
Abstract
In addition to promoting the survival, proliferation, and differentiation of immature erythroid cells, erythropoietin and the erythropoietin receptor have recently been shown to modulate cellular signal transduction pathways that extend beyond the erythropoietic function of erythropoietin. In particular, erythropoietin has been linked to the prevention of programmed cell death in neuronal systems. Although this work is intriguing, the underlying molecular mechanisms that serve to mediate neuroprotection by erythropoietin are not well understood. Further analysis illustrates that erythropoietin modulates two distinct components of programmed cell death that involve the degradation of DNA and the externalization of cellular membrane phosphatidylserine residues. Initiation of the cascades that modulate protection by erythropoietin and its receptor may begin with the activation of the Janus tyrosine kinase 2 protein. Subsequent downstream mechanisms appear to lead to the activation of multiple signal transduction pathways that include transcription factor STAT5 (signal transducers and activators of transcription), Bcl-2, protein kinase B, cysteine proteases, mitogen-activated protein kinases, proteintyrosine phosphatases, and nuclear factor-κB. New knowledge of the cellular pathways regulated by erythropoietin in neuronal environments will potentially solidify the development and initiation of therapeutic strategies against nervous system disorders.
Keywords
Erythropoietin (EPO) is a glycoprotein with a carbohydrate content contributing to nearly one half of its molecular weight. The molecule has four glycosylated chains, which include three N-linked and O-linked acidic oligosaccharide side chains. The glycosylated chains confer some of the biological activity to the EPO molecule, which is stabilized by the carbohydrate chains. The oligosaccharides in EPO may protect the protein structure from oxygen radical destruction (Uchida et al., 1997). In addition, the N- and O-linked chains may be necessary for the cellular secretion of EPO (Krantz, 1991). Biological activity of EPO also is determined by two disulfide bonds formed between cysteines at positions 7 and 160 and at positions 29 and 33 of the molecule. Reduction of the sulfhydryl groups can result in irreversible loss of the biological activity of EPO, but restoration of the sulfhydryl group function can restore 85% of the biological activity of EPO (Wang et al., 1985).
Erythropoietin was originally identified as the principal regulator of erythroid progenitor cells, which are responsible for the formation of red blood cells. Erythropoietin is initially produced in the liver, but shortly after birth, EPO production is subsequently shifted to the kidney. Circulating EPO binds to its receptor (EPOR) expressed on erythroid progenitor cells and results in the stimulation of erythropoiesis. The induction of erythropoiesis yields a progressive improvement in the supply of oxygen to tissue. In addition, tissue oxygen supply functions as a critical feedback mechanism and becomes a significant modulator of EPO production and erythropoiesis (Sasaki et al., 2000). Any impairment in the production of EPO will result in the deficiency of circulating erythrocytes and severe anemia (Jelkmann, 1992). As a result, EPO has been widely used in the treatment of anemia during renal failure and hematologic malignancy (Littlewood, 2001).
Interestingly, the role of EPO extends far beyond the production of red blood cells. Several organs throughout the body use EPO for a variety of functions (Table 1). For example, the uterus uses EPO for angiogenesis that is required to compensate for the loss of blood vessels during the estrous cycle (Yasuda et al., 1998). Activation of the EPOR present in vascular endothelial cells and smooth muscle cells stimulates the proliferation and migration of endothelial cells, resulting in angiogenesis (Anagnostou et al., 1994; Ammarguellat et al., 1996). More recent work has begun to elucidate the role of EPO and its receptor in the brain. In particular, work that has demonstrated the production of EPO and the expression of the EPOR in brain tissue strongly suggests a significant role for EPO in the central nervous system (CNS). In this respect, this review will focus on evidence that targets EPO as a neuroprotectant for CNS disease and outline some of the novel signal transduction systems of EPO that may form the basis for future treatments against neuronal injury.
Multifold production and expression sites of erythropoietin and the erythopoietin receptor
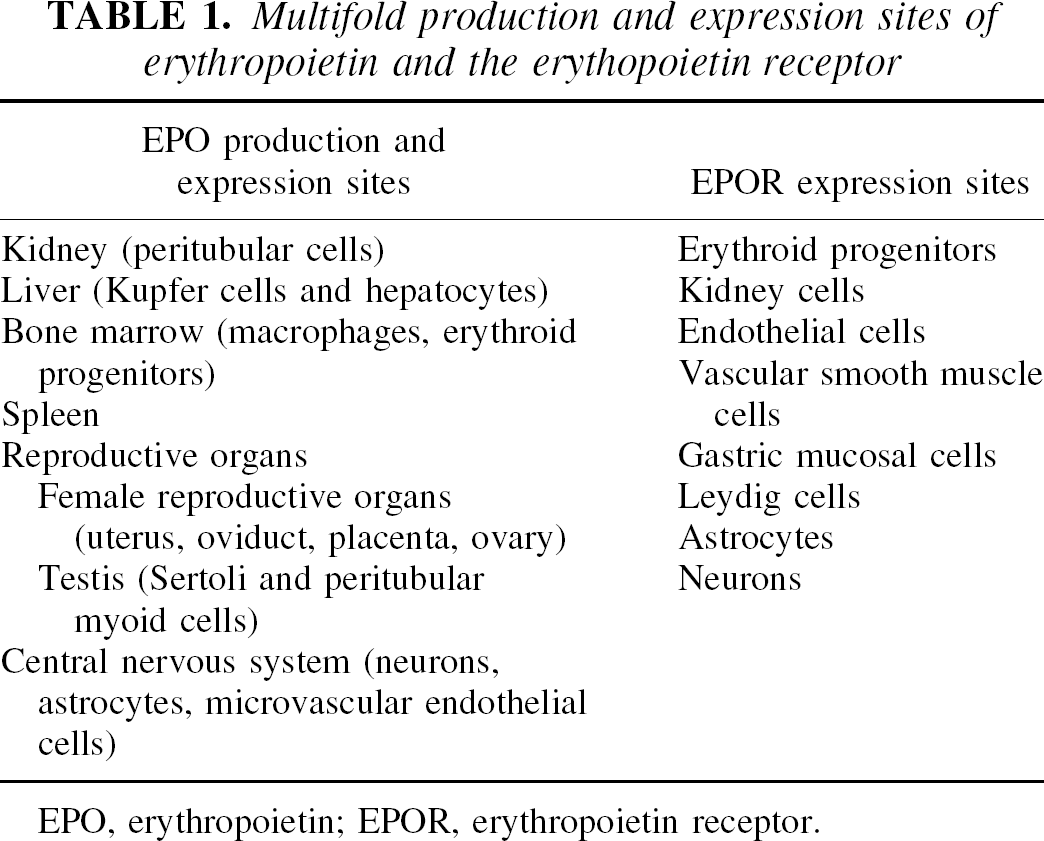
EPO, erythropoietin; EPOR, erythropoietin receptor.
EXPRESSION OF ERYTHROPOIETIN AND THE ERYTHROPOIETIN RECEPTOR IN THE CENTRAL NERVOUS SYSTEM
Both EPO and the EPOR are functionally expressed in the nervous system of rodents, primates, and humans. In the mouse, EPO is present in the hippocampus, capsula interna, cortex, and midbrain areas (Digicaylioglu et al., 1995). In cultured cortical neurons of rats, the expression of EPOR has been demonstrated by immunostaining and reverse transcription–polymerase chain reaction (Morishita et al., 1997). Cytochemical immunostaining has revealed the expression of EPO and the EPOR also in rat hippocampal neurons (Ahmad et al., 2001; Chong et al., 2001) (Fig. 1). In primates, messenger RNA (mRNA) of EPO and the EPOR gene is present in the hippocampus, amygdala, and temporal cortex of the primate brain (Marti et al., 1996). If one focuses on the human CNS, EPO and the EPOR are present in both astrocytes and neurons (Juul et al., 1999). The level of EPO in the human nervous system is variable, with elevated production during gestation and reduced production after birth.
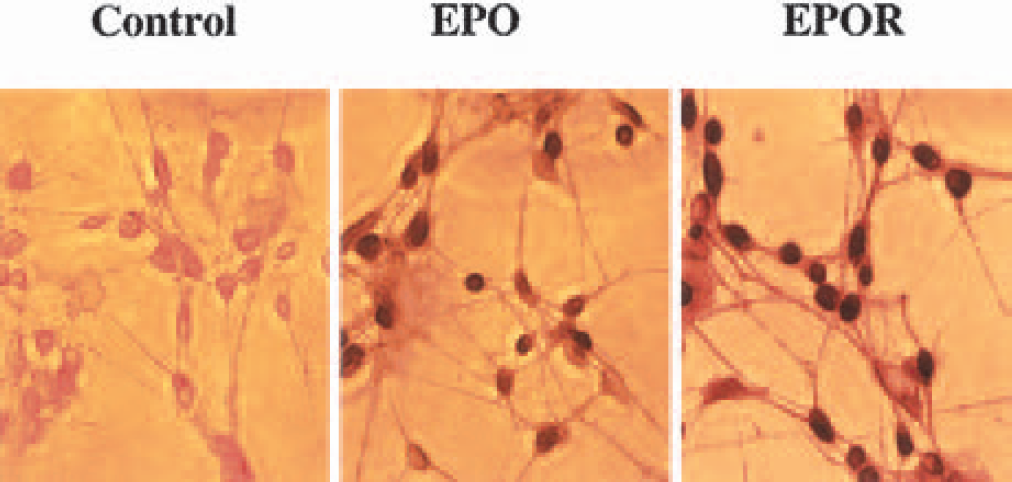
Erythropoietin (EPO) and its receptor (EPOR) are expressed in rat hippocampal neurons. Hippocampal neuronal cultures were subjected to immunocytochemical detection for EPO and the EPOR by using a rabbit primary polyclonal anti-EPO and anti-EPOR antibody. Biotinylated horse anti-rabbit antibody was used as a secondary antibody. Detection of EPO and the EPOR was performed with 3,3′-diaminobenzidine. Absence of the primary antibody was used as a negative control.
GENERATION OF ERYTHROPOIETIN PRODUCTION IN THE CENTRAL NERVOUS SYSTEM
Oxygen deficiency results in the induction of EPO in brain tissues. Accumulation of EPO mRNA and the EPO protein in response to hypoxia has been observed in cultured astrocytes (Marti et al., 1996). Astrocytes, incubated under the condition of 1% oxygen, have shown over 100-fold time-dependent erythropoietin mRNA accumulation. Erythropoietin and the EPOR are also inducible by hypoxia in cultured hippocampal neurons (Lewczuk et al., 2000). Investigations with in vivo animal models can illustrate a broader upregulation of EPO and the EPOR during hypoxia. Focal cerebral ischemia results in a significant increase in expression of EPO and the EPOR in neurons, astrocytes, and cerebral microvascular endothelial cells in mice (Bernaudin et al., 1999). In a similar manner, ischemic insults in the human brain lead to the elevated expression of both EPO and the EPOR in microvessels and astrocytes (Siren et al., 2001).
Generation of EPO by hypoxia is dependent on the hypoxia-inducible factor 1 (HIF-1). HIF-1 is a heterodimeric transcription factor containing two subunits, HIF-1α and HIF-1β. HIF-1β is a constitutively expressed subunit, but HIF-α is an oxygen-labile subunit that undergoes rapid degradation via the ubiquitin–proteasome pathway under normoxic conditions (Huang et al., 1998). Hypoxia can impair the degradation of HIF-1α by blocking its association with the von Hippel-Lindau protein that targets HIF-1α for proteolysis (Maxwell et al., 1999). In the absence of degradation, HIF-1α translocates to the nucleus and dimerizes with HIF-1β to form a stable HIF-1 complex. This complex then binds to a conserved sequence (5′RCGTG3′) near the 5′ end of the hypoxia-response enhancer of the EPO gene and upregulates EPO gene transcription (Bunn et al., 1998). Although some HIF-1 variants may not always be associated with hypoxia (Drutel et al., 2000), the expression of HIF-1 rapidly increases and plateaus within 5 hours in the brain after a hypoxic insult (Stroka et al., 2001). In addition to a change in response to low oxygen states, HIF-1 expression can be suppressed in the presence of heme-binding ligands such as carbon monoxide and nitric oxide (NO) (Huang et al., 1999). These studies suggest that, similarly to the hemopoietic system, HIF-1 may function as a cellular oxygen sensor in the nervous system that confers an adaptive response to decreased oxygen tension through the transcription and translation of EPO.
ANIMAL MODELS OF ERYTHROPOIETIN IN THE CENTRAL NERVOUS SYSTEM
Given the link between the generation of low oxygen tension states and the cellular response of EPO to prevent tissue injury, initial experimental studies have examined the potential role of EPO in the nervous system during cerebral ischemia. The development of recombinant human EPO (rhEPO) has assisted these research efforts to investigate the efficacy and underlying mechanisms of EPO protection in the CNS (Egrie et al., 1985). Initial reports included the infusion of EPO into the lateral ventricle of gerbils subjected to the occlusion of the common carotid arteries (Sakanaka et al., 1998). Infusion of EPO prevented ischemic-induced learning disability and rescued hippocampal CA1 neurons from degeneration. Reduction in infarct volume and improved navigation ability also have been reported in rats after EPO administration (Sadamoto et al., 1998). In mice subjected to occlusion of the middle cerebral artery, a significant reduction in infarct volume was observed with administration of a bolus intraventricular infusion of recombinant EPO 24 hours before the onset of focal cerebral ischemia (Bernaudin et al., 1999).
These initial in vivo studies substantiated a cytoprotective role for EPO in the CNS, but one that required direct administration of EPO to the injury site. Clinical utility of EPO for nervous system disorders could therefore be limited if systemic administration was not efficacious. Further work eventually crossed this hurdle through the demonstration that large glycosylated molecules such as EPO may cross the blood–brain barrier during periods of cerebral injury (Dame et al., 2001). Systemic administration of EPO has been shown to attenuate secondary hippocampal neuronal injury after global cerebral ischemia (Calapai et al., 2000). In addition, cortical ischemic neuronal damage as a result of subarachnoid hemorrhage was reduced with the systemic administration of EPO (Alafaci et al., 2000). Current experimental models and the efficacy of EPO during cerebral ischemia are further illustrated in Table 2.
Neuroprotective studies of erythropoietin in vivo and in vitro

EPO, erythropoietin; ↑ and ↓, positive and negative outcomes, respectively; CCAO, Common carotid artery occlusion; icv, intracerebroventricular injection; ip, intraperitoneal injection; MCAO, middle cerebral artery occlusion; NMDA, N-methyl-
CELLULAR MODELS OF ERYTHROPOIETIN IN THE CENTRAL NERVOUS SYSTEM
Experimental in vivo (animal) models serve an important purpose for the assessment of EPO as a potential protectant in the CNS. Experimental in vitro models that focus on the cellular mechanisms of injury not only identify potential cellular targets, but also serve to develop specific cytoprotective mechanisms used by EPO for future drug development. Recent studies have further supported a protective role for EPO that is broad in nature against a variety of insults.
Erythropoietin can prevent neuronal injury during reduced or absent oxygen tension, hypoglycemia, glutamate toxicity, and free-radical injury. Erythropoietin and EPOR have been shown to be expressed in both cortical and hippocampal neurons (Morishita et al., 1997; Chong et al., 2001). Under some injury paradigms, endogenous cellular EPO can be depleted, supporting a role for additional exogenous EPO administration during neurodegenerative disorders (Ahmad et al., 2001; Chong et al., 2001). During the onset of hypoxia or excitotoxicity, administration of EPO can result in a significant reduction in neuronal cell death in cultured hippocampal neurons (Morishita et al., 1997; Chong et al., 2001). The protection conferred by EPO may, under some circumstances, require the generation of new proteins. In many respects, neuroprotection by EPO is quite robust in nature; it can, for instance, prevent the degeneration of cortical neurons during hypoxia and hypoglycemia administration (Lewczuk et al., 2000).
Although neuronal protection by EPO is efficacious against more upstream insults such as hypoxia and hypoglycemia, enhanced survival by EPO may be dependent on specific downstream mediators of cell injury, such as glutamate, extracellular calcium, and NO. During experimental paradigms with excitotoxicity, EPO can protect cortical neurons in a dose-dependent manner. Interestingly, neuroprotection by EPO during excitotoxicity may require a transient increase in intracellular calcium concentration, because a decrease in intracellular calcium can negate any protection by EPO during glutamate administration (Morishita et al., 1997). Such observations concerning a potential synergistic function for neuronal protection by intracellular calcium are not unique and have been reported with other cellular receptor systems, such as those that involve G-protein–related pathways (Maiese et al., 1999). Neuronal injury during NO exposure also can be prevented by EPO, suggesting that the mechanism of EPO protection functions below the level of hypoxic and excitotoxic insults (Chong et al., 2001). In addition, further work has shown a specific ‘therapeutic window‘ for the application of EPO (Ahmad et al., 2001; Chong et al., 2001). Administration of EPO to hippocampal neuron cultures up to 6 hours after NO exposure can provide significant protection against free-radical injury. Yet treatment with EPO that is delayed as long as 12 hours after NO exposure offers no significant increase in survival. These observations may suggest that EPO requires an incubation period for the sufficient induction of necessary signal transduction pathways required for the protection of neurons. Other studies provide support for this hypothesis by showing that incubation periods of several hours are required for EPO neuronal protection during some insults, such as excitotoxicity (Morishita et al., 1997). Application times that occur after the induction of specific signal transduction pathways of cell injury appear to render EPO ineffective as a cytoprotectant.
DOWNSTREAM “PATHWAYS OF PROTECTION” FOR ERYTHROPOIETIN
Programmed cell death: Membrane asymmetry and DNA degradation
Cellular self-destruction known as programmed cell death (PCD) or apoptosis plays a significant role during neuronal degeneration. Following one of the original morphologic descriptions of PCD (Kerr et al., 1972), several general biochemical and physiologic features of PCD have been identified. These processes include the loss of plasma membrane asymmetry, nuclear chromatin condensation, and DNA fragmentation. In the neuronal population, PCD is considered to be a significant contributor of cellular injury during a variety of neurologic disorders such as stroke (Johnson et al., 1995), trauma (Satake et al., 2000), and Alzheimer disease (Shimohama, 2000). Experimental models that use injury models of free-radical exposure (Maiese et al., 2001), β-amyloid deposition (Kajkowski et al., 2001), or ultraviolet radiation (Kimura et al., 1998) provide further evidence that the majority of neurons succumbs through PCD.
Recently, at least two independent pathways have been shown to lead to cellular PCD. One initial pathway involves the externalization of membrane phosphatidylserine residues (Chang et al., 2000; Maiese and Vincent, 2000). Membrane phospholipids are asymmetrically distributed across the cellular membrane bilayer with the membrane phosphatidylserine residues positioned in the inner leaflet of cells under normal conditions. Exposure of phosphatidylserine residues is believed to occur before genomic DNA degradation and serves to “tag” injured cells for phagocytosis (Vincent and Maiese, 1999a; Chang et al., 2000; Lin et al., 2000). An additional role for membrane phosphatidylserine externalization in the vascular cell system is the activation of coagulation cascades. The externalization of phosphatidylserine in platelets or endothelial cells can promote the formation of a procoagulant surface (Bombeli et al., 1997). In contrast to the early externalization of membrane phosphatidylserine residues, the cleavage of genomic DNA into fragments is a delayed event that occurs late during PCD. The degradation of genomic DNA through the activity of endogenous neuronal endonucleases is considered to be a committed event that results in neuronal demise (Okamoto et al., 1993; Tominaga et al., 1993; Vincent and Maiese, 1999b; Vincent et al., 1999a,b; Love et al., 2000). Because DNA fragmentation and phosphatidylserine externalization can each lead to cellular injury, the mechanisms that induce these individual processes may be important targets for neuroprotective strategies.
Erythropoietin can offer neuronal protection at two distinct levels during apoptosis. Application of EPO during anoxia or NO exposure can prevent the early exposure of membrane phosphatidylserine residues and also inhibit the later stages of genomic DNA destruction (Chong et al., 2001) (Fig. 2). Similar to cell culture survival studies with anoxia or NO, posttreatment paradigms visualized in “real time” with living neuronal systems also have shown a “window of opportunity” for preventing the further progression of membrane phosphatidylserine residue exposure once an injury has been initiated (Vincent and Maiese, 1999a; Maiese and Vincent, 2000; Maiese, 2001). Thus, in one aspect EPO protects neurons on an immediate basis by maintaining the integrity of genomic DNA and preventing acute neuronal degeneration. In addition, the maintenance of membrane phosphatidylserine asymmetry by EPO also provides a more long-term protection by inhibiting the destruction of cells by phagocytes (Savill, 1997).
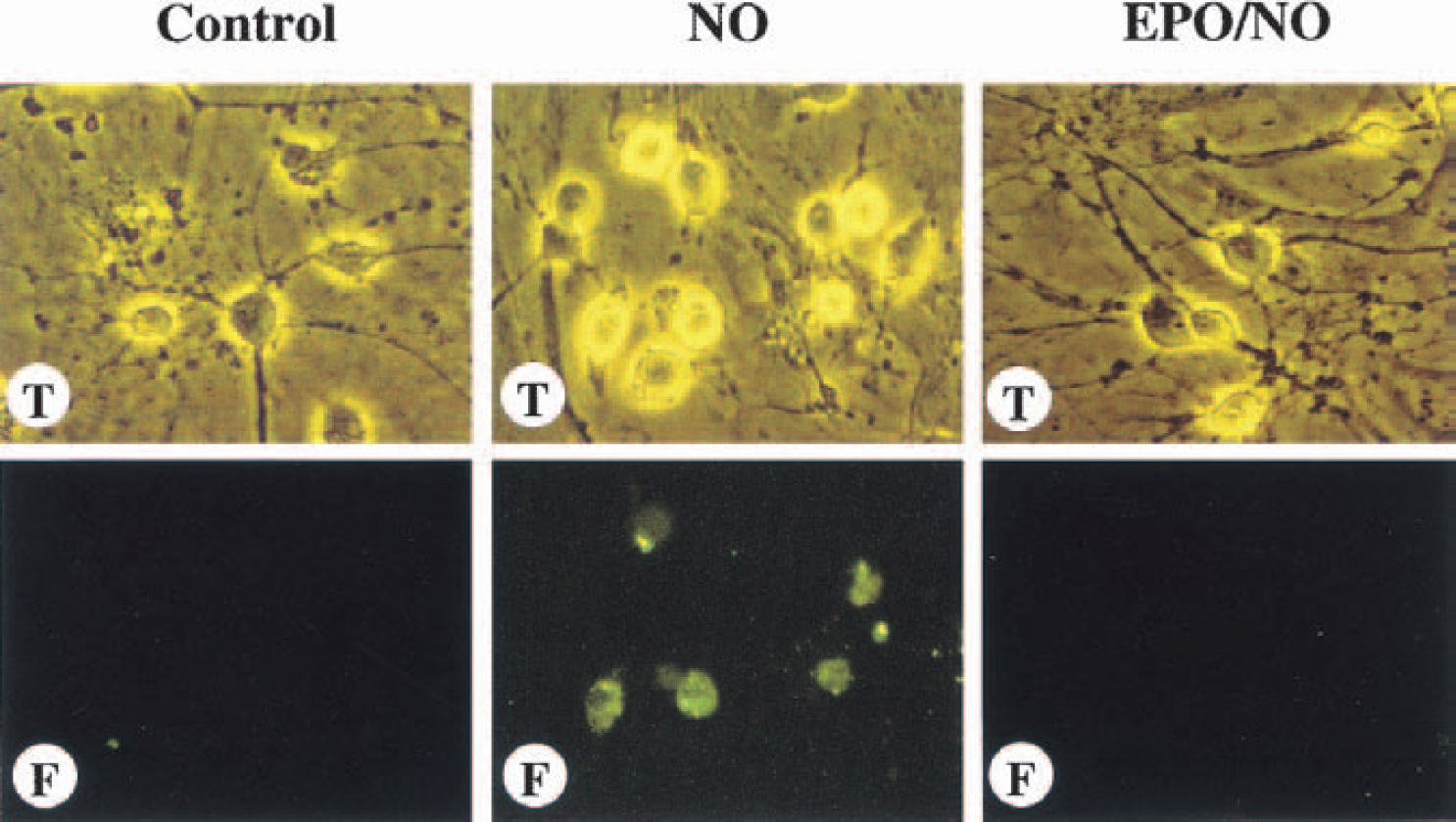
Erythropoietin prevents nitric oxide (NO)–induced externalization of membrane phosphatidylserine (PS) residues. A representative image of externalization of membrane PS residues of rat hippocampal neurons is illustrated. Neurons were labeled with annexin V phycoerythrin 24 hours after exposure to a NO donor (NOC-9, 300 μmol/L) for 5 minutes. A representative microscopy field of neurons was imaged using transmitted (T) light and corresponding fluorescence (F) image of the same microscopy field was obtained using 490-nm excitation and 585-nm emission wavelengths to locate externalized PS residues. No externalized PS residues were evident in the untreated control culture field under the fluorescent light (Control, F). Twenty-four hours after NO exposure, neurons revealed PS membrane externalization (NO, F). Pretreatment with erythropoietin (EPO; 10 ng/mL) 1 hour before NO application reduced PS membrane externalization in neurons (EPO/NO, F).
Erythropoietin and Janus tyrosine kinase 2
Cellular signal transduction initiated by EPO begins with the activation of its membrane receptor, EPOR. The EPOR is part of the type 1 super-family of cytokine receptors and is activated via homodimerization. This receptor family shares a common domain structure consisting of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular domain. The extracellular domain is necessary for the initial binding of EPO, and the intracellular domain is responsible for the transduction of intracellular signaling (Mulcahy, 2001).
The cytoplasmic portion of the EPOR contains a Box-1 motif that specifically binds to and activates Janus tyrosine kinase 2 (Jak2) by phosphorylation. It is believed that inhibition of PCD by EPO is critically dependent on the EPOR Box-1 domain (Joneja and Wojchowski, 1997). Investigations that use cells with dominant Jak2 deletions show that EPO is no longer effective in inhibiting apoptosis (Zhuang et al., 1995). Construction of cortical neurons with a biologically incompetent Jak2 also abrogates EPO-mediated prevention of neuronal PCD (Digicaylioglu and Lipton, 2001). After the activation of Jak2, EPO modulates neuronal survival and PCD through a variety of downstream signals that range from the level of signal transducer and activator of transcription (STAT) proteins to cysteine protease executioner proteins (Fig. 3).
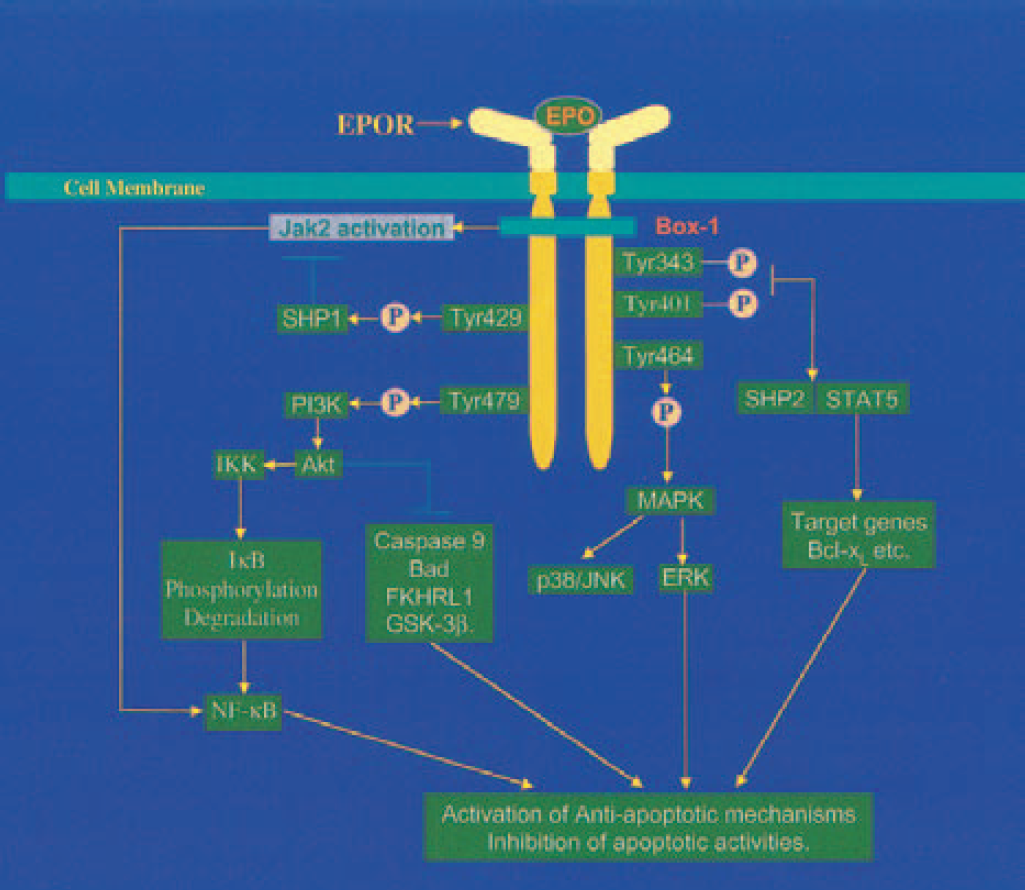
Schematic of the signal transduction pathways of erythropoietin (EPO) and the erythropoietin receptor (EPOR). Binding of EPO to the EPOR results in the phosphorylation of the Janus tyrosine kinase 2 (Jak2). The activated Jak2 mediates the phosphorylation of tyrosine sites located in the intracellular portion of EPOR leading to the recruitment and activation of signal transducer and activator of transcription 5 (STAT5), phosphoinositol 3 kinase (PI3K), mitogen-activated protein kinases (MAPKs), SH2-containing protein–tyrosine phosphatases (SHPs), and transcription factor NF-κB. The downstream events involve the production of antiapoptotic proteins such as Bcl-xL, inhibitor of apoptotic proteins (IAPs), and the inhibition of cysteine protease activity. SHP1 acts as a negative regulator of the EPOR and Jak2 activity.
Erythropoietin and STAT5
The STAT proteins are latent cytosolic transcription factors and direct substrates of Janus tyrosine kinases. Activation of Jak kinases results in tyrosine phosphorylation and dimerization of STATs. The activated STATs then translocate to the nucleus and bind to specific DNA sequences in the promoters of responsive genes to promote gene transcription. Two isoforms of STAT5, known as STAT5a and STAT5b, are associated with separate tyrosine residues (tyrosine 343 and tyrosine 401) of the intracellular domain of the EPOR that are required for STAT activation (Gobert et al., 1996).
Activation of STAT5 is one cellular pathway that can modulate EPO protection against PCD. In erythroleukemic cell lines, EPO-dependent cell survival is accompanied by sustained STAT5 DNA-binding activity. Stable expression of the truncated STAT5a has been shown to enhance STAT5 DNA-binding activity and reduce the induction of apoptosis (Bittorf et al., 2000). On the converse side, induction of PCD can be observed in cells that lack STAT5 (STAT5a−/− and STAT5b−/−) functionality (Socolovsky et al., 2001). In addition, STAT5a−/− and −5b−/− fetal liver erythroid progenitors show higher levels of apoptosis and are less responsive to the presence of EPO (Socolovsky et al., 1999).
Erythropoietin and Bcl-2/Bcl-xL
The Bcl-2 family is a group of proteins that modulate the induction of apoptosis. Several Bcl-2 family members have been identified that are functionally categorized into two separate subsets. One set consists of “antiapoptosis” members (Bcl-2, Bcl-xL) and a second set contains “proapoptosis” members (Bax, Bad, Bak, Mcl,-2). Both Bcl-2 and Bcl-xL block neuronal apoptosis by preventing mitochondrial cytochrome c release as well as modulating intracellular calcium levels (Murphy and Fiskum, 1999). In contrast, the proapoptosis member Bax oligomerizes after release from Bcl-2 binding and subsequently triggers the release of cytochrome c from mitochondria (Green, 2000). Ultimately, it is the fine balance between activity of the proapoptotic and the antiapoptotic Bcl-2 family members that can determine the fate of a cell (Oltvai and Korsmeyer, 1994).
As a survival factor, EPO mediates some of its actions through the regulation of the Bcl-2 family. Exogenous administration of EPO can increase the expression of Bcl-2 in hemopoietic cell lines (Klampfer et al., 1999). Erythropoietin also can induce Bcl-x L expression primarily through STAT5 activation. In response to EPO stimulation, STAT5 can bind to the +Bcl-xL promoter to induce Bcl-x L expression and prevent PCD (Socolovsky et al., 1999). At least in erythroid cells, the Bcl-2 member Bcl-xL has been shown to be strongly expressed and necessary for EPO to prevent apoptosis in the later stages of erythroid progenitor cell life (Gregoli and Bondurant, 1997). In addition, expression of Bcl-2 and Bcl-xL has been shown to be dependent on EPO. For example, in a murine erythroid progenitor cell line, EPO can specifically maintain the expression of Bcl-2 and Bcl-xL. In the absence of EPO, Bcl-2 and Bcl-xL are downregulated, and apoptotic cell death results (Silva et al., 1996). Given that ischemic neuronal damage may be prevented through Bcl-2 and Bcl-xL expression (Guegan et al., 1999), it is conceivable that EPO may provide neuronal protection through the enhancement of antiapoptotic Bcl-2 family members.
Erythropoietin, protein kinase B, mitochondrial membrane permeability, and cysteine proteases
Protein kinase B, also known as Akt, is a common mediator of cell survival via its antiapoptotic effects. The downstream components of the Akt pathway include Bad (Li et al., 2001), caspase-9 (Cardone et al., 1998), the forkhead transcription factor (FHKRL1) (Brunet et al., 1999), and glycogen synthase kinase-3β (GSK3β) (Cross et al., 1995). Each of the cellular systems targeted by Akt is inactivated by phosphorylation resulting in the blockade of PCD. Phosphorylation of Bad precipitates an interaction with the cytosolic 14-3-3 protein, resulting in the liberation of the antiapoptotic protein Bcl-2/Bcl-xL (Li et al., 2001). The phosphorylation of FKHRL1 leads to its association with the cytosolic 14-3-3 protein and the subsequent retention of FKHRL1 in the cytoplasm, rendering it unable to regulate its target genes in the nucleus for the induction of PCD (Brunet et al., 1999). Overexpression of GSK3β can trigger apoptosis. Yet direct inhibition of GSK3β activity by Akt can block induction of PCD (Yu et al., 2001).
Inhibition of PCD by Akt also has been closely associated with the modulation of mitochondrial membrane potential. Preservation of mitochondrial integrity can be a key determinant for a cell to recover from toxic insults. A subsequent drop in mitochondrial membrane potential has been suggested to be an important trigger for the release of cytochrome c, a critical determinant of cell survival (Kroemer and Reed, 2000). Injury in cells results in rapid depolarization of mitochondrial membrane potential, cytoplasmic increase of cytochrome c, and apoptosis, manifested by DNA fragmentation and phosphatidylserine exposure (Chong et al., 2002). Usually residing exclusively in the inner membrane space of the mitochondria, cytochrome c, once released into the cytosol, can activate both upstream and downstream caspases such as caspase-8, caspase-9, and caspase-3 through the apoptotic protease-activating factor 1 (Apaf-1) (Slee et al., 1999). Protein kinase B may serve to prevent mitochondrial membrane depolarization and the subsequent release of cytochrome c (Kennedy et al., 1999).
Intimately linked to alterations in mitochondrial membrane potential is the modulation of cysteine protease activity. Caspases are a family of cysteine proteases that cleave their substrates after aspartic residues. These proteins are usually synthesized as inactive zymogens that are cleaved into subunits during the initiation of apoptosis. The caspases are then activated by forming heteromeric complexes with accessory molecules such as a death factor receptor. Caspases can be functionally categorized into three groups. Group I is the cytokine-processing caspases, which include caspase-1, −4, −5, −11, −12, and −13. Group II caspases comprise caspase-3, −6, and −7 and are termed executioner or effector caspases, which cleave crucial cellular protein substrates leading to cell destruction. Group III members include caspase-2, −8, −9, and −10 and are described as initiator caspases, which activate downstream executioner caspases, resulting in an amplification of caspase activity (Wolf and Green, 1999).
Separate pathways are involved in caspase activation leading to PCD. The first pathway occurs before alterations in mitochondrial membrane potential. This pathway, termed the extrinsic pathway, is initiated by death receptor activation at the cell surface and results in enhanced caspase-8 and −10 activities. As a result, Bid is cleaved by caspase-8 and translocates to mitochondria to release cytochrome c through the Bax subfamily of Bcl-2 proteins. This leads to the subsequent activation of executioner caspases (Green, 2000). The second pathway is mediated by caspase-9 after the release of mitochondrial cytochrome c. Cytochrome c binds to Apaf-1, followed by activation of capase-9 (Cain et al., 2000). The active caspase-9 can then activate executioner caspases −3 and −7 (Fig. 4).
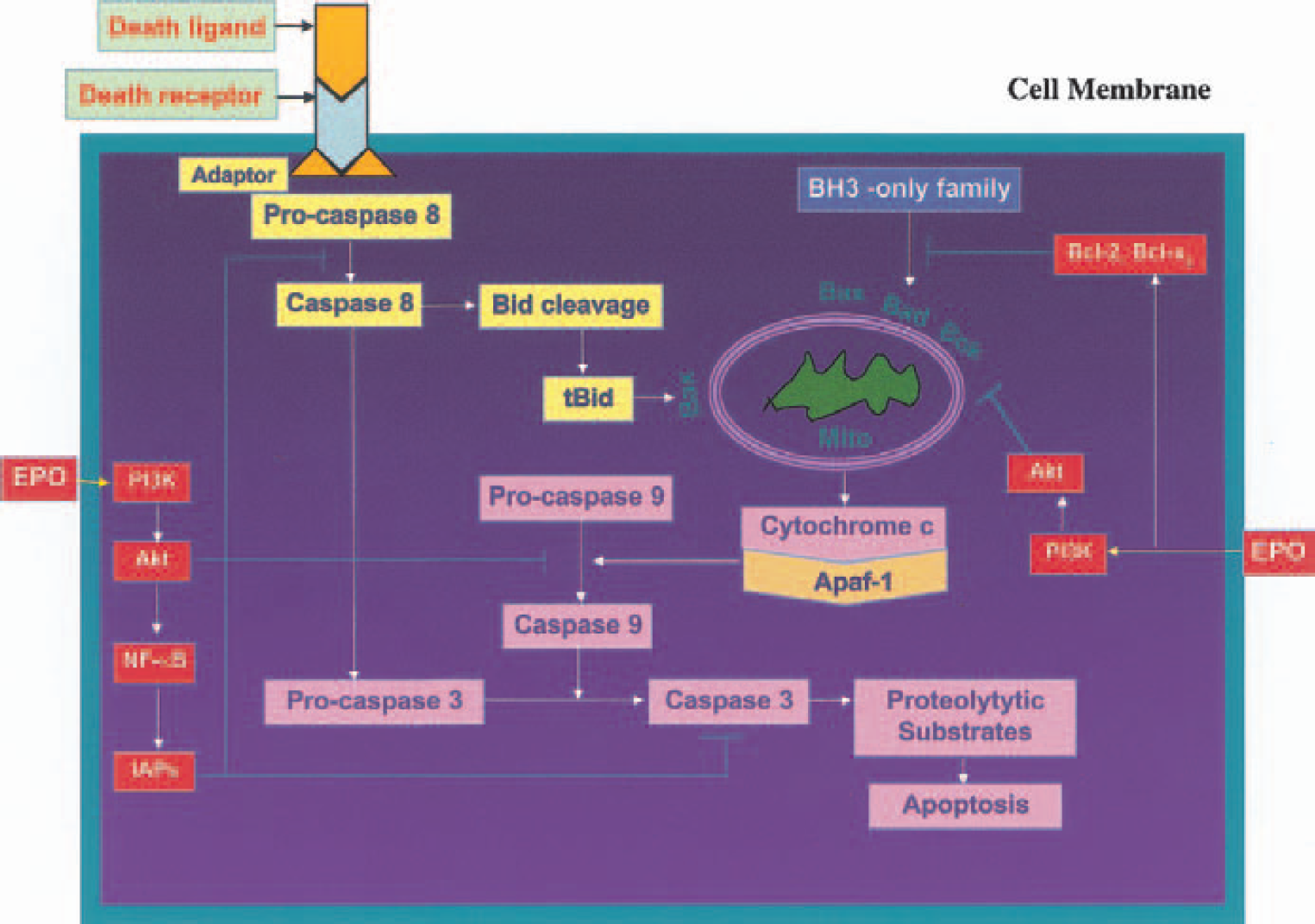
Erythropoietin (EPO) intervention during caspase activation. Pathways that lead to caspase activation are illustrated. One pathway is initiated by membrane death receptor activation resulting in the recruitment of adaptor proteins. Subsequent activation of caspase-8 and caspase-3 ensues. Caspase-8 can then cleave Bid to yield a truncated Bid (tBid) that translocates to the mitochondria (Mito) to release cytochrome c. Another pathway for caspase activation involves mitochondrial membrane depolarization. The mitochondrial pathway is associated with the BH3-only members of Bcl-2 family that facilitate cytochrome c release. Once released, cytochrome c binds to Apaf-1, followed by activation of caspase-9, which subsequently activates the executioner caspase-3. Erythropoietin can affect caspase activation through the PI3K/Akt and Bcl-2/Bcl-xL pathways.
Caspase-1 and caspase-3 function as death proteases to catalyze the specific cleavage of many key cellular proteins (Brown and Borutaite, 1999; Yabuki et al., 2000; Lin and Maiese, 2001; Maiese, 2001). In particular, caspase-1–like proteases promote DNA degradation through the activation of other proteins, such as protein kinase C (Emoto et al., 1995) and caspase-3 (Enari et al., 1996). Caspase-3 is a prominent mediator of genomic DNA degradation. Experimental models that use caspase-3 gene deletions exhibit little or no DNA fragmentation after toxic cellular insults (Keramaris et al., 2000). Direct inhibition of caspase-1- and caspase-3–like activities also can protect neuronal cells against injury (Maiese et al., 2001).
Phosphorylation of Akt by EPO is dependent on the activation of phosphatidylinositide-3-kinase (PI3K) and Jak2. Activation of Jak2 promotes the phosphorylation of tyrosine residues in the intracellular portion of the EPOR (Nguyen et al., 2001). Phosphorylation of the last tyrosine of the EPOR initiates binding of the 85-kd regulatory subunit of PI3K, a heterodimer consisting of a 110-kd catalytic subunit and an 85-kd regulatory subunit. As a result of the binding of the 85-kd regulatory subunit, the 110-kd catalytic subunit becomes active and leads to the phosphorylation of Akt (Miura et al., 1994).
Central to the ability of EPO to prevent cellular PCD is the activation of Akt by EPO. In primary human erythroid progenitors, PCD is blocked through the activation of Akt (Uddin et al., 2000). During anoxia or free-radical exposure, expression of the active form of Akt (phospho-Akt) is increased (Maiese, 2001; Chong et al., 2002). This upregulation of phospho-Akt during injury paradigms seems to be vital for EPO protection, because prevention of Akt phosphorylation abrogates cellular protection by EPO (Akimoto et al., 2000). Erythropoietin may use Akt to prevent PCD at either a pre- or a postmitochondrial level. Activation of Akt can prevent the release of cytochrome c from mitochondria (Kennedy et al., 1999). In addition, Akt can inhibit caspase-9- and caspase-3–like activities downstream of cytochrome c and before the activation of caspase-9 (Zhou et al., 2000). Feedback of these systems, which modulate the half-life of Akt, also can exist. Activity of Akt can be eliminated by caspase-3 induction, because caspase-3 has been shown to cleave Akt leading to the inhibition of Akt kinase activity (Bachelder et al., 2001).
Erythropoietin and mitogen-activated protein kinases
The mitogen-activated protein kinases (MAPKs) are serine/threonine kinases that modulate cell differentiation, growth, and death. Phosphorylated MAPKs can translocate to the cell nucleus to regulate both the activation of transcription factors and the subsequent expression of genes. The MAPKs consist of three distinct components: the extracellular signal-related kinases (ERKs), the c-Jun-amino terminal kinases (JNKs), and the p38 kinase.
Increased activity of the MAPKs p38 and JNK can sometimes lead to cell injury. The MAPKs p38 and JNK have been shown to increase the activity of both caspase-1 and caspase-3 (Ko et al., 2000; MacFarlane et al., 2000). The function of p38 and JNK during injury paradigms in neuronal systems, however, is not entirely evident. The MAPK p38 and possibly JNK are believed to have a role during cellular stress and during neurodegenerative diseases (Hensley et al., 1999). Injury paradigms with anoxia and free-radical injury can result in a significant increase in the phosphorylation of p38 and JNK (Lin et al., 2000; Lin and Maiese, 2001; Chong et al., 2002). Other work has linked p38 activation to Bax translocation during free-radical exposure (Ghatan et al., 2000) and to endothelial injury during cytokine administration (Yue et al., 1999).
Because EPO can enhance cell survival during a variety of injury paradigms, it is possible that the protective mechanisms of EPO may involve the cellular pathways of the MAPKs. The phosphorylation of ERK has been suggested to account for the protection of EPO. In some cell lines, activation of ERK can block proapoptotic pathways (Nagata and Todokoro, 1999). Erythropoietin also can phosphorylate and increase the activity of JNK and p38 (Jacobs-Helber et al., 2000). This enhancement of JNK and p38 activity is believed to mediate both erythroid proliferation and differentiation (Nagata et al., 1998). It is important to note, however, that the modulation of JNK and p38 activity by EPO may be cell specific. In hippocampal neurons and other cell systems during toxic insults, protection by EPO does not appear to require the phosphorylation of p38 or JNK, suggesting that cellular protection by EPO against PCD may be independent of the modulation of p38 and JNK activity (Jacobs-Helber et al., 2000; Chong et al., 2001).
Erythropoietin and protein–tyrosine phosphatases
Approximately 80 protein–tyrosine phosphatases (PTPases) have been identified and form a family of receptor-like and cytosolic enzymes (Schaapveld et al., 1997). In this family, a subgroup of cytoplasmic PTPases, characterized by containing two SH2 NH2-terminal domains to their catalytic phosphatase domain and referred to as SHP, are intimately involved in several cellular activities, such as cytoskeletal maintenance, cell division, and cell differentiation (Feng et al., 1994). Of these, two particular SHP proteins known as SHP1 (SH-PTP1, PTP1C, HCP, and SHP) and SHP2 (SH-PTP2, PTP1D, Syp, PTP2C, and SH-PTP3) have been linked to trophic factor signaling (Yamauchi et al., 1995; You and Zhao, 1997), cell growth (Yi et al., 1993), MAPK activity (Pani et al., 1995), and chemotactic responses (Kim et al., 1999). Of particular interest is the observation that both SHP1 and SHP2 are expressed in the brain cortex, cerebellum, and the hippocampus (Suzuki et al., 1995; Horvat et al., 2001). During neuronal injury paradigms, such as ischemia or trauma, both SHP1 and SHP2 protein expression can be decreased (Horvat et al., 2001).
Although the tyrosine phosphatases SHP are used in tyrosine kinase signaling pathways as regulators of cell growth and development, the role of SHP and its downstream signaling pathways during neuronal injury is not well understood. Recently, loss of SHP function has been associated with enhanced neuronal injury and the induction of PCD. Interestingly, cells that are deficient in SHP function are without overt evidence of anatomical or physiologic disability. Yet during injury paradigms, cells that are without intact SHP function suffer from early induction of genomic DNA degradation and membrane phosphatidylserine exposure (Maiese, 2001). In addition, overexpression of SHP can inhibit PCD in some cell lines (Chauhan et al., 2000). These observations suggest that SHP may be neuroprotective during neuronal degeneration.
Despite the potential role for SHP as a neuroprotectant, however, its relation with EPO appears to be more closely linked with the modulation of EPO signal transduction activity. For example, SHP1 can inhibit the activity of EPO. The phosphorylation of SHP1 is associated with the tyrosine residue, Tyr 429, of the EPOR and serves to negatively regulate EPO signal transduction pathways. After Jak2 activation and phosphorylation of the EPOR, SHP1 binds to the phosphorylated EPOR via its N-terminal SH2 domain, resulting in the dephosphorylation of EPOR and Jak2 (Jiao et al., 1996). Jak2 is subsequently inactivated by dephosphorylation leading to the downregulation of the EPO signal transduction cascade.
Erythropoietin and nuclear factor-κB
The transcription factor nuclear factor-κB (NF-κB) is activated in response to cytokines and stress stimuli (Janssen-Heininger et al., 2000; Aoki et al., 2001; Korn et al., 2001). In resting cells, NF-κB is held captive by proteins of the IκB family and sequestered in the cytoplasm. Stimulation by tumor necrosis factor-α (TNF-α) or oxidative stress results in the activation of the IκB kinase complex, which phosphorylate IκB, ensuring that it is “ubiquitinated” by the addition of a ubiquitin group, degraded, and has released the bound NF-κB. The liberated NF-κB can then translocate to the nucleus and transcriptionally activate target genes (Kyriakis, 2001).
Nuclear factor-κB functions as an antiapoptotic factor through its induction of genes that inhibit PCD. Nuclear factor-κB has been shown to induce the expression of the inhibitor of apoptosis (IAP) protein family (c-IAP1, c-IAP2, and X-chromosome–linked IAP [xIAP]). These proteins specifically inhibit the active forms of caspase-3, caspase-7, and caspase-9 (Reed, 2001). Induction of c-IAP1 and c-IAP2 activity by NF-κB also suppresses TNF-α–initiated apoptosis through the inhibition of caspase-8 activity (Wang et al., 1998). In addition, Gadd45β, a member of the Gadd45 family associated with cell cycle regulation and DNA repair (Zhan et al., 1999), has been identified as a downstream target of NF-κB. The induction of Gadd45β protein by TNF-α is NF-κB dependent and responsible for the downregulation of JNK activation and suppression of apoptosis (De Smaele et al., 2001). Nuclear factor-κB also may prevent apoptosis through the direct activation of Bcl-xL (Chen et al., 2000).
Neuronal protection by EPO is dependent, in part, on the activation of NF-κB. Erythropoietin, in conjunction with the activation of Akt, can promote IκB kinase activity, resulting in the degradation of IκB and the subsequent liberation of NF-κB (Romashkova and Makarov, 1999). After the activation of NF-κB, inhibitor of apoptosis proteins xIAP and c-IAP2 are generated. Loss of NF-κB activity negates the neuroprotective effects of EPO, suggesting that activation of NF-κB is necessary for EPO protection in the nervous system. In some cells, such as neurons, NF-κB activation may require the Jak2 signaling pathway (Digicaylioglu and Lipton, 2001); in other experimental systems, however, EPO induction of NF-κB activation is not dependent on the Jak2 pathway (Bittorf et al., 2001).
CONCLUSIONS
The hemopoietic factor EPO offers a novel approach for elucidating the cellular mechanisms responsible for neuronal development and survival. Erythropoietin relies on a broad array of cellular signal transduction pathways to prevent neuronal injury. Initial protection resides with the prevention of two distinct pathways of PCD that involve the degradation of genomic DNA and the exposure of membrane phosphatidylserine residues.
Subsequently, neuronal protection by EPO is configured in a series of signal transduction cascades. Initially, EPO activates the Jak2 and STAT5 proteins. A second level links the modulation of the Bcl-2 family, Akt, mitochondrial membrane potential, and cysteine protease activity. The final series uses the protein–tyrosine phosphatases SHP and NF-κB to modulate EPOR function and the family of IAP proteins.
Under some clinical and experimental conditions, EPO may not always represent a viable therapeutic agent (Wiessner et al., 2001; Casadevall et al., 2002). Future work, therefore, should continue to identify specific cellular targets of EPO that can promote recovery during injury. In this way, a more complete understanding of the cellular pathways that lead to disorders of the nervous system should ensue, for the development of new therapeutic treatments.