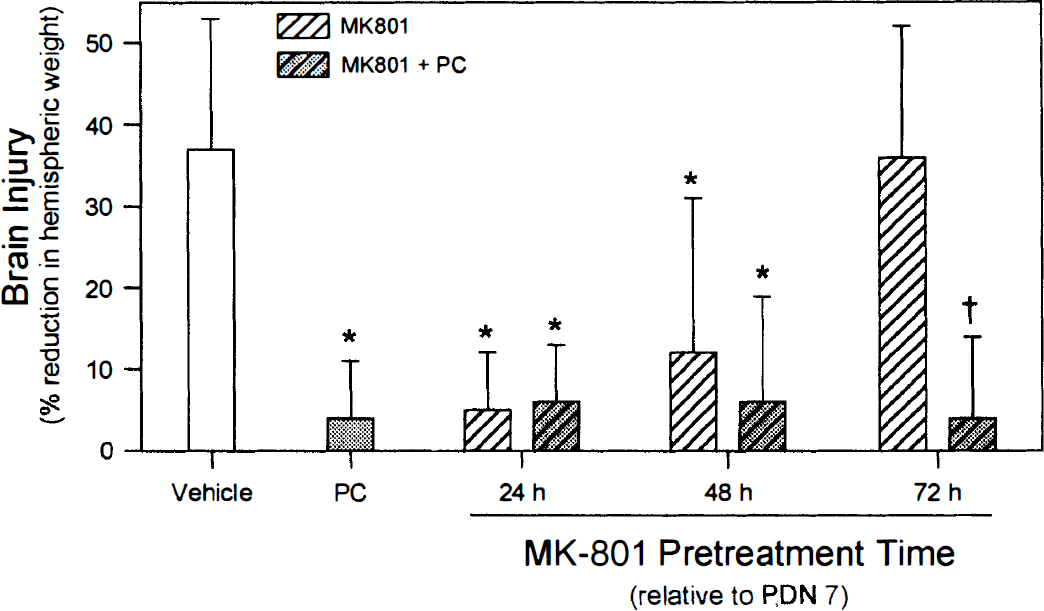
Pretreatment [48 hours before preconditioning (PC); 72 hours before hypoxia-ischemia] with the noncompetitive NMDA receptor antagonist MK-801 (1 mg/kg intraperitoneally) is without effect on preconditioning-induced protection against hypoxic-ischemic brain injury. Resultant brain injury (means and standard deviations) in three separate study groups is shown: MK-801 was administered 30 minutes, 24 hours, or 48 hours before PC on PND 6 (i.e., 24, 48, or 72 hours before hypoxia-ischemia on PND 7, respectively). Brain injury in nonpreconditioned animals receiving MK-801 at 24, 48, and 72 hours before hypoxia-ischemia on PND 7 is represented by the open hatched bars; note that the protective effect of MK-801 lasted at least 48 hours after its administration. The shaded hatched bars represent brain injuries in preconditioned animals receiving MK-801 at the same pretreatment times. Brain injury shown in vehicle-treated animals (vehicle) and preconditioned animals without MK-801 pretreatment (PC) represents the total mean from each respective subgroup in each of the three studies. *P < 0.05 versus vehicle-treated animals; †P < 0.05 versus MK-801 pretreatment without PC.
DISCUSSION
In the central nervous system, brief periods of preconditioning hypoxia or sublethal ischemia can confer protection against injury resulting from longer periods of focal or global ischemia that typically cause severe injury (Chen and Simon, 1997). Despite the availability of a number of reproducible small animal models of ischemic tolerance, little is known regarding the mechanistic basis for this robust protective response other than a possible role for heat shock proteins. We reported previously that the severe unilateral injury of the PND 7 rat pup brain resulting from unilateral carotid ligation and subsequent exposure to hypoxia, the basis of a well established model of perinatal ischemic brain injury (Rice et al., 1981; Hagberg et al., 1997), can be completely prevented if animals are exposed to short-term hypoxia 24 hours earlier (Gidday et al., 1994). We used this model of ischemic tolerance to test the hypothesis that NO production during the hypoxic preconditioning stimulus is necessary to activate the protective pathways that will ultimately render the brain resistant to the lethal hypoxic-ischemic insult imposed the next day. We also tested the related hypothesis that it is the nNOS isoform, secondary to a hypoxia-induced stimulation of NMDA receptors, that serves as the enzymatic source of NO. We found that neither selective nNOS inhibition nor NMDA receptor antagonism affected the magnitude of preconditioning-induced protection. Inhibition of iNOS with AG was also without effect on the development of ischemic tolerance. However, blockade of preconditioning with the nonselective NOS inhibitor L-NA suggests that NO production by the eNOS isoform is necessary for tolerance induction either as a permissive factor or in the direct triggering of the endogenous protective mechanisms.
Our measures of constitutive NOS activity indicate that L-NA significantly inhibited NOS during the period of hypoxic preconditioning. In fact, this inhibition persisted 24 hours later, even though nonpreconditioned animals pretreated on PND 6 were not protected relative to animals treated with the same dose of L-NA immediately before hypoxia-ischemia (Hamada et al., 1994). One interpretation suggested by these findings is that compensatory changes might occur in response to prolonged periods of NOS inhibition. This was recently shown for the normally nNOS-dependent hyperemic response to NMDA-a response blocked by acute but not chronic L-NA administration (Pelligrino et al., 1996). Along these same lines, one might consider that the possible compensation occurring with prolonged nNOS inhibition may not occur in association with extended eNOS blockade. Thus, vasoconstriction and other adverse effects of eNOS inhibition may persist, whereas the beneficial effects of nNOS inhibition may not. In any event, the key point is that the acquisition of tolerance was blocked by inhibition of NOS during preconditioning hypoxia, which suggests an “adequate” level of NO production needs to be maintained during or shortly after hypoxic preconditioning for the induction of ischemic tolerance in our model. Since the nonselective nature of L-NA-mediated NOS inhibition (Fukuto and Chaudhuri, 1995) does not allow for the identification of the particular NOS isoform (nNOS, eNOS, or iNOS) or the NO-producing cell type (neurons, astrocytes, or endothelial cells) involved in preconditioning the neonatal rat brain, we also tested other isoform-selective inhibitors of NOS.
We originally hypothesized that NMDA receptor stimulation during hypoxic preconditioning might activate nNOS within the extensive processes of nNOS-positive neurons and the NO so formed might then signal the induction of preconditioning mechanisms in surrounding neurons. In other words, the same anatomic relationship that forms the basis for the widespread hemodynamic and cytotoxic actions of NO in brain under physiologic and pathophysiologic conditions, respectively, could also provide NO a widespread influence in the setting of preconditioning. However, we found the nNOS-selective inhibitor 7-NI (Wang et al., 1995; Southan and Szabó, 1996; Moore and Handy, 1997) to be without any effect on the development of ischemic tolerance. This observation implies that NO derived from nNOS is not required for the protective effect induced by hypoxic preconditioning. Although 7-NI is known to exhibit a short-acting pharmacokinetic profile, we showed directly that the dose of 7-NI we used (40 mg/kg) was still sufficient enough to reduce NOS activity in the PND 6 brain rapidly and substantially (55 to 81%) 0.5 to 3.5 hours after its administration, coincident with the period of hypoxic preconditioning. In addition, we demonstrated that this same dose of 7-NI attenuated brain injury robustly when administered to nonpreconditioned PND 7 animals just prior to hypoxia-ischemia; this latter result is consistent with the hypothesis that the neuronal isoform of NOS is detrimental in the setting of cerebral ischemia in both adult animals (Huang et al., 1994; Yoshida et al., 1994) and in this PND 7 rat model (Ferriero et al., 1996).
The inability of both a competitive and a noncompetitive NMDA receptor antagonist to affect preconditioning outcome is also not consonant with our hypothesis that nNOS activation secondary to a hypoxia-induced stimulation of NMDA receptors is required for the induction of ischemic tolerance in our model. Like our 7-NI results, these findings were unanticipated for several reasons. First, exposure of PND 7 rat pups to moderate hypoxia similar to our preconditioning stimulus increases brain glutamate levels, albeit modestly and transiently (Krajnc et al., 1996). Second, the link between NMDA receptor stimulation, calcium, and NOS activation is well established (Garthwaite and Boulton, 1995; Zhang and Snyder, 1995). Third, MK-801 blocks the development of ischemic tolerance in adult gerbils (Kato et al., 1992). Fourth, reports in the literature suggest that the systemic doses of MK-801 and CPP we used were sufficient to antagonize NMDA receptors, given the significant reductions in PND 7 rat brain injury afforded by these compounds following hypoxia-ischemia (McDonald et al., 1987) and intracerebral NMDA injections (Van Lookeren Campagne et al., 1994; McDonald et al., 1989). The complete cerebroprotection against hypoxic-ischemic injury that we witnessed in nonpreconditioned animals in the present study with 1 mg/kg MK-801, even up to 48 hours after its administration, indicates that NMDA receptor blockade was likely during the period of preconditioning hypoxia. These findings lead us to conclude that NMDA receptor stimulation is not required for the induction of ischemic tolerance in this model. Of course, this does not exclude a role for α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors or metabotropic glutamate receptors in the mechanistic pathway of tolerance induction; this possibility awaits further study.
We also examined the possibility that the establishment of a state of ischemic tolerance following the preconditioning hypoxia stimulus requires iNOS induction, as L-NA can also inhibit this isoform to varying degrees depending on experimental conditions. These experiments were prompted by several findings. First and foremost, iNOS mRNA and protein are basally expressed in parenchymal blood vessels throughout the PND 7 brain, in contrast to the adult brain, wherein basal microvessel iNOS expression is absent (Galea et al., 1995). This constitutive vascular iNOS, or the traditional iNOS in astrocytes (Murphy et al., 1993), could provide the framework for a widespread influence of NO in terms of signaling or initiating tolerance mechanisms in adjacent neurons. Rat cortical iNOS mRNA and iNOS enzymatic activity increases robustly within 12 hours of middle cerebral artery occlusion (Iadecola et al., 1996), which is less than the 24 hours or greater time periods typically required in most models between exposure to the preconditioning stimulus and the manifestation of a state of ischemic tolerance (Chen and Simon, 1997). However, the impotency of AG on the development of preconditioning-induced protection in our model, even though the same dose of AG protected against hypoxic-ischemic injury in nonpreconditioned animals, argues against the possibility that NO produced by the iNOS isoform is mechanistically important in ischemic tolerance.
Blockade of tolerance induction with the nonselective NOS inhibitor L-NA, but not with the nNOS-selective inhibitor 7-NI and the iNOS inhibitor AG, suggests, by exclusion, that the eNOS isoform serves as the NO source required for hypoxic preconditioning. Endothelial cells comprising the cerebral microcirculation are ubiquitously juxtaposed to nearly all resident neuronal and glial cells and thus have a large spatial domain of influence. They are also directly exposed to changes in blood PaO2. There are two ways in which endothelially derived NO could participate in the preconditioning process: NO production by endothelial cells could increase in response to an up-regulation of eNOS secondary to the hypoxic preconditioning stimulus; conversely, no additional increase in endothelial NO production above baseline may occur during hypoxic preconditioning, but the ongoing production of NO by these cells may still be necessary to permit an independent mechanism of preconditioning to be fully operational.
In the first instance, an eNOS-dependent increase in NO production above basal levels, the magnitude of which would likely be dependent on the duration and intensity of the sublethal hypoxic/ischemic preconditioning stimulus, could signal a series of adaptive changes in neuronal gene expression at concentrations below those implicated in neuronal cytotoxicity secondary to severe ischemia (Iadecola, 1997). Indeed, NO can affect immediate early gene expression (Peunova and Enikolopov, 1993; Morris, 1995; Pilz et al., 1995), enzyme and receptor sensitivity (Lipton and Stamler, 1994), and the synthesis of heat shock proteins (Malyshev et al., 1995), which are putatively important in some models of ischemic tolerance (Chen and Simon, 1997). If such an up-regulation of eNOS occurs in response to hypoxic preconditioning, we would not have detected it in our assay of total constitutive NOS catalytic activity for two reasons: First, the assay we used measures soluble NOS activity (nNOS) and not particulate NOS activity (eNOS), and second, eNOS represents only a small fraction of total brain NOS (Huang et al., 1994). Although focal cerebral ischemia is associated with a fairly rapid increase in eNOS activity (Zhang et al., 1993; Nagafuji et al., 1995), the effects of hypoxia on eNOS activity in vivo are less convincing. In fact, exposure of adult rats to 4-hour hypobaric hypoxia does not up-regulate eNOS expression in cerebellum (Prabhakar et al., 1996) and hypoxic vasodilation in the newborn piglet brain occurs independently of NO formation (Armstead, 1995; Leffler et al., 1997).
It is also possible the L-NA-mediated block of preconditioning may be “indirect” as a result of a reduction in the tonic influence of eNOS-generated NO on the activity of other biochemical pathways required for the development of tolerance. Such a “permissive effect” of NO may depend on the maintenance of a certain level of enzyme phosphorylation by cyclic GMP-dependent kinases and/or on the ADP ribosylation and S-nitrosylation of transcription factors and other enzymes (Zhang and Snyder, 1995) including cyclooxygenase, NADH-ubiquinone oxidoreductase, cis-aconitase, glyceraldehyde-3-phosphate dehydrogenase, and iron-sulfur-containing enzymes (Zhang and Snyder, 1995). The resultant L-NA-induced alterations in the metabolic status of the cell may no longer permit the optimal functioning of another independent mechanism more directly involved in inducing the ischemia-tolerant state. Such a permissive mechanism is akin to that proposed for NO in mediating hypercapnic vasodilation (Iadecola and Zhang, 1996).
With respect to the relevance of our findings in this neonatal model of ischemic tolerance to the adult brain, ontogenic studies of both NOS expression and NMDA receptor activity in the developing rat brain suggest some important similarities and differences among these age groups. For example, NMDA receptor-mediated excitotoxic pathways underlie brain injury in response to hypoxia-ischemia in the PND 7 rat in a mechanistic fashion similar to that which is operative in the adult brain; if anything, NMDA receptor-dependent functions may, in fact, be more predominant in the PND 7 brain than in the adult (McDonald and Johnston, 1990). With respect to NOS, the level of eNOS expression in rat cerebral blood vessels during early pre- and postnatal life is equivalent to that seen in adults (Keilhoff et al., 1996). In the pig, NO-dependent cerebrovascular regulation appears to assume increasing significance as the animals mature (Zuckerman et al., 1996). There are age-dependent differences in NOS distribution and activity as well: The regional distribution of constitutive brain NOS activity changes somewhat during maturation, together with a shift from particulate to cytosolic NOS; on the other hand, it appears that it is the same NOS protein in all instances, and that the inhibitory effects of L-NA on both fractions are the same (Matsumoto et al., 1993). Northern and western blot results, as well as NOS immunoreactivity and NADPH-diaphorase histochemistry studies, specifically indicate that nNOS isoforms are present in rat brain as early as embryonic day 10 (Keilhoff et al., 1996). nNOS protein expression in cerebrum tends to increase after birth, reaching adult levels by PND 14 (Keilhoff et al., 1996; Lizasoain et al., 1996), consistent with a role for NO in cell maturation, synaptic connectivity, and reshaping of the developing central nervous system.
Considerable interest surrounds the physiologic role of NO in the coupling of cerebral blood flow and metabolism, in NMDA-mediated neurotoxicity, in diverse paracrine signaling roles, and in forms of synaptic plasticity such as long-term potentiation (Garthwaite and Boulton, 1995). We propose that NO's integral role in preconditioning is a previously unrecognized aspect of NO function in brain. Indeed, studies of ischemic preconditioning in other tissues support a critical role for NO in mediating both immediate (Vegh et al., 1992) and delayed (Bolli et al., 1997) protection in heart, liver (Peralta et al., 1997), and intestine (Hotter et al., 1996). Our results also lead us to conclude that eNOS-dependent production of NO by cerebral endothelial cells may serve not only in a vascular homeostatic role by increasing tissue perfusion and inhibiting leukocyte and platelet adherence but also as a signal to surrounding neurons and glia that is required in some as yet undefined way to promote delayed, relatively long-lasting adaptive responses that increase their resistance to ischemia. Further studies are required to elucidate the NO-dependent mechanisms that are required for such robust neuroprotection.
Footnotes
Acknowledgements
The authors thank Nimish Shah for technical assistance on these studies.