
Abstract
This study assessed the mechanism(s) by which the autoregulatory vasodilation of rat pial artery in response to acute hypotension during the acute phase of subarachnoid hemorrhage (SAH) was markedly blunted. Increased superoxide production from the cerebral vessels in response to NAD(P)H at 24 hours after SAH +NG-nitro-
Delayed cerebral vasospasm after subarachnoid hemorrhage (SAH) is known to be associated with impaired vasodilator and vasoconstrictor mechanisms in cerebral vessels and to begin on approximately day 3 or thereafter, reaching a maximum on days 6 to 8 in humans (Sobey and Faraci, 1998). The alteration of cerebral microcirculation is considered to be a possible contributing cause of cerebral ischemia (Kassell et al., 1985), and a marked autoregulatory disturbance of CBF has been observed in patients with SAH (Voldby et al., 1985; Yundt et al., 1998). After SAH in humans, decreased CBF in association with raised intracranial pressure with hydrocephalus and development of prolonged cerebral vasospasm was documented (Heilbrun et al., 1972; Voldby et al., 1985; Dirnagl et al., 1989).
Activation of tyrosine kinase with tyrosine phosphorylation plays a crucial role in signal transduction, including cell growth and differentiation and in modulation of K+ channel activity (Hollenberg, 1994; Xiong et al., 1995). Marked membrane depolarization of cerebral vascular smooth muscles following SAH was reported from 30 minutes up to 24 hours, most likely as a result of inhibition of potassium channels (Waters and Harder, 1985; Harder et al., 1987). Reactive oxygen species (ROS), including superoxide anion, play an important role in regulating vascular tone and in intracellular signaling of the vessels. In signaling pathways, endothelial and smooth muscle cells express a ROS-generating NAD(P)H-dependent oxidase containing p22phox and gp91phox, which are a membrane-bound component of the phagocyte-type NAD(P)H oxidase (Jones et al., 1996; Meyer et al., 1999). On activation of this enzyme, several cytosolic proteins, including Rac, p47phox, p67phox, and p40phox, translocate to the membrane and associate with the membrane-bound subunits. The importance of membrane depolarization was emphasized in the production of endothelial superoxide production (Al-Mehdi et al., 1998). Membrane depolarization of cultured human umbilical vein endothelial cells was shown to increase the tyrosine phosphorylation of the proteins in accordance with a significant increase in membrane association of the small G-protein Rac (Sohn et al., 2000). Rac protein is required for NAD(P)H oxidase activation in phagocytes (Mizuno et al., 1992; Heyworth et al., 1993) and nonphagocytes (fibroblast cells;Sundaresan et al., 1996). Given the known role of small GTP-binding protein Rac and ROS in signal transduction, Rac protein may function to regulate the production of intracellular ROS in cerebral vessels after SAH.
Investigators using SAH models of delayed vasospasm rarely refer to the cerebrovascular responsiveness to hypotension during the acute phase after SAH. Furthermore, little is known about the cellular mechanisms underlying superoxide formation during the early stage after SAH. We therefore examined whether the autoregulatory vasodilation is diminished during the acute phase in rat pial artery after SAH plus
MATERIALS AND METHODS
Preparation of animals
All animal studies carefully conformed to the guidelines outlined in the Guide for Animal Experiments edited by the Korean Academy of Medical Sciences, and were approved by the Animal Experimental Committee of College of Medicine, Pusan National University.
A single-hemorrhage rat model was used for SAH. Male Sprague-Dawley rats (250 to 300 g) were anesthetized with anesthesia cocktail composed of 1 mg/kg acetopromazine, 5 mg/kg xylazine, and 50 mg/kg ketamine. With the animal in a supine position on the stereotactic frame (Stoelting, Wood Dale, IL, U.S.A.), arterial catheter was inserted into femoral artery for withdrawing and sampling of the arterial blood. A small sagittal incision was made in the neck, and the atlanto-occipital membrane was exposed. The cisterna magna was pierced with a 27-gauge needle and 0.3 mL cerebrospinal fluid (CSF) was aspirated. Then, 0.3 mL autologous whole blood was injected into the cisterna magna. For the sham-operated control group, 0.3 mL of normal saline was injected into the cisterna magna in the same manner. Free access to food and water was allowed after recovery from anesthesia.
Measurement of autoregulatory vasodilation
Twenty-four hours after SAH, rats were anesthetized with urethane (1 g/kg) and placed on a heating pad (Homeothermic Blanket System; Harvard Apparatus, South Natick, MA, U.S.A.) to maintain a constant rectal temperature (37 ± 0.5°C). Following a tracheostomy, the animal was mechanically ventilated with room air by a respirator (model 683, Harvard Apparatus) after immobilization with 5-mg/kg gallamine triethiodide. The mean Paco2 was monitored with end-tidal CO2 analyzer (CapStar-100; IITC Life Science, Woodland Hills, CA, U.S.A.). Catheters were placed in a carotid artery for measurement of systemic arterial blood pressure (Statham P23D pressure transducer; Gould, Cleveland, OH, U.S.A.). The blood was collected before and after installation of a cranial window for blood gas and pH determination (i-STAT Portable Clinical Analyzer; Abbott Laboratories, East Windsor, NJ, U.S.A.).
The animal head was then fixed in a prone position with a stereotaxic apparatus (Stoelting, Wood Dale, IL, U.S.A.) and a square shape (5 × 5 mm) burr hole was made over the right parietal cortex. The dura was resected with caution. Pial precapillary microvessels were visualized through the cranial window, where prewarmed artificial CSF saturated with a mixed gas of 95% O2 and 5% CO2 was constantly suffused over the cortical surface at 0.3 mL/min. Cerebral microvessels were allowed to equilibrate for 60 to 90 minutes after installation of the cranial window. The image of pial microvessels was captured with a CCD videocamera (VDC 3900; Sanyo, Japan) through a stereomicroscope (model SMZ-2T; Nikon, Japan). It was fed to a television monitor for direct observation, and the caliber was measured using a Width analyzer (C3161; Hamamatsu Photonics, Hamamatsu, Japan). The composition (in mmol/L) of the artificial CSF was as follows: 132 NaCl, 2.9 KCl, 1.4 MgCl2, 24.6 NaHCO3, 1.2 CaCl2, 6.7 urea, and 3.7 d-glucose (pH 7.4). The intracranial pressure was maintained constant at 5 to 6 mm Hg throughout the experiment by adjusting the height of the free end of the plastic tubing, which was connected to the outlet of the window. Only one arteriole was observed under the window in each rat.
Protocol for in vivo experiments
For inducing hypotension, the femoral blood was withdrawn to the reservoir 1 mL every 2 minutes while the arterial blood pressure was monitored. In the case of infusion, 1 mL of blood in the reservoir was infused back every 2 minutes. The decrease in blood pressure at each step was maintained for 2 minutes, and changes in vessel diameter during the last minute were measured.
Measurement of superoxide anion
Superoxide production in cerebral vessels after SAH was measured as an index for NAD(P)H oxidase activity. After removal of brain samples, cerebral vessels including anterior, middle, and posterior cerebral arteries, as well as the basilar artery were immediately isolated and immersed in ice-cold physiological salt solution (pH 7.4). Cerebral vessels isolated were placed in 200 μL of HEPES buffer containing lucigenin (bis-N-methylacridinium nitrate, 5 μmol/L) and were placed into the luminometer (Microlumat LB96P; EG & G Berthold, Germany). After dark adaptation, NADH and NADPH (100 μmol/L, each) were added to the vial. Counts were then recorded every 30 seconds for 10 minutes, and the respective background count was subtracted. Chemiluminescence was expressed as counts per second per milligram dry weight.
p22phox/gp91phox Expression
The expression of the NAD(P)H oxidase subunits p22phox and gp91phox was determined by RT-PCR. The cerebral vessels were homogenized in Trizol reagent (Gibco BRL; Life Technologies, Rockville, MD, U.S.A.), and chloroform was added to the lysates. After centrifugation, the upper layer was taken and subjected to isopropanol precipitation. Final total RNA concentration and purity was assessed by measurement of absorbance at 260 and 280 nm. First-strand cDNA synthesis was performed by using 1 μg of the total RNA with 500 ng of random hexamers (Promega, Madison, WI, U.S.A.) and DEPC-H2O. Reaction mixtures were preincubated at 65°C for 5 minutes, followed by cooling on ice for 10 minutes, before addition of 10 mmol/L of each dNTP, 5xRT buffer (250 mmol/L Tris-HCl (pH 8.3), 375 mmol/L KCl, 15 mmol/l MgCl2 and 50 mmol/L DTT), 33 U of the RNase inhibitor RNasin, and 100 U MMLV reverse transcriptase (Promega), and performed at 42°C for 60 minutes. PCR primers for amplification of p22phox were designed based on the sequences obtained (sense, 5′-GCT CAT CTG TCT GCT GGA GTA-3′ antisense, 5′-ACG ACC TCA TCT GTC ACT GGA-3′) (Zalba et al., 2000). PCR was carried out in a total volume of 50 μL containing reverse transcription reaction, 10x Taq polymerase buffer (500 mmol/L KCl, 100 mmol/L Tris-HCl [pH 9.0 at 25°C], 1.0% Triton X-100, 25 mmol/L MgCl2, 100 pmol primers, and 1 U Taq polymerase [Promega]). The conditions were 35 cycles of denaturation at 94°C (1 minute), annealing at 50°C (1 minute), and extension at 72°C (2 minute), followed by a 10-minute extension reaction at 72°C. The primer for gp91phox was based on Bayraktutan et al. (2000) (sense, 5′-CCT ATG ACT TGG AAA TGG AT-3′; antisense, 5′-CAG AGC CAG TAG AAG TAG AT-3′). The conditions were 35 cycles of denaturation at 94°C, annealing at 54°C (1 minute), and extension at 72°C (1 minute), followed by a 10-minute extension reaction at 72°C.
Western blotting for gp91phox and Rac translocation
The result of gp91phox mRNA expression by RT-PCR and Rac protein translocation were confirmed by Western blot analysis. The vessels isolated were centrifuged (250 g for 5 minutes, 4°C). After the supernatant was discarded, the pellet was resuspended in 1 mL of extraction buffer (25 mmol/L Tris-HCl, 2.0 mmol/L EGTA, 50 mmol/L 2-mercaptoethanol, 0.005% leupeptin, 0.25 mol/L sucrose, and 1.0 mmol/L phenylsulfonyl fluoride, pH 7.45). The suspension was homogenated for 10 seconds 3 times, then ultracentrifuged at 100,000 g at 4°C for 60 minutes. The supernatant of the sample was collected as the cytosolic fraction. One milliliter of the extraction buffer containing 0.1% Triton-X was added to the pellet of the sample and incubated at 4°C for 60 minutes. Thereafter, it was ultracentrifuged at 100,000 g at 4°C for 60 minutes. The supernatant containing the membrane fraction was used. Each sample (50 μg of total protein) was loaded into an SDS-PAGE gel, and transferred to nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, U.S.A.). The expression of gp91phox protein was determined using specific monoclonal antibody against neutrophil NAD(P)H oxidase subunit, gp91phox (donated by Dr. Mark T. Quinn), and the membrane-located Rac protein was determined by specific monoclonal antibody against Rac (Upstate Biotechnology, Lake Placid, NY, U.S.A.). The immunoreactive bands were visualized using chemiluminescent reagent of the Supersignal West Dura Extended Duration Substrate Kit (Pierce, Rockford, IL, U.S.A.). The signals of the bands were quantified using the Calibrated Imaging Densitometer (GS-710; Bio-Rad Laboratories, Hercules, CA). The protein concentration of the lysate was determined using the Bio-Rad DC assay kit (Bio-Rad Laboratories).
Protein tyrosine kinase assay
To determine whether the tyrosine phosphorylation is involved in depolarization-induced activation of endothelial superoxide production, protein tyrosine kinase activity was assayed. Cerebral vessels were quickly removed at 3, 12, 24, and 96 hours after SAH. Then, the samples were homogenized in extraction buffer (20 mmol/L Tris, pH 7,4, 50 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.2 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL pepstatin, 0.5 μg/mL leupeptin, 0.2 mmol/L Na3VO4, 5 mmol/L mercaptoethanol). Cell debris was removed by centrifugation at 16,000 g for 80 minutes and supernatant was used for enzyme-linked immunosorbent assay. The contents of protein tyrosine kinase in the homogenated cerebral vessels were measured using immunoassay kit (QIA28; Oncogene Research Products, Cambridge, MA, U.S.A.). Briefly, extract aliquots were incubated in the presence of compounds with Mg2+ and ATP. Phosphorylation of an immobilized substrate was recognized by a horseradish peroxidase—labeled, phosphotyrosine-specific antibody. The conversion of tetramethylbenzidine as a substrate of horseradish peroxidase was monitored spectrophotometrically at 450 nm with a reference at 540 nm. Protein tyrosine kinase activity was expressed as unit per milligram protein.
Drugs
L-NAME, β-nicotinamide adenine dinucleotide (phosphate) reduced form NAD(P)H, genistein, diphenyleneiodonium, allopurinol, sulfaphenazole, and indomethacin were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Genistein, diphenyleneiodonium, and allopurinol were dissolved in dimethylsulfoxide to make a stock solution of 1 mmol/L and diluted when used. Clostridium difficile toxin B was purchased from Calbiochem (San Diego, CA, U.S.A.).
Statistical analysis
Results are expressed as means ± SD. The comparison of mean arterial blood pressure—dependent arterial diameter changes between SAH and sham-operated control groups was analyzed by repeated measures analysis of variance, and followed by Tukey's multiple comparison tests as a post hoc comparison. Student's t-test was used for analyzing values between the data of vehicle and inhibitor-treated groups. P < 0.05 was accepted as statistically significant.
RESULTS
Mean levels of blood gas, pH and baseline pial arterial diameter of SAH, SAH +
Physiologic variables: mean arterial blood pressure and blood gas analyses
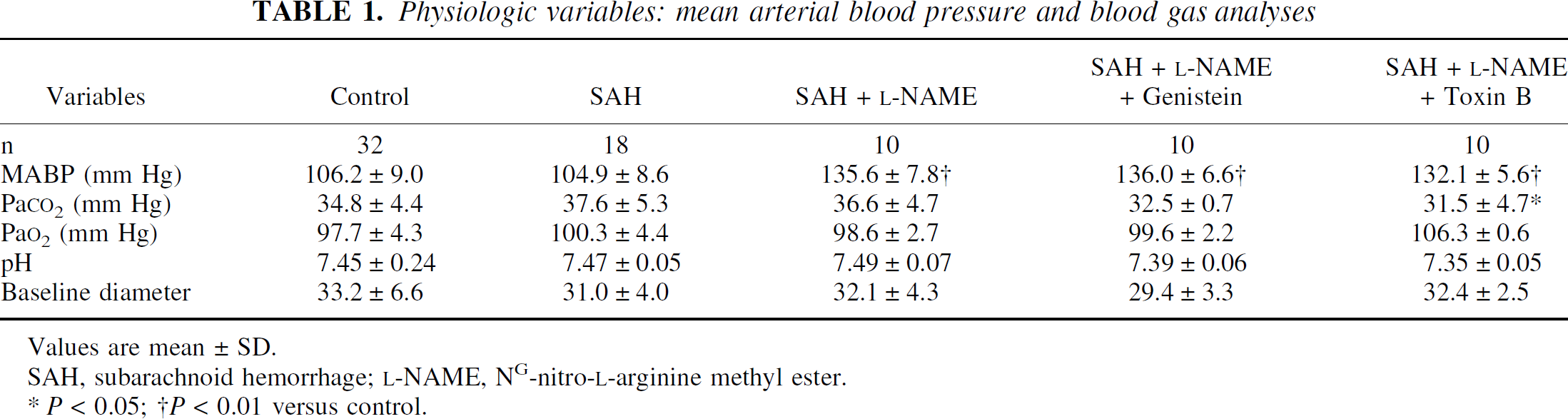
Values are mean ± SD.
SAH, subarachnoid hemorrhage;
P < 0.05;
P < 0.01 versus control.
Autoregulatory vasodilation following subarachnoid hemorrhage
At 24 hours after SAH, the autoregulatory vasodilation of the pial artery in response to acute hypotension was markedly blunted in 6 of 10 rats. When SAH rats were treated with
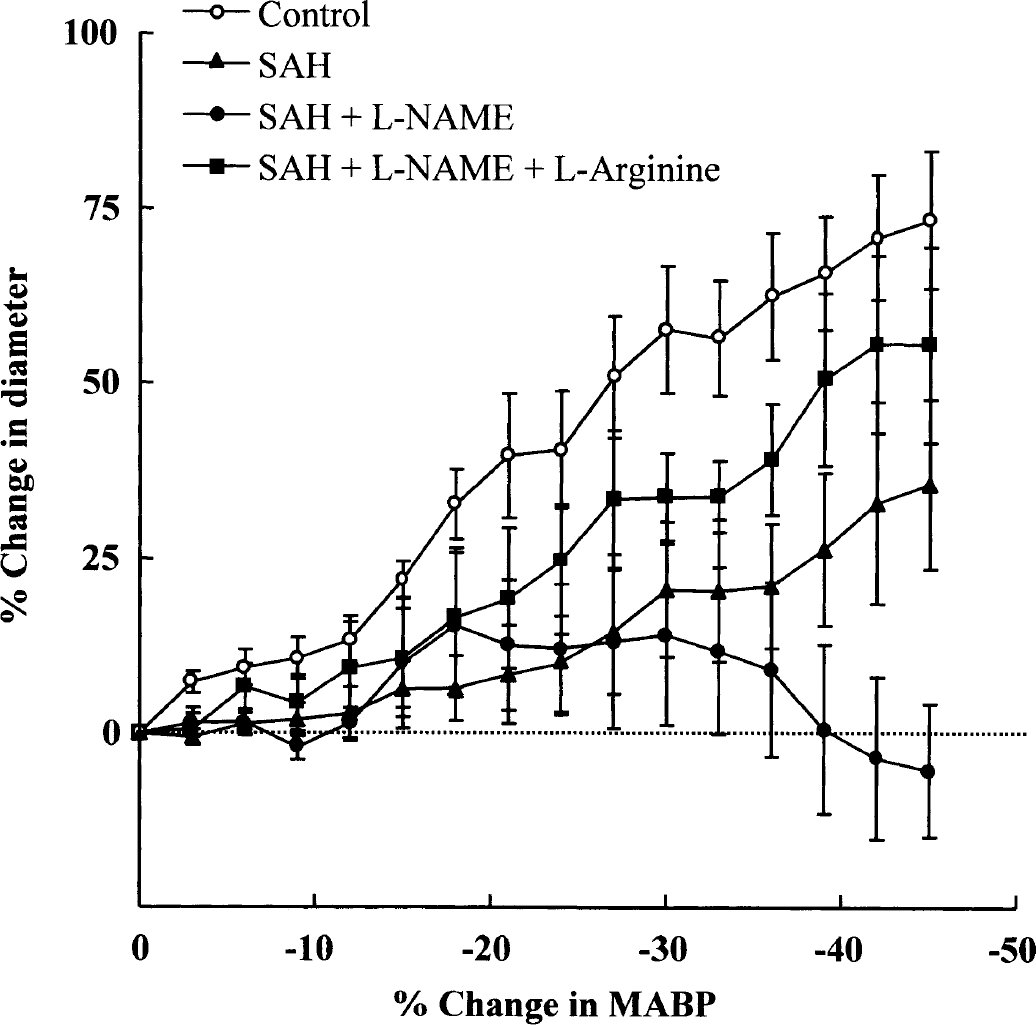
Graph showing a significant reduction in the autoregulatory vasodilator response to change in arterial blood pressure at 24 hours after subarachnoid hemorrhage (SAH; n = 10) and SAH +
The autoregulatory vasodilations to hypotension, which were markedly blunted after SAH +
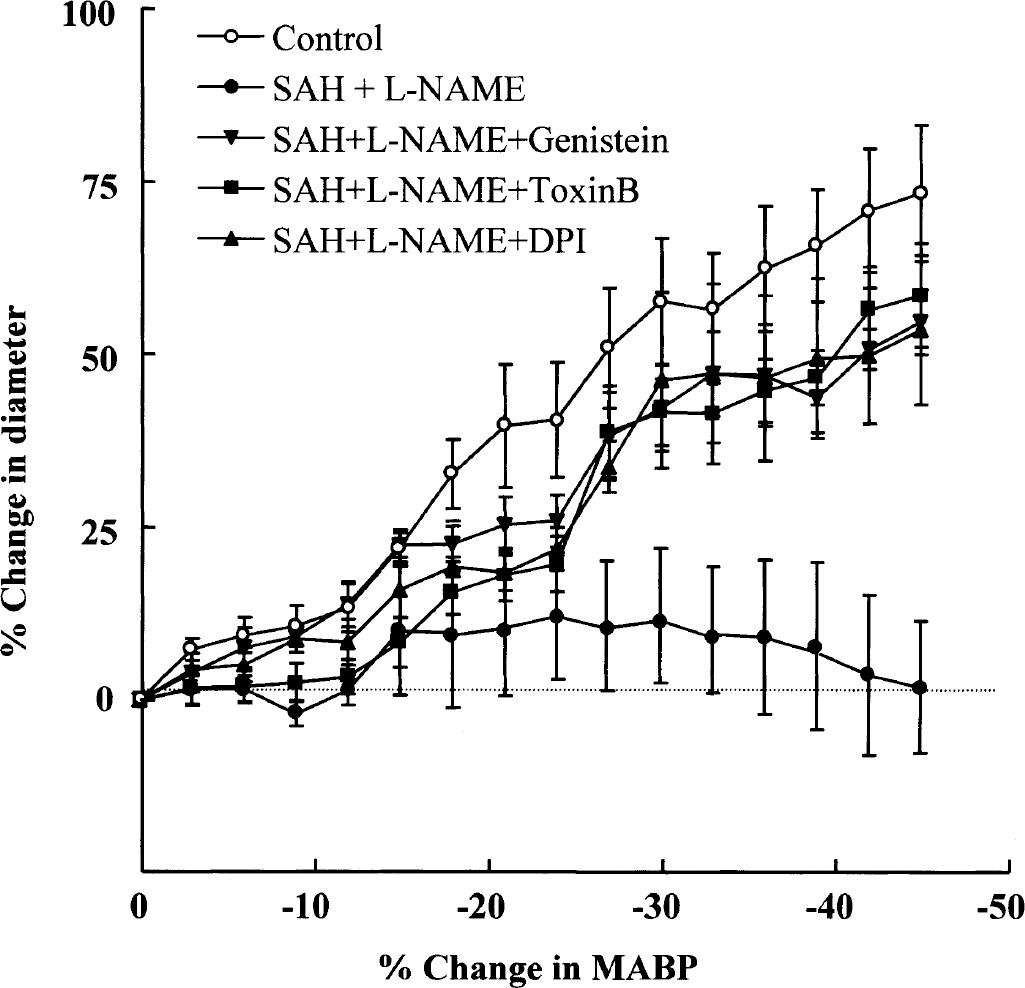
Graph showing a reduction in the autoregulatory vasodilator response to change in arterial blood pressure at 24 hours after subarachnoid hemorrhage (SAH) +
NAD(P)H oxidase—dependent superoxide generation
The production of superoxide anion from cerebral vessels in response to NAD(P)H (control, 5.2 ± 1.12 counts/s/mg tissue) was significantly elevated at 12 hours (11.6 ± 4.7 counts/s/mg tissue, P < 0.05) and maximized at 24 hours (16.5 ± 2.3 counts/s/mg tissue, P < 0.01) after intracisternal administration of whole blood (Fig. 3, inset). Superoxide production measured at 24 hours was significantly increased either by intraperitoneal administration of
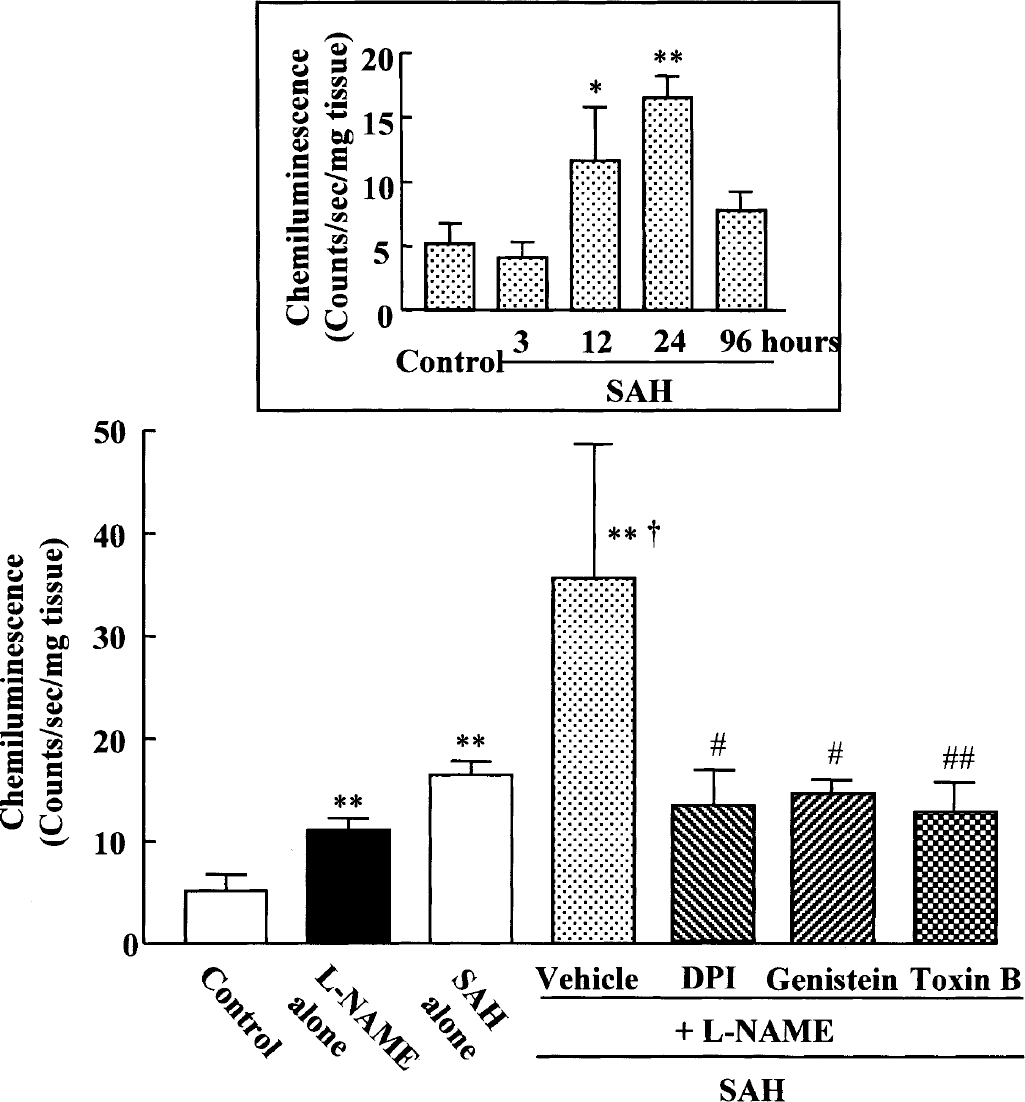
NAD(P)H oxidase—dependent superoxide generation in cerebral vessels following subarachnoid hemorrhage (SAH) +
SAH-induced superoxide production (16.5 ± 1.3 counts/s/mg tissue) of cerebral vessels measured at 24 hours was not affected by intracisternal administration of allopurinol (10 μmol/L, an inhibitor of xanthine oxidase), sulfaphenazole (10 μmol/L, a selective cytochrome P450 2C9 inhibitor), and indomethacin (10 μmol/L, a cyclooxygenase inhibitor), but by diphenyleneiodonium (10 μmol/L, 6.5 ± 2.6 counts/s/mg tissue, P < 0.01), suggesting that the predominant source of superoxide anion in cerebral vessels of SAH rat is NAD(P)H-dependent oxidase (Table 2).
Effect of specific enzyme inhibitors on NADH/NADPH-dependent superoxide anion production in cerebral vessels of control and subarachnoid hemorrhage rats
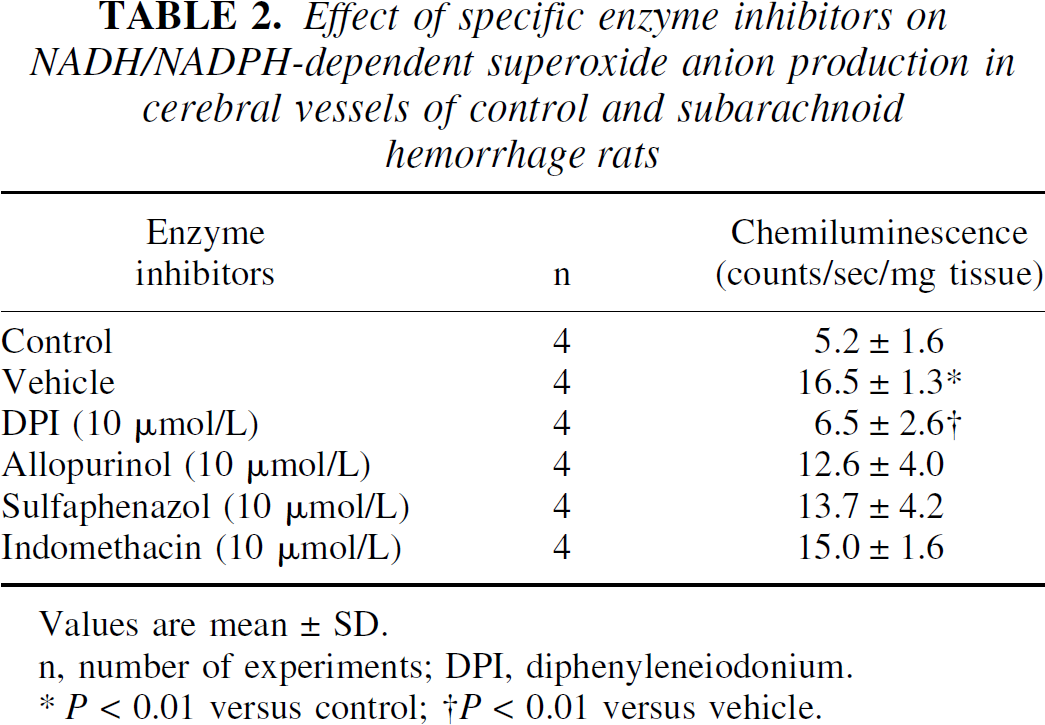
Values are mean ± SD.
n, number of experiments; DPI, diphenyleneiodonium.
P < 0.01 versus control;
P < 0.01 versus vehicle.
Expression of NAD(P)H oxidase subunits
Expression of the NAD(P)H oxidase subunits, p22phox, gp91phox and Rac protein, was examined in the cerebral vessels isolated at 24 hours after SAH. The potential of hemorrhagic bleeding to stimulate the expression of p22phox and gp91phox mRNA was studied by RT-PCR. The expression of p22phox was little altered during 3 to 96 hours throughout after SAH in comparison to control animals (Fig. 4). In contrast, the expression of gp91phox mRNA (17.4 ± 21.5% of β-actin) was markedly enhanced from 12 (73.6 ± 24.8%, P < 0.05) to 24 hours (74.5 ± 48.6%, P < 0.05). At 96 hours after SAH, the expression was declined (Fig. 5).
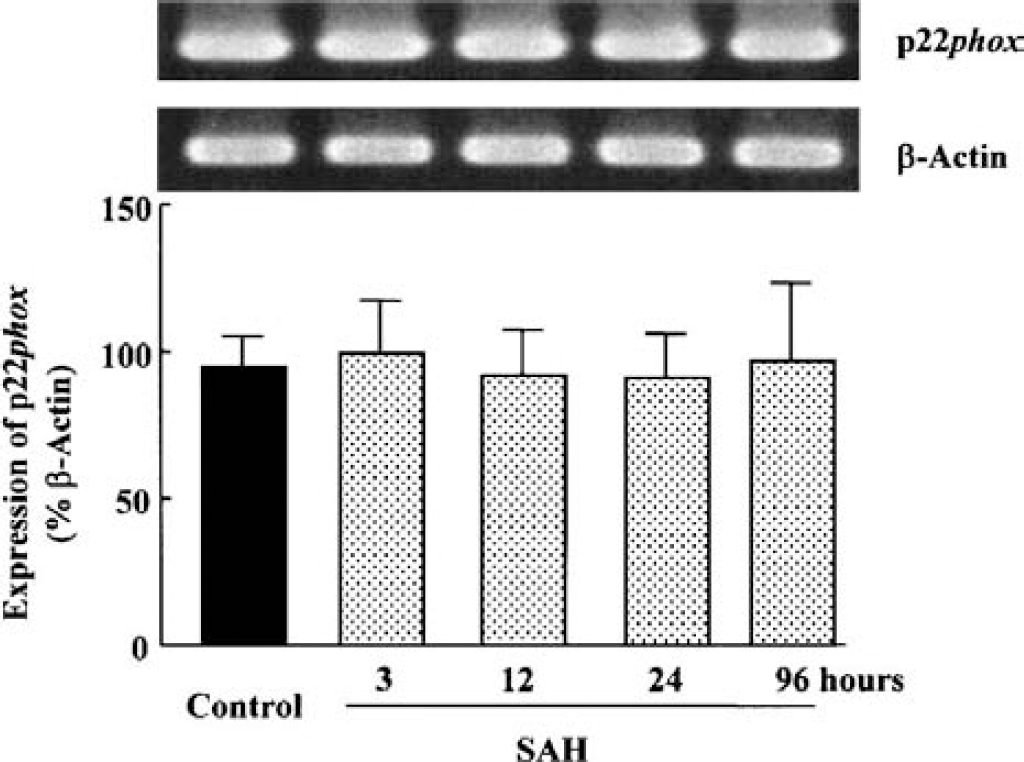
p22phox mRNA expression of NAD(P)H oxidase subunits analyzed by RT-PCR in cerebral vessels after subarachnoid hemorrhage (SAH).
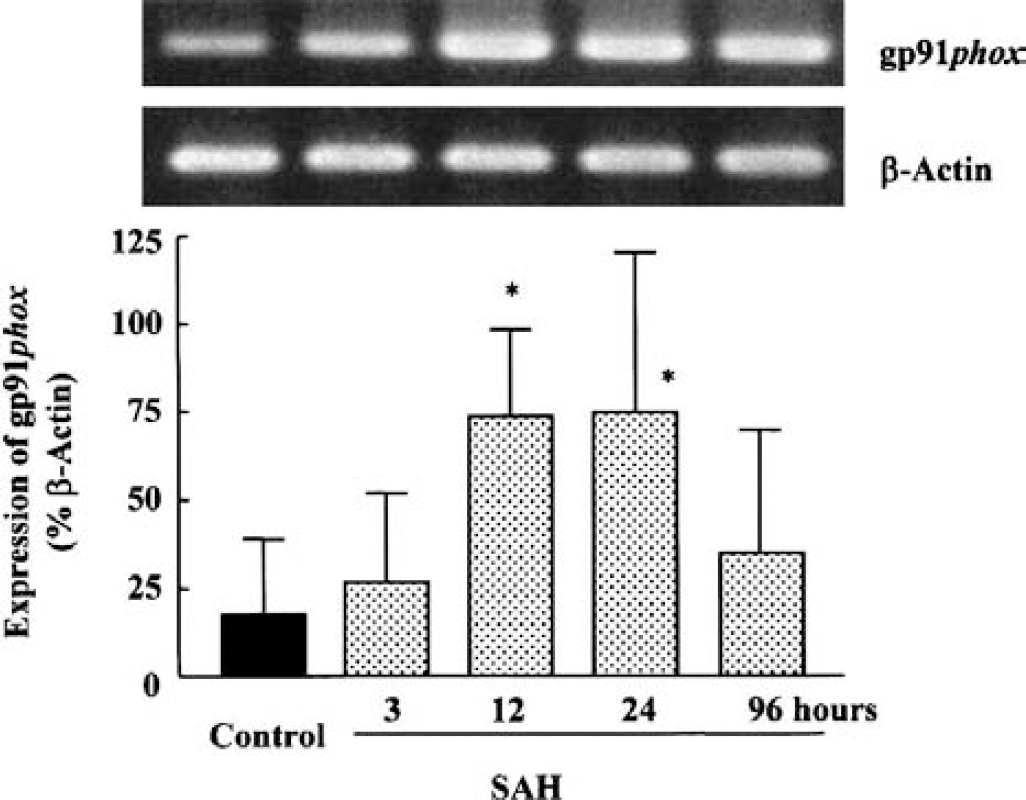
Time-dependent increase in gp91phox mRNA expression of NAD(P)H oxidase subunits analyzed by RT-PCR in cerebral vessels after subarachnoid hemorrhage (SAH).
The increased expression of gp91phox mRNA by SAH +
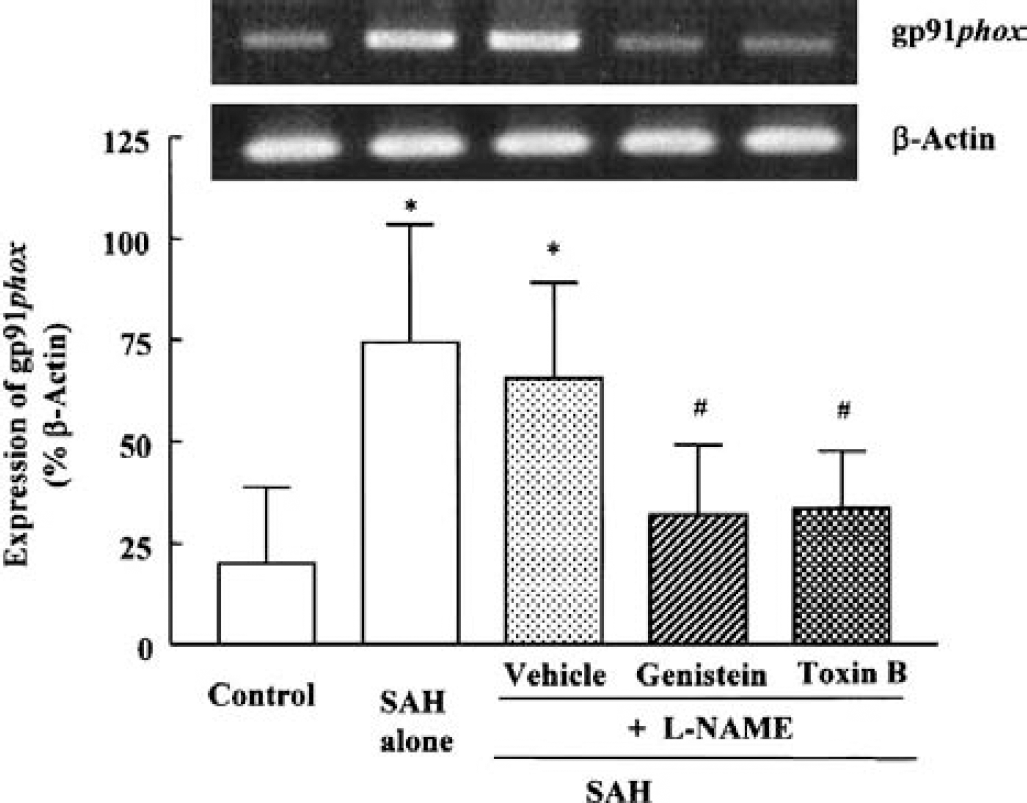
Effect of genistein (10 μmol/L) and Clostridium difficile toxin B (1 ng/mL) on the increased mRNA expression for gp91phox in cerebral vessels after subarachnoid hemorrhage (SAH) +
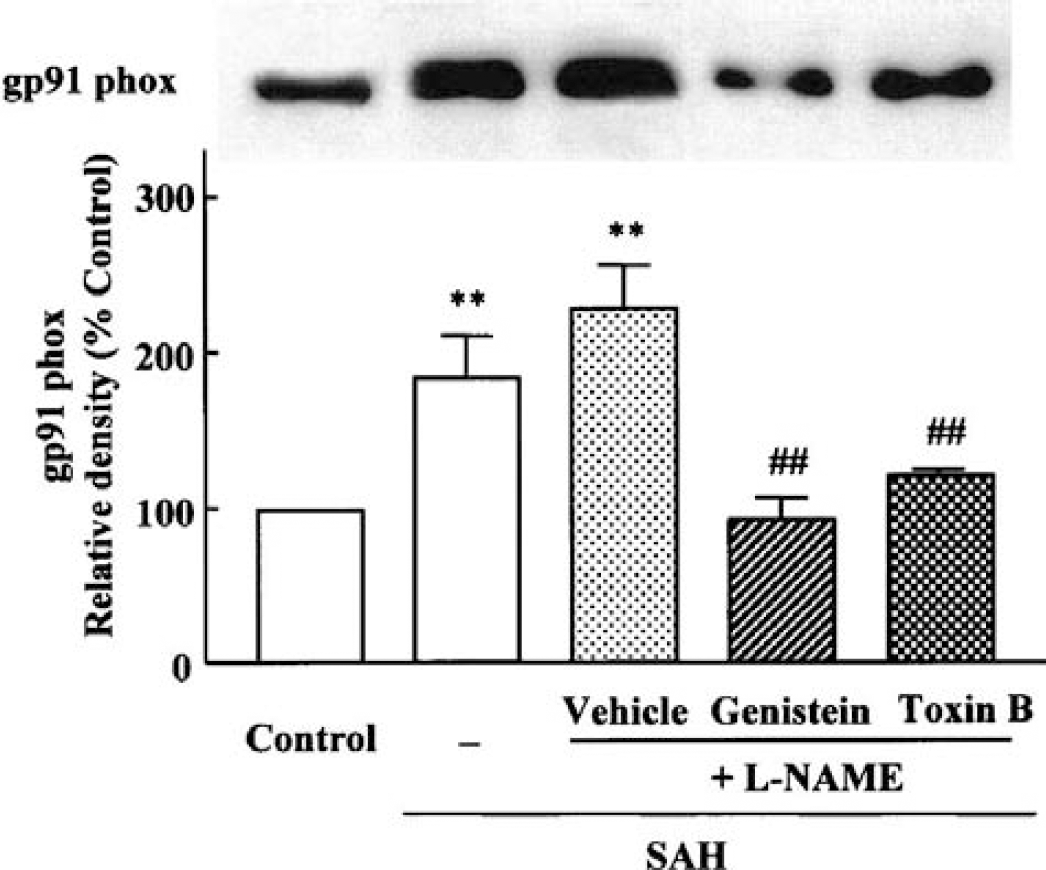
Effect of genistein (10 μmol/L) and Clostridium difficile toxin B (1 ng/mL) on the increased gp91phox determined by Western blot analysis. Both genistein and toxin B significantly inhibited the increased amount of gp91phox protein.
Western blot analysis for translocated expression of Rac, a small GTP-binding protein, was conducted. After SAH, membrane translocation of Rac protein was markedly enhanced at 24 hours (141.5 ± 28.2%, P < 0.05). At 96 hours after SAH, the expression declined (Fig. 8, inset). SAH-induced increase in membrane translocation of Rac protein was modestly increased by treatment with
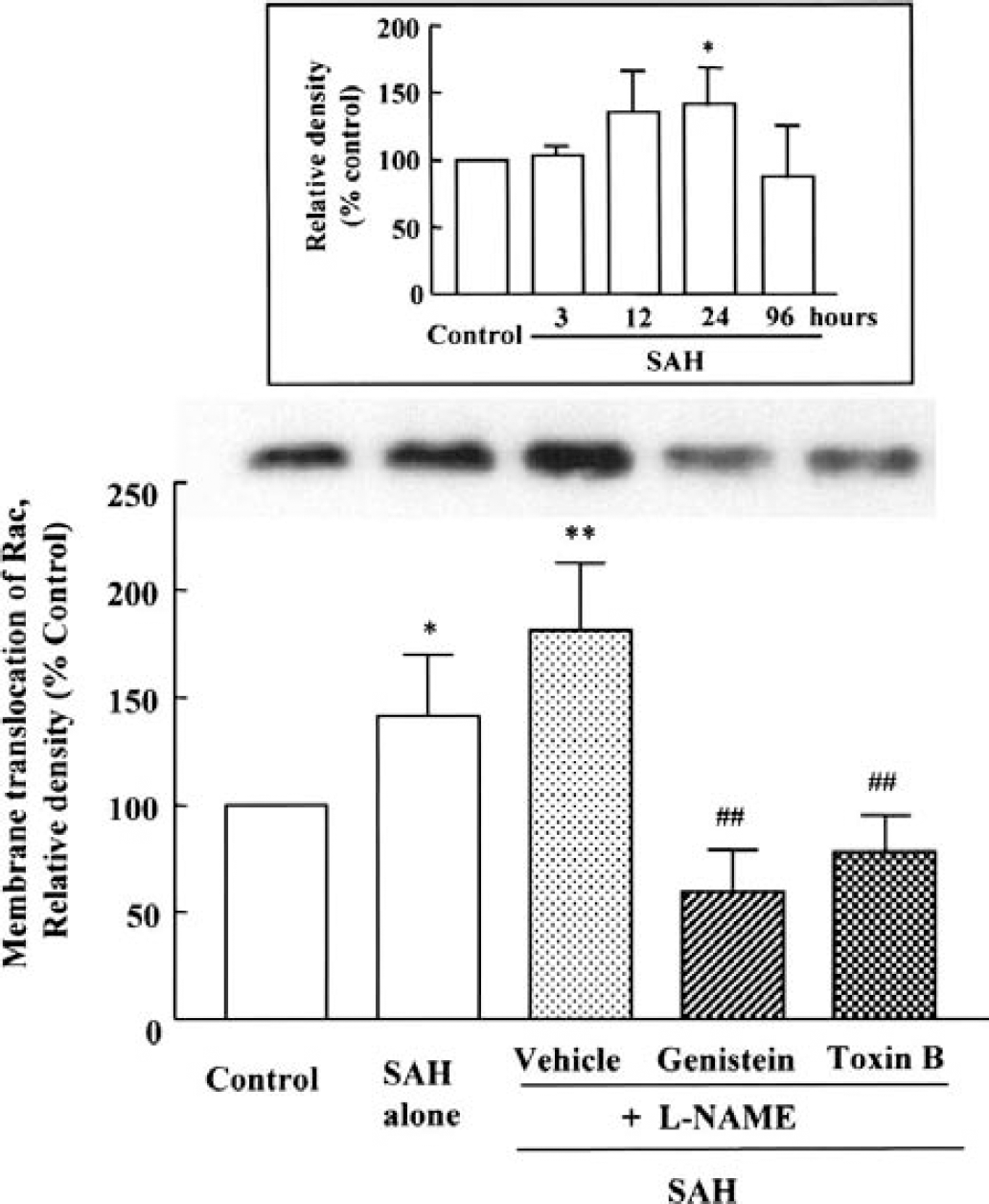
Increased membrane translocation of Rac, a small GTP-binding protein, in cerebral vessels after subarachnoid hemorrhage (SAH) +
Protein tyrosine kinase assay
Protein tyrosine kinase activity (control, 0.93 ± 0.14 U/mg protein) significantly increased from 3 hours and maximized at 12 hours (2.11 ± 0.18 U/mg protein, P < 0.01) after SAH, and thereafter, slightly declined at 24 hours (1.47 ± 0.20 U/mg protein, P < 0.01). SAH-activated tyrosine kinase activity was not affected by
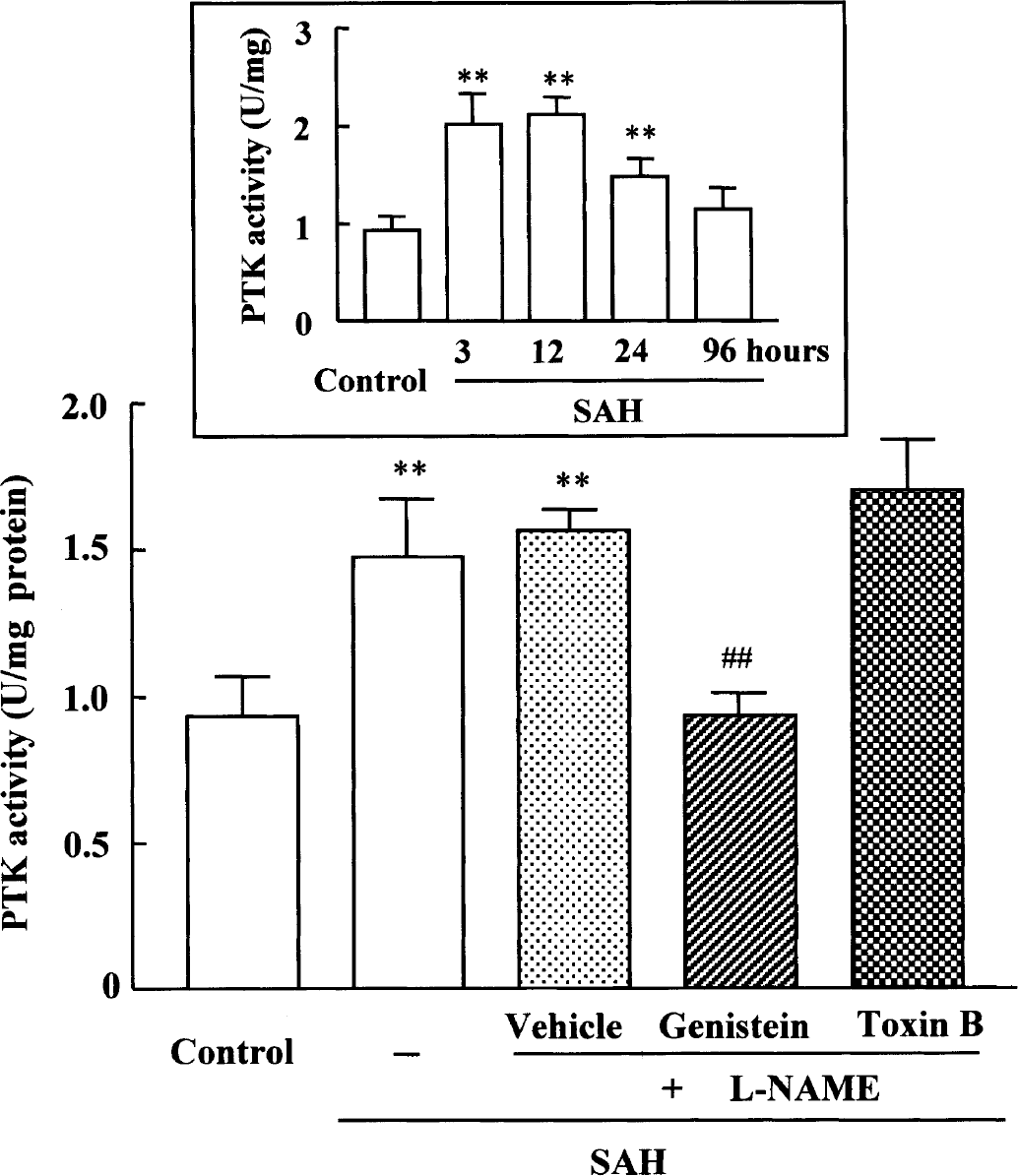
Effect of genistein and Clostridium difficile toxin B on the increased protein tyrosine kinase activity in cerebral vessels after subarachnoid hemorrhage (SAH) +
DISCUSSION
The major findings of the present study were that (1) the blunted autoregulatory vasodilation to acute hypotension of the rat pial artery observed during acute phase of SAH was effectively recovered by genistein and C. difficile toxin B as well as by diphenyleneiodonium, (2) NAD(P)H oxidase activation—induced increases in superoxide production in cerebral vessels were markedly reduced by not only genistein but also by toxin B as they were by diphenyleneiodonium, (3) SAH-induced increases in both gp91phox expression and membrane translocation of Rac protein were strongly inhibited by genistein and toxin B, and (4) SAH-activated protein tyrosine kinase activity was significantly blocked by genistein, but not by toxin B.
A marked autoregulatory disturbance of CBF has been observed in patients with SAH (Voldby et al., 1985; Yundt et al., 1998). The alteration of the cerebral microcirculation was reported as a possible contributing cause of cerebral ischemia (Kassell et al., 1985). Changes in CBF, especially during hypotension, are largely dependent on the capacity of the autoregulatory vasodilation in response to hypotension and to the reactivity of arterioles. Solomon et al. (1985) documented a 40% decrease in CBF at 60 minutes after experimental SAH in rats, while rats with saline injection showed only a 15% decrease. Nihei et al. (1991) reported that cerebral vasospasm does not occur in small pial arteries. Our results also showed that mean baseline diameters were little altered between groups of SAH and SAH +
As the etiological factor for cerebral vasospasm, subarachnoid blood clots, especially the hemolysates, were demonstrated to enhance tyrosine phosphorylation of proteins in canine basilar artery (Marton et al., 1996; Patlolla et al., 1999). In our results, reduced autoregulatory vasodilation to gradual hypotension in the pial artery of SAH rats was restored under intracisternal treatment with genistein (10 μmol/L) and C. difficile toxin B (1 ng/mL) as well as with diphenyleneiodonium (10 μmol/L). Genistein is a natural flavonoid tyrosine kinase inhibitor derived from the fermentation broth of Pseudomonas sp, which has no discernible effect on protein kinase C and protein kinase A (Di Salvo et al., 1993). C. difficile toxin B was reported to glucosylate Rac1, which markedly diminished its ability to support the activity of superoxide-generating NADPH oxidase of phagocytes (Sehr et al., 1998). Tyrosine phosphorylation plays a modulatory role in regulation of several K+ channel subunits (Huang et al., 1993). Most recently, Hong et al. (2001) demonstrated that, in the pial arterioles, the autoregulatory vasodilation and vasodilation to calcitonin gene-related peptide and levcromakalim (ATP-sensitive K+ channel opener), which were suppressed after fluid percussion injury, were wholly restored after pretreatment with genistein, suggesting that activation of tyrosine kinase links to impairment of autoregulatory vasodilation.
It is believed that oxyhemoglobin releases superoxide in its conversion to methemoglobin (Misra and Fridovich, 1972), and oxyhemoglobin released from erythrocytes in the subarachnoid space elicits a role of potent trigger of vasospasm (Steele et al., 1991). Vascular smooth muscle was demonstrated to be depolarized following SAH (Waters and Harder, 1985). In line with these results, Kamii et al. (1999) recently investigated changes in arterial diameter after SAH in transgenic mice overexpressing CuZn-SOD (SOD1), in that the diameter of the middle cerebral artery in SOD-transgenic mice was significantly larger than that in nontransgenic mice on day 3 after SAH, and they concluded that SOD is effective on the amelioration of vasospasm after SAH. Our results also showed that SAH caused enhanced production of an NAD(P)H oxidase—mediated superoxide in cerebral vessels at an early stage. NAD(P)H oxidase as the major source of ROS has been identified in endothelial cells (Mohazzab et al., 1994) and vascular smooth muscle cells (Griendling et al., 1994) of the vessel wall. Nevertheless, it remains unclear in this ex vivo study what cells of cerebral vessels specifically provide an NAD(P)H oxidase—mediated superoxide production after SAH.
Endothelial cells express the flavocytochrome b558 subunits, gp91phox and p22phox, as well as the cytosolic factors, p47phox and p67phox and a small G protein Rac, whereas vascular smooth muscle cells contain p22phox and p47phox, but expression of p67phox and gp91phox was, however, not demonstrated (Ushio-Fukai et al., 1996; Görlach et al., 2000). Our results showing the apparent time-dependent expression of gp91phox and the suppression by inhibitors (genistein and toxin B) in the cerebral vessels after SAH suggest an involvement of endothelial cells rather than smooth muscle in the production of superoxide anion. Quinn et al. (1993) have addressed the possibility that Rac protein plays a role in assembly of the active NAD(P)H oxidase complex of the human neutrophils, in that translocation of Rac from cytosol to the plasma membrane correlates with NAD(P)H oxidase activation in human neutrophils after stimulation with formyl—methionyl—leucyl—phenylalanine.
The increase in protein kinase C—dependent smooth muscle contraction plays an important role in causing vasospasm after SAH (Sako et al., 1993; Zhang, 2000). However, a number of reports regarding involvement of protein kinase C in the development of vasospasm were from the experiments conducted during the later stage (from days 3 to 7 or thereafter) or from the double hemorrhage model (Weir et al., 1978; Dietrich and Dacey, 2000). Sohn et al. (2000) documented that depolarization-induced endothelial superoxide production involved tyrosine phosphorylation but not protein kinase dependency. In agreement with their reports, our results showed that in cerebral vessels 24 hours after exposure to whole blood, the NAD(P)H oxidase—induced superoxide production was inhibited by tyrosine kinase inhibitor genistein but not by protein kinase C inhibitor staurosporine (data not shown). Moreover, the expression of gp91phox and the translocation of Rac were inhibited by genistein, as was the protein tyrosine kinase activity, but toxin B could not inhibit protein tyrosine kinase activity, suggesting that tyrosine phosphorylation precedes the translocation of Rac and the expression of gp91phox. It is likely that these findings may contribute to the impairment of the autoregulatory vasodilation in response to acute hypotension in the cerebral microvessels.
Taken together, SAH rats treated with
Footnotes
Acknowledgments
Dr. Mark T. Quinn, Department of Microbiology, Montana State University, Bozeman, Montana, USA, generously donated specific monoclonal antibody against gp91phox. We thank Jonathan Kaskin for his helpful comments on the English-version of the manuscript.