
Abstract
BH3-only proteins are a subfamily of proapoptotic Bcl-2 proteins that act upstream of the mitochondrially mediated cell death pathway, and their association with the pathogenesis of brain ischemia remains largely unknown. The authors explored the temporal profiles of the expression levels and subcellular localization of BH3-only proteins in permanent middle cerebral artery occlusion (MCAO) by Western blot analysis. They observed an increased mitochondrial distribution of Bim at 3 to 6 hours of MCAO that appeared unrelated to transcriptional upregulation, as assessed by semiquantitative reverse transcription—polymerase chain reaction. At 3 to 6 hours of MCAO, Bim immunoreactivity was enhanced in neurons and oligodendrocytes in the ischemic regions. The increased mitochondrial localization of Bim coincided with a marked cytochrome c release and preceded the peak of caspase-9 activation. The authors observed an association of Bim with the dynein intermediate chain, a major component of the dynein motor complex, in the brain using a coimmunoprecipitation assay. Cerebral ischemia induced a time-dependent significant decrease in dynein expression, which started at 3 hours of MCAO. The authors deduced that the liberation of Bim from the dynein motor complex is a likely mechanism for the increased mitochondrial localization of Bim. During MCAO, Bad did not show any change in phosphorylation state or subcellular localization.
There is accumulating evidence that the cell death mechanisms that have been elucidated through studies of apoptosis are involved in ischemic neuronal death (for review, see Mattson et al., 2000). For example, caspases, a family of cysteine proteases related to the execution of apoptosis in mammals, are implicated in the cerebral damage induced by focal ischemia (Hara et al., 1997). Namura et al. (1998) characterized in detail the activation of caspase-3, a terminal caspase, in transient focal cerebral ischemia. In addition to these pioneering studies, we have shown that ischemia-induced oligodendroglial death is mediated by caspases (Shibata et al., 2000). Among the mechanisms for activating caspases, the Bcl-2 family proteins play crucial roles by regulating the release of apoptogenic factors, such as cytochrome c and Smac/DIABLO, from the mitochondria (Du et al., 2000; Kroemer and Reed, 2000; Verhagen et al., 2000). The release of cytochrome c into the cytosol leads to the activation of caspase-9 in the presence of Apaf-1 (Li et al., 1997). The subsequent activation of downstream caspases by caspase-9 culminates in cell demise (Kroemer and Reed, 2000). Many studies have demonstrated that this mitochondrially mediated cell death—inducing pathway is extremely important in the neuronal damage caused by ischemic stroke as well as in neurodegenerative diseases like amyotrophic lateral sclerosis (Graham et al., 2000; Kroemer and Reed, 2000; Guéegan et al., 2001). In particular, Bcl-2–overexpressing transgenic mice are resistant to cerebral ischemia (Martinou et al., 1994), and, conversely, antisense bcl-2 treatment exacerbates ischemic brain damage (Chen et al., 2000). These findings unambiguously indicate that the Bcl-2 family members play a role in brain ischemia and therefore are promising therapeutic targets for ischemic stroke.
The Bcl-2 family proteins are classified into antiapoptotic molecules, which include Bcl-2 and Bcl-XL, and proapoptotic ones, such as Bax, Bak, and Bid (Gross et al., 1999). In the latter group, there is a subfamily referred to as the “BH3-only proteins,” whose known members are mammalian Bad, Bid, Blk, Hrk/DP5, Bim, Noxa, Bik, and C elegans EGL-1 (Huang and Strasser, 2000). On the induction by certain cell death—related stimuli, these Bcl-2 proteins translocate to the mitochondria, where they function upstream of Bax and/or Bak to execute cell demise. The activity of the BH3-only proteins is normally controlled by transcriptional upregulation or a variety of posttranslational modifications, such as cleavage and phosphorylation (Huang and Strasser, 2000; Oda et al., 2000). Although Bim was originally found to play an important role in the apoptosis of hematopoietic cells (Bouillet et al., 1999; Shinjyo et al., 2001), the spectrum of roles for Bim-associated cell death is expanding. Recent studies have shown that Bim is important in the apoptosis of sympathetic neurons after the withdrawal of trophic factors (Putcha et al., 2001; Whitfield et al., 2001). Moreover, Bim-deficient mice exhibit reduced programmed cell death in the thoracic and lumbar dorsal root ganglionic cells at the embryonic stage (Putcha et al., 2001). Bim is present in the adult central nervous system (O'Reilly et al., 2000); there is, however, no in vivo evidence that it plays an important role in a pathogenic state of the brain.
Here, we investigated the temporal profiles of the expression and subcellular distribution of Bim and Bad during permanent middle cerebral artery occlusion (MCAO). We observed that Bim was translocated to the mitochondria during ischemia and that this translocation coincided with the release of cytochrome c and preceded the peak of caspase-9 activation. We also found that Bim is associated with the dynein motor complex in the brain. Cerebral ischemia induced a significant decrease in dynein expression, which concurred with the enhanced mitochondrial localization of Bim. We deduce that the ischemia results in the dissociation of Bim from the dynein motor complex and the subsequent translocation of Bim to the mitochondria. Meanwhile, we did not find any significant change in the expression level or subcellular localization of Bad. This is the first in vivo study demonstrating a pathogenic role for Bim in the diseased brain.
MATERIALS AND METHODS
Animals
Fifty-five adult male C57/B6 mice (body weight, 15.3 to 21.5 g) were purchased from Charles River Laboratory (Kanagawa, Japan), and were used in the present study. They were given food and water ad libitum before and after surgery. All the experimental protocols pertinent to animals were given prior approval as meeting the Animal Experimentation Guidelines of the School of Medicine, Keio University, by the Laboratory Animals Care and Use Committee.
Surgical procedures
Mice were anesthetized with 1 to 2% isoflurane (Forane; Abbott Laboratories, North Chicago, IL, U.S.A.) in a nitrous oxide/oxygen mixture (70%/30%). The rectal temperature was maintained at 36.5°C to 37.5°C using a thermostat-controlled heating pad during surgery. Permanent focal cerebral ischemia was produced by occluding the middle cerebral artery (MCA), using a nylon thread according to Hara et al. (1996). A flexible fiberoptic probe was attached to the exposed skull overlying the left MCA territory, for the continuous cerebral blood flow measurement by laser Doppler flowmeter (ALF21R; Advance, Tokyo, Japan). Successful occlusion was verified by the laser Doppler flowmeter in all cases. After the surgery, the anesthesia was terminated. Neurologic function was evaluated according to Hara et al. (1996). Only the mice that manifested overt right-sided hemiparesis were used in the following experiments. In sham-operated animals, all the procedures except the insertion of the nylon monofilament were carried out.
Western blot analysis
The sham-operated mice and mice ubjected to 1, 3, 6, 12, or 24 hours of MCAO were killed by excess sodium pentobarbital (Dainippon Pharmaceuticals, Osaka, Japan; over 100 mg/kg, intraperitoneally). Ischemic brain tissue, including the ischemic core and penumbra, and the corresponding portion of brain tissue from the sham-operated animals were quickly excised. The samples were homogenized in 7 volumes of ice-cold lysate buffer (200 mmol/L HEPES [pH 7.5], 250 mmol/L sucrose, 1 mmol/L dithiothreitol, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, 5 μg/mL pepstatin, 2 μg/mL aprotinin) in a Teflon homogenizer (800 rpm, 30 strokes). The procedure for subcellular fractionation follows. The homogenates were spun at 3,000 g for 10 minutes. The pellet was subsequently suspended in 10 mol/L sucrose solution, followed by centrifugation at 40,000 g for 60 minutes. The resultant jellylike pellet was used as the nuclear fraction (Nishi, 1994). The 3,000 g supernatant was spun at 8,000 g for 20 minutes, and the pellet was designated as the mitochondrial fraction. The supernatant was further spun at 54,000 g for 60 minutes. The resultant pellet and supernatant were used as the microsome fraction and cytosolic fraction, respectively. The protein concentration of each sample was determined using the Bradford assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
For the mitochondrial and cytosolic samples, an aliquot of 40 μg of protein was subjected to 12.5% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For the nuclear samples, 20 μg of protein was used and SDS-PAGE was performed in the same manner. The separated protein was electrically transferred onto a polyvinylidene difluoride membrane (Immobilon-P; Millipore Corporation, Bedford, MA, U.S.A.). For immunoblotting, the following primary antibodies were used: a rabbit polyclonal anti-Bax antibody (1:1,000; Cat# sc-493; Santa Cruz Biotechnologies, Santa Cruz, CA, U.S.A.), a rabbit polyclonal anti-Bak antibody (1:500; Cat# 06-536; Upstate Biotechnology, Lake Placid, NY, U.S.A.), a mouse monoclonal anti—Bcl-2 antibody (1:500; Cat# sc-7382; Santa Cruz Biotechnology), a goat polyclonal anti-Bim antibody (1:500; Cat# sc-8267; Santa Cruz Biotechnology), a rabbit polyclonal anti-Bad antibody (1:500; Cat# sc-943-G; Santa Cruz Biotechnology), a mouse monoclonal anti-phospho[Ser112]-Bad antibody (1:500; Cat# 9296S; Cell Signaling Technologies, Beverly, MA, U.S.A.), a mouse monoclonal anti-bovine cytochrome oxidase (COX) subunit IV antibody (1:1,000; Cat# A-6431; Molecular Probes, Eugene, OR, U.S.A.), a mouse monoclonal anti–α-tubulin antibody (1:2,000; Cat# T9026; Sigma, St. Louis, MO, U.S.A.), a mouse monoclonal anti—cytochrome c antibody (1:1,000; Cat# 556433, clone 7H8.2C12; PharMingen, San Diego, CA, U.S.A.), a rabbit polyclonal anti—caspase-9 antibody (1:500; Cat# AAP-109; StressGen Biotechnologies, Victoria, Canada), a mouse monoclonal anti-cytoplasmic dynein (74-kd intermediate chain) antibody (1:1,000; Cat# MAB1618; Chemicon International, Temecula, CA, U.S.A.), a rabbit polyclonal anti—c-Jun antibody (1:500; Cat# sc-1694; Santa Cruz Biotechnology), and a rabbit polyclonal anti—phospho[Ser73]-c-Jun antibody (1:500; Cat# 9271; New England Biolabs, Beverly, MA, U.S.A.). The secondary antibodies used were alkaline phosphatase-conjugated anti—immunoglobulin G (IgG) antibodies (anti-mouse IgG, Code Number 115-055-146; anti-rabbit IgG, Code Number 111–055–144; anti-goat IgG, Code Number 705-055-147; all from Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.). The immunoreactive bands were visualized by alkaline phosphatase staining performed in a solution containing 100 mmol/L Tris (pH 9.5), 100 mmol/L NaCl, 50 nmol/L MgCl2, 0.78 mmol/L nitrobluetetrazolium (NBT), and 0.40 mmol/L 5-bromo-4-chloro-3-indolyl phosphate. In some cases, the band intensity was measured using National Institutes of Health image software (version 1.61).
Histologic analysis
Mice were killed as previously described. They were then transcardially perfused with ice-cold 0.9% saline solution, then with 4% paraformaldehyde–0.1 mol/L phosphate-buffered saline (PBS; pH 7.4). Frozen sections were prepared, and stored at −80°C until use.
Some sections were used for hematoxylin and eosin staining to assess tissue damage. The neuronal density in the left cerebral cortex and striatum was calculated, and was expressed as the mean + SD cells/mm3. The regions of interest were set at the lateral striatum and the parietal cortex at the level of 1.10 mm anterior to bregma (Paxinos and Franklin, 2001). The statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Bonnferroni/Dunn post hoc tests. A P value less than 0.01 was considered statistically significant.
For immunostaining, a goat polyclonal anti-Bim antibody (1:100; Santa Cruz Biotechnology) or a mouse monoclonal anti-adenomatous polyposis coli (APC) antibody (1:20; Cat# OP80; Oncogene Research Products, Boston, MA, U.S.A.) were used, and avidin—biotin—peroxidase immunostaining procedures were performed for immunoreactivity visualization using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.) according to the manufacturer's instructions. The terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling (TUNEL) assay was performed with the In Situ Cell Death Detection Kit, POD (Cat# 1684817; Boehringer Mannheim Corp., Indianapolis, IN, U.S.A.) in accordance with the manufacturer's instructions.
For double-immunostaining of Bim and APC, the sections were successively treated with 0.3% H2O2–0.1 mol/L PBS, 0.1% Triton X-100–0.1 mol/L PBS, and 4% skim milk–0.1 mol/L PBS. The primary antibodies were applied at 4°C overnight (anti-Bim antibody, 1:100; anti-APC antibody, 1:20). The sections were treated with Texas red dye—conjugated anti-mouse IgG antibody (Code Number 715-075-150; Jackson ImmunoResearch Laboratories) and peroxidase-conjugated anti-goat IgG antibody (Code Number 705-035-147; Jackson ImmunoResearch Laboratories) at dilutions of 1:200 and 1:500, respectively. The visualization of Bim immunoreactivity was carried out with tyramide signal amplification using TSA Fluorescence Systems (NEM Life Science Products, Boston, MA, U.S.A.) according to the manufacturer's instructions. The nuclear staining was performed by incubating the sections in 20 μmol/L bisbenzimide (Cat# B-2883; Sigma)–0.1 mol/L PBS at room temperature for 5 minutes. For double-immunostaining of APC immunohistochemistry and TUNEL, APC immunostaining was performed using the mouse monoclonal anti-APC antibody (1:20; Oncogene Research Products) and the Texas red dye—conjugated anti-mouse IgG antibody (1:200; Jackson ImmunoResearch Laboratories), followed by the TUNEL method using the In Situ Cell Death Detection Kit, Fluorescein (Boehringer Mannheim Corp.) in accordance with the manufacturer's instructions. Nuclear counterstaining was performed using bisbenzimide (Sigma) as previously described. The specimens were examined using a fluorescence microscope (Eclipse E800; Nikon, Tokyo, Japan). For double staining of Bim and NeuN, a neuronal marker, Bim and NeuN immunoreactivity was visualized using the avidin—biotin—peroxidase reaction with diaminobenzidine as chromogen and the alkaline phosphatase reaction, respectively. An anti-NeuN antibody was purchased from Chemicon International (Cat# MAB377).
Semiquantitative reverse transcription—polymerase chain reaction
After administration of excess sodium pentobarbital, sham-operated mice and mice subjected to 3 or 24 hours of MCAO were perfused transcardially with ice-cold 0.9% saline solution for 3 minutes to minimize the number of remaining intravascular blood cells in the tissues, including the brain. The brain was removed from the skull immediately. The brain region corresponding to the MCA territory was separated and frozen in liquid nitrogen. Isolation of total RNA from the brain samples was carried out using TRIZOL LS Reagent (Cat# 10296; Life Technologies, Rockville, MD, U.S.A.). RNA content was measured with a spectrophotometer (DU 640; Beckman Instruments, Fullerton, CA, U.S.A.). Complementary DNA (cDNA) was synthesized from the RNA samples using the SUPERSCRIPT First-Strand Synthesis System for RT-PCR (Cat# 11904-018; Life Technologies) according to the manufacturer's protocols. First-strand cDNA was synthesized using random hexamers and the SUPERSCRIPT II reverse transcriptase. The method of polymerase chain reaction (PCR) for Bim cDNA follows. Two-microliter cDNA aliquots were mixed with 1.5 mmol/L MgCl2, 200 μmol/L dNTP, 2 U Taq polymerase, and 0.2 μmol of the following primers: forward 5′-CTGAGTGTGACAGAGAAGGTGG-3′ and reverse 5′-GTGGTCTTCAGCCTCGCGGT-3′. PCR was run at 95°C for 5 minutes, followed by 33 cycles at 94°C for 1 minute, 60°C for 1 minute, 72°C for 1 minute, and was finished with the final extension at 72°C for 5 minutes. Our preliminary studies showed that 33 cycles was most appropriate for semiquantification for Bim cDNA. The cDNA of a housekeeping gene, β-actin, was amplified as a control under identical conditions using the following primers: forward 5′-ATCCGTAAAGACCTCTATGC-3′ and reverse 5′-AACGCAGCTCAGTAACAGTC-3′. The PCR products were separated by polyacrylamide gel electrophoresis, and visualized with SYBR Green I (Cat# S-7563; Molecular Probes).
Caspase-9–like protease activity assay
Sham-operated mice and mice subjected to 1, 3, or 6 hours of MCAO were killed with excess sodium pentobarbital (over 100 mg/kg, intraperitoneally). The ischemic brain tissues were removed as previously described. The brain tissues were homogenized in 7 volumes of a protease inhibitor—free lysis buffer (200 mmol/L M HEPES [pH 7.5], 250 mmol/L sucrose, 1 mmol/L dithiothreitol, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA) using a Teflon homogenizer. The homogenates were spun at 54,000 g for 60 minutes, and the resultant supernatants were stored at −80°C until use. The caspase-9–like protease assay was performed using a fluorogenic substrate, Ac-LEHD-7-amino-4-trifluoromethyl coumarin (Ac-LEHD-AFC) (Caspase-9 Substrate I, Fluorogenic; Cat# 218765; Calbiochem-Novabiochem, San Diego, CA, U.S.A.) according to Enari et al. (1996), with some modifications. After the addition of 12.5 μmol/L Ac-LEHD-AFC, the brain homogenates were diluted 10-fold in lysis buffer and incubated at 37°C for 60 minutes. Quantification of released free AFC was performed at an excitation of 380 nm and an emission of 460 nm using a CytoFluor Series 400 (PerSeptive Biosystems, Framingham, MA, U.S.A.). In parallel, the protein concentrations of the samples were determined by the Bradford assay. The amount of released free AFC was normalized to the protein concentration in each sample. The normalized amount of released AFC was considered to represent the caspase-9–like protease activity. The values were expressed as a percentage + SD of the control level obtained from the sham-operated mice. Statistical analysis was performed using one-way ANOVA followed by Bonferroni/Dunn post hoc tests. A P value less than 0.01 was considered statistically significant.
Immunoprecipitation
To assess the association of Bim with cytoplasmic dynein in vivo, we performed a coimmunoprecipitation assay. The brain homogenates prepared from sham-operated mice and those subjected to 1 and 3 hours of MCAO were used. Protein (300 μmg diluted in 1 mL of lysis buffer) was precleared with 0.25 μg of normal IgG together with 20 μl of resuspended Protein A/G-PLUS Agarose beads (Cat# sc-2003; Santa Cruz Biotechnology). After centrifugation at 1,000 g for 5 minutes, the supernatant was transferred to a fresh tube, and either 3 μl of mouse monoclonal anti-cytoplasmic dynein (74-kd intermediate chain) antibody (Chemicon International) or 10 mL of goat polyclonal anti-Bim antibody (Santa Cruz Biotechnology) was added, followed by incubation at 4°C for 60 minutes. After adding 20 μl of resuspended Protein A/G-PLUS Agarose beads, the samples were incubated at 4°C in a rotating device overnight. After centrifugation at 1,000 g for 5 minutes, the pellets were recovered and washed three times with 0.1 mol/L PBS. The pellets were then subjected to Western blot analysis in duplicate as described previously. A goat polyclonal anti-Bim antibody (Santa Cruz Biotechnology) and a mouse monoclonal anti-cytoplasmic dynein (74-kd intermediate chain) antibody were used at dilutions of 1:200 and 1:1,000, respectively, as primary antibodies. The immunoreactive bands were visualized by alkaline phosphatase staining.
RESULTS
Ischemic tissue damage induced by middle cerebral artery occlusion
The insertion of a nylon monofilament reproducibly resulted in an abrupt decrease in cerebral laser Doppler flow measured over the left MCA territory, which was less than 20% of the control value (data not shown). The extent of the change in laser Doppler flow is consistent with previous studies that used a similar murine MCAO model (Hara et al., 1996; Morikawa et al., 1998, Hermann et al., 2001). Hence, it was confirmed that the MCA territory was reproducibly subjected to ischemia in each animal used in the present study. Consistently, the cerebral cortex and lateral striatum on the ischemic side at 24 hours of MCAO were pathologically characterized by ischemic changes, such as pyknotic nuclei of neurons and perineuronal vacuolation (data not shown). Furthermore, at this time point, numerous remaining cells in the same brain regions exhibited DNA fragmentation detectable by the TUNEL method (data not shown).
Expression and subcellular localization of Bcl-2 family proteins during middle cerebral artery occlusion
A proapoptotic BH3-only protein, Bim, was scarcely found in the mitochondrial fraction in the normal state (Fig. 1A). After MCAO, the level of Bim expression in the mitochondrial fraction significantly increased to 278.5 + 38.0% (n = 3, P h 0.001) at 3 hours and 172.2 + 27.9% (n = 3, P h 0.01) at 6 hours as compared with the control level (Fig. 1A). The apparent molecular mass of Bim as identified by Western blotting was approximately 28 to 29 kd (Fig. 1A). There are three isoforms of Bim protein, namely, BimS, BimL, and BimEL, which are produced by alternative splicing (O'Connor et al., 1998; O'Reilly et al., 2000; Putcha et al., 2001). Based on its apparent molecular mass, the Bim protein recognized on our Western blots was considered to be BimEL. There were no apparent Bim-immunoreactive bands in Western blot using cytosolic samples (data not shown). We examined the expression of Bim in normal and ischemic brains at the mRNA level using semiquantitative RT-PCR. The RT-PCR demonstrated that all three splice variants exist in the brain under normal circumstances (Fig. 1B), suggesting that the protein expression of both BimS and BimL is likely to be negatively regulated by posttranscriptional mechanisms. Our results showed that the RNA expression level of BimEL was low compared with the other two variants (Fig. 1B). Three hours of MCAO did not appear to affect the relative mRNA expression pattern of the Bim splice variants (Fig. 1B). The semiquantitative RT-PCR showed that there was no significant increase in the amount of BimEL mRNA at 3 hours of MCAO, compared with the control state (Fig. 1B). It has recently been elucidated that c-Jun—mediated transcriptional control of Bim mRNA expression is involved in neuronal death (Putcha et al., 2001; Whitfield et al., 2001). Therefore, we checked the effect of ischemia on the phosphorylation at Ser73, a well-established hallmark of c-Jun activation (Pulverer et al., 1991; Smeal et al., 1991, Herdegen et al., 1998), by Western blot analysis using the nuclear samples. There was no enhanced Ser73 phosphorylation of c-Jun during MCAO (Fig. 1C). In combination with the aforementioned RT-PCR data, it is inferred that the increased Bim expression in the mitochondria at 3 hours of MCAO was not attributable to transcriptional upregulation.
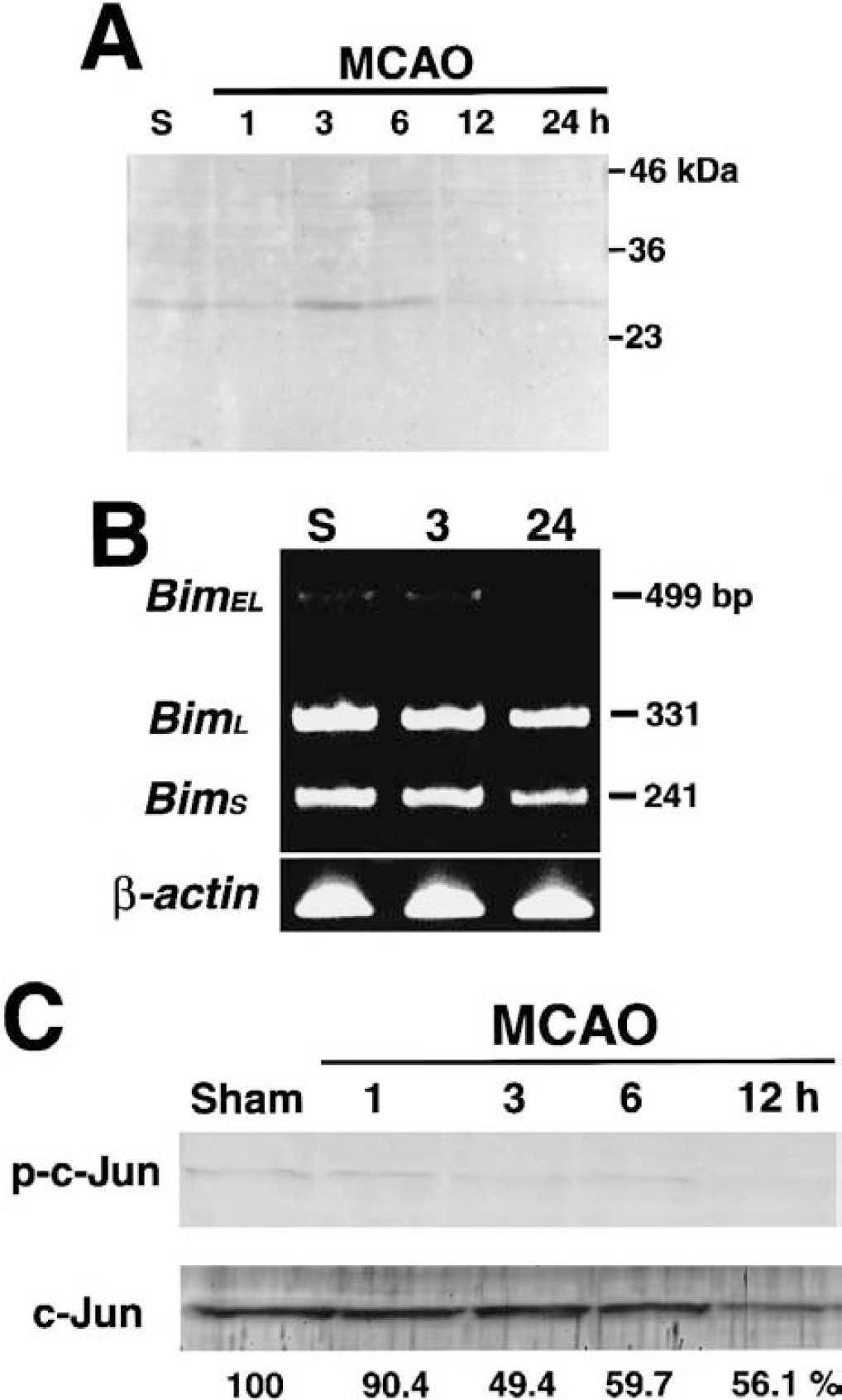
Temporal profiles of Bim protein expression.
Another proapoptotic BH3-only protein, Bad, was detected only in the cytosol at any time point examined (Fig. 2). Its expression level decreased after 6 hours of MCAO. Furthermore, we did not observe the phosphorylation of Bad at Ser112 during MCAO (data not shown). Bax expression was observed in both the cytosolic and mitochondrial fractions (Fig. 2). During ischemia, the mitochondrial Bax exhibited a gradual decrease (Fig. 2). The expression of Bak was restricted to the mitochondrial fraction before and during ischemia (Fig. 2). The expression level of Bak exhibited a slight increase after 6 hours of MCAO (Fig. 2). An antiapoptotic molecule, Bcl-2, was consistently present in the mitochondrial fraction before and during MCAO (Fig. 2). The successful fractionation of the cytosolic and mitochondrial fractions in our preparations was confirmed by the selective presence of the respective markers (α-tubulin for cytosol and COX subunit IV for mitochondria) in the corresponding fractions (Fig. 2).
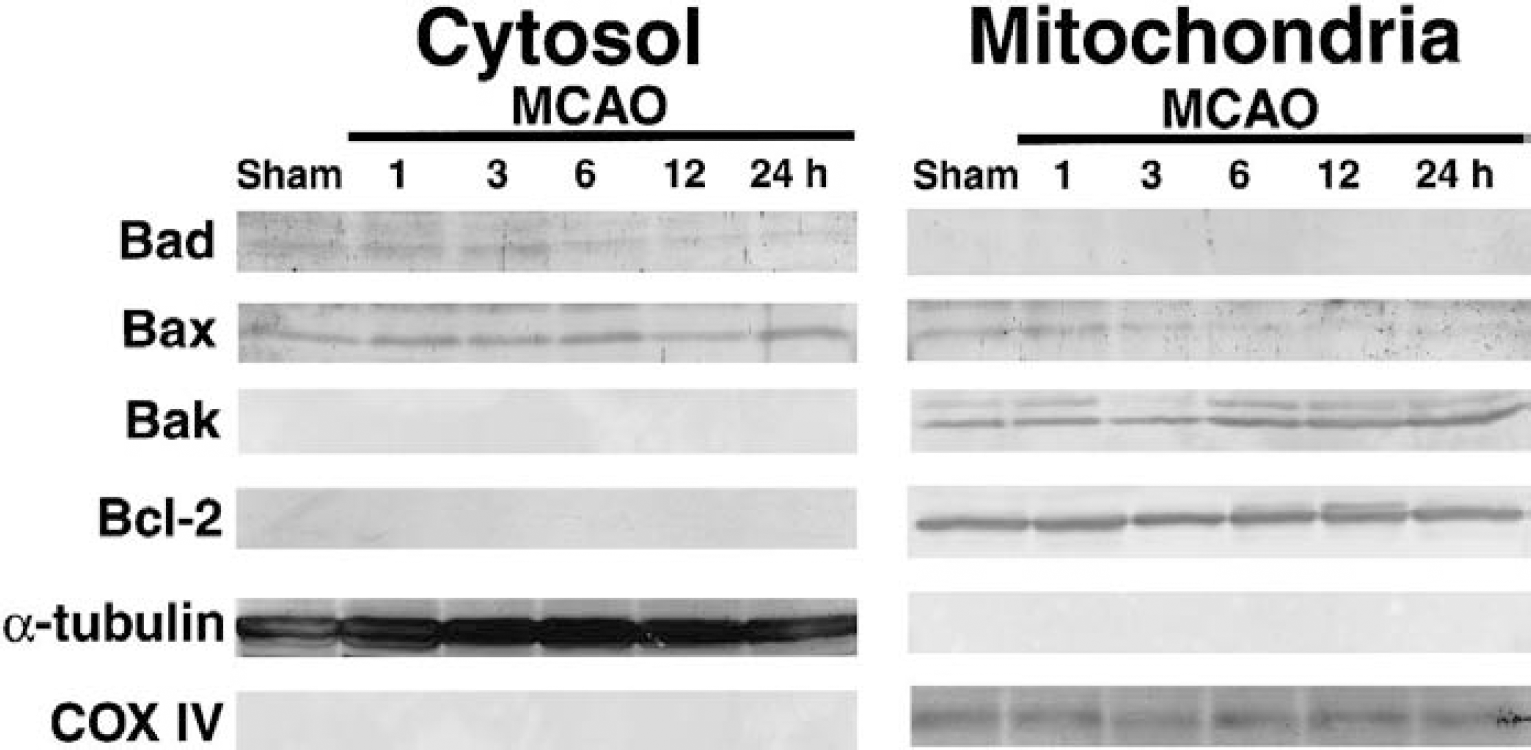
Temporal profiles of expression and subcellular localization of Bcl-2 family proteins other than Bim during permanent middle cerebral artery occlusion (MCAO). The immunoreactivity of Bad was restricted to the cytosol during MCAO, indicating that there was no significant translocation to the mitochondria. The immunoreactivity of cytosolic Bad exhibited a gradual decrement during MCAO. Bax was recognized in both the cytosolic and mitochondrial fractions. During MCAO, there was a gradual reduction in mitochondrial Bax, especially after 3 hours of MCAO. Bak resided in the mitochondria during MCAO, and its expression increased after 6 hours of MCAO. Bcl-2 was also localized to the mitochondria, but without any significant alteration in its expression level during MCAO. The markers for the cytosolic (α-tubulin) and mitochondrial fractions (cytochrome oxidase subunit IV) verified the separation of the fractions and the equal loading of protein samples in each lane. The results are representative of two or three independent experiments.
Bim expression in neurons and oligodendrocytes in the ischemic brain
Immunohistochemical analysis did not detect any significant staining of Bim in the normal brain (data not shown). However, Bim immunoreactivity was apparent in the cerebral cortex and lateral striatum at 3 and 6 hours of MCAO (Fig. 3A). Based on the morphologic features, the majority of Bim-positive cells appeared to be neurons (Fig. 3A). Double immunostaining using antibodies against Bim and NeuN, a neuronal marker, demonstrated that most Bim-positive cells were neurons, although there were a small number of nonneuronal (NeuN-negative) cells immunoreactive for Bim (Fig. 3B). Moreover, Bim-positive cells were also identified in the white matter in the left MCA territory at the same time points (Fig. 3C). This staining was specific, because there was no significant Bim immunoreactivity in the contralateral corresponding region (Fig. 3D). The double immunostaining of Bim and an oligodendroglial marker, APC, demonstrated that the Bim-positive cells in the white matter were oligodendrocytes (Figs. 3E and 3F). To examine whether oligodendrocytes in the same region died after ischemia, we performed double staining of APC immunohistochemistry and TUNEL. Most oligodendroglial cells were positive for TUNEL at 24 hours of MCAO (Figs. 3G, 3H and 3I). Hence, the upregulated Bim in oligodendrocytes was likely to be relevant to cell death. Consistent with the results of the Western blot analysis, Bim immunoreactivity was not detected at 12 or 24 hours of MCAO (data not shown).
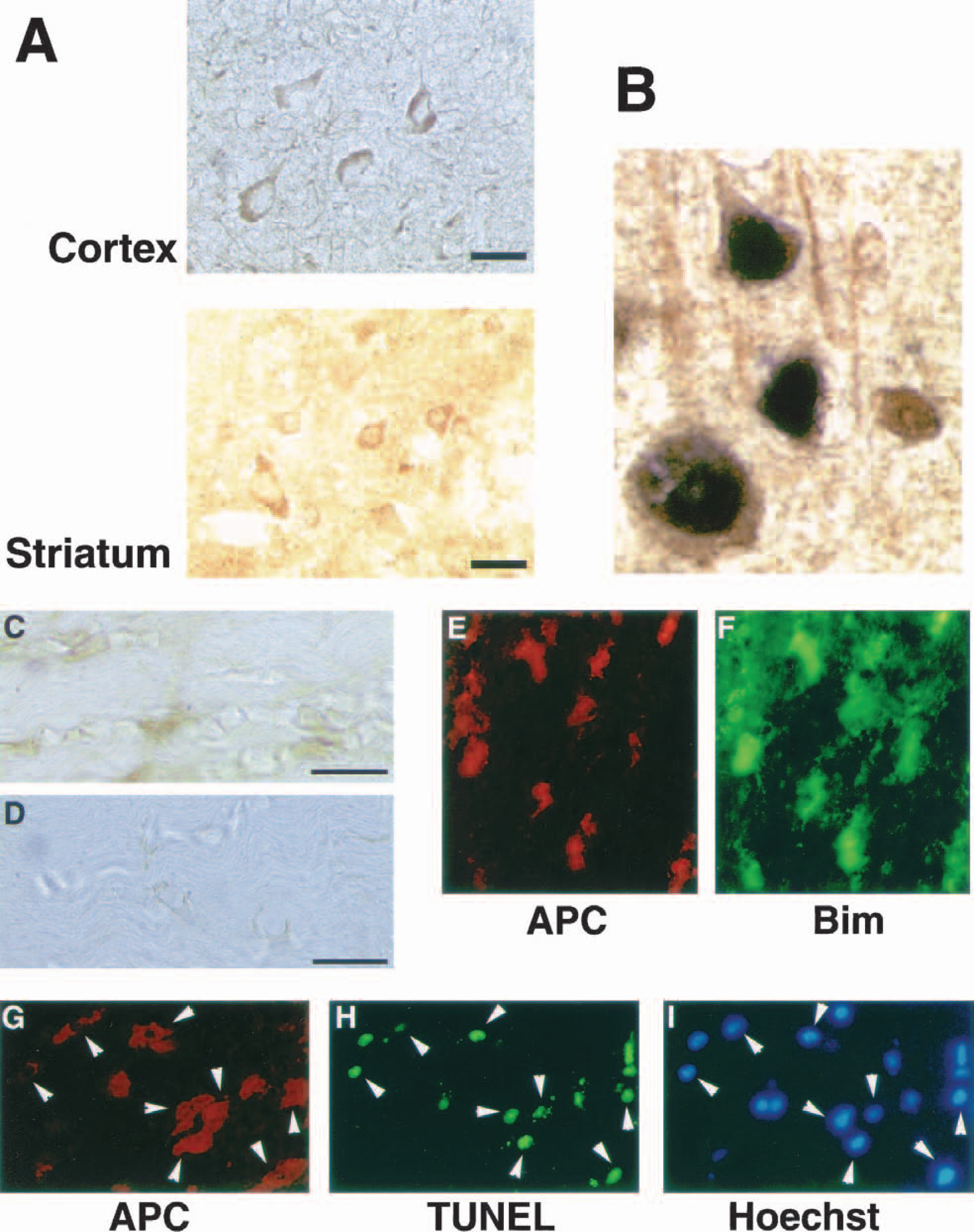
Immunohistochemical analysis of Bim in the brain.
Cytochrome c release and caspase-9 activation during middle cerebral artery occlusion
One of the most important events initiated by the proapoptotic Bcl-2 family members is the release of cytochrome c from mitochondria into the cytosol. To elucidate the temporal relation between the mitochondrial distribution of Bim and cytochrome c release, we assessed the release of cytochrome c by Western blotting for up to 6 hours of MCAO. There was a marked increase in cytochrome c immunoreactivity in the cytosol at 3 hours of MCAO (Fig. 4A). In line with this finding, there was a significant decrease in the mitochondrial level of cytochrome c between 1 and 3 hours of MCAO (91.2 + 10.3% at 1 hour of MCAO, 56.4 + 9.5% at 3 hours of MCAO [P h 0.001, ANOVA, compared with control], 67.5 + 9.8% at 6 hours of MCAO [P h 0.001, ANOVA, compared with control], n = 4 at each time point, Fig. 4A). The close temporal relation between the increased mitochondrial localization of Bim and the cytochrome c release strongly implies that the association of Bim with the mitochondrial membrane could trigger the cytochrome c release at around 3 hours of MCAO. It is known that released cytochrome c binds to Apaf-1, which leads to the cleavage and activation of caspase-9 (Zou et al., 1997). Therefore, we investigated the activation of caspase-9 by Western blotting and an enzyme activity assay. In Western blot analysis of the cytosolic fraction, the noncleaved pro-form (46 kd) of caspase-9 decreased after MCAO, and the cleaved form of caspase-9 (36 kd) appeared after 3 hours of MCAO, showing that caspase-9 activation occurred after 3 hours of MCAO (Fig. 5A). Meanwhile, there was no significant alteration in caspase-9 immunoreactivity in the mitochondrial fraction during MCAO (Fig. 5A). Because we detected no significant immunoreactivity for COX subunit IV in the cytosolic samples on the Western blot, we could exclude the possibility of significant contamination of cytoplasmic samples with mitochondrial materials (Fig. 5B). In keeping with these Western blot data, caspase-9–like protease activity increased after ischemia, and rose to 741.1 + 403.4% (n = 8, P h 0.0001) of the control level at 6 hours of MCAO (Fig. 5C). This temporal profile of caspase-9 activation seemed quite reasonable, because the peak of caspase-9 activation was preceded by the ischemia-induced cytochrome c release into the cytosol.
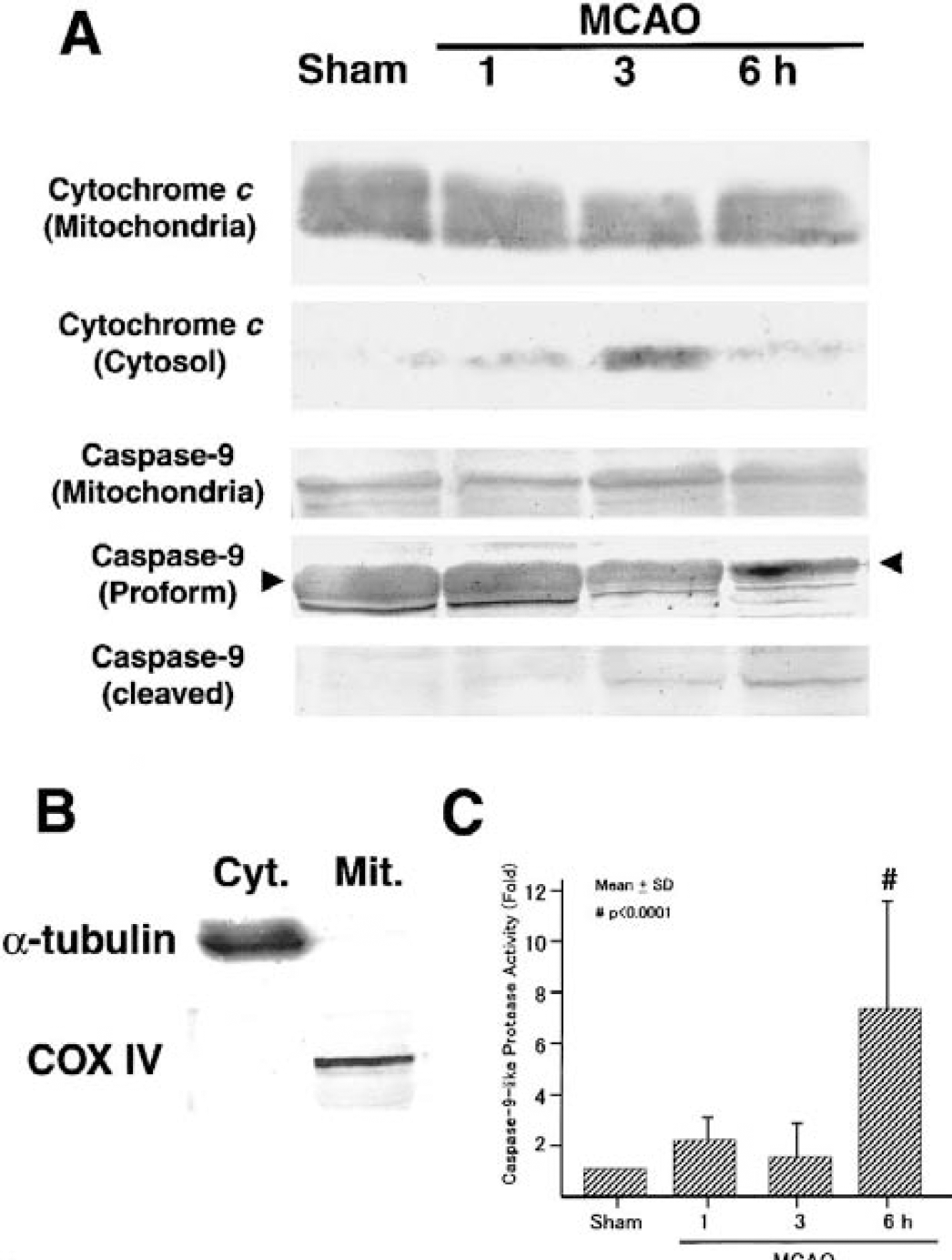
Cytochrome c release and caspase-9 activation during middle cerebral artery occlusion (MCAO).
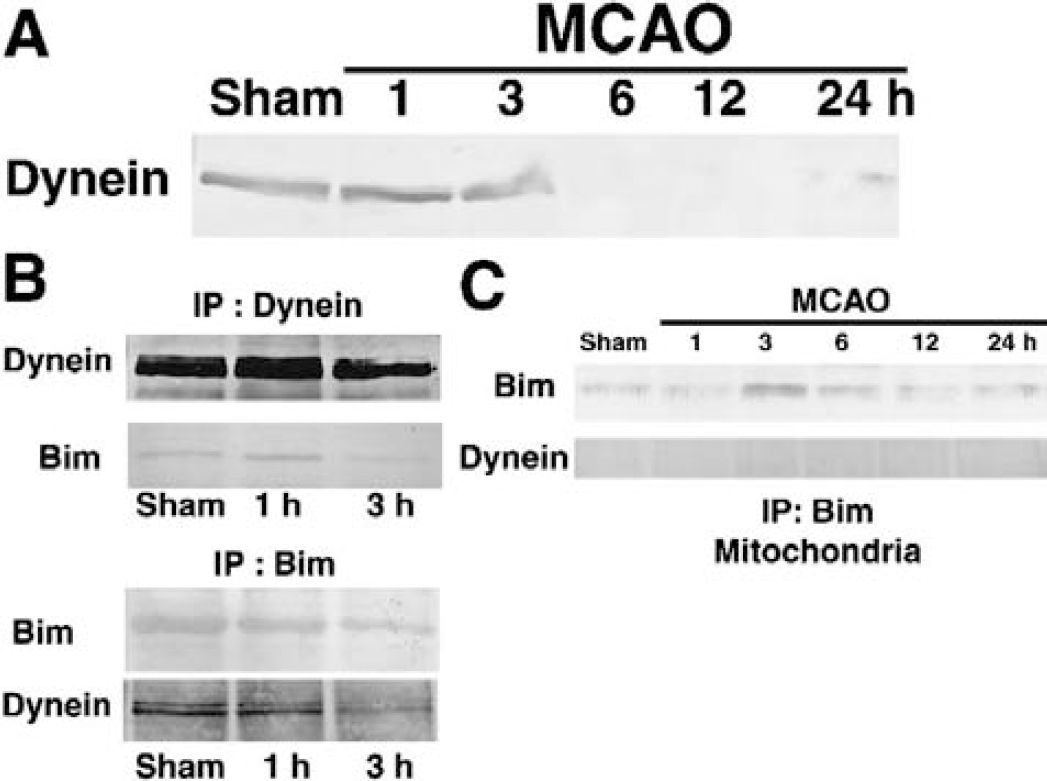
Expression of cytoplasmic dynein and its association with Bim.
Association of Bim and cytoplasmic dynein in vivo
A recent in vitro study showed that, under normal conditions, Bim is associated with the dynein motor complex through an interaction with the LC8 cytoplasmic dynein light chain component, and that certain cell death—related stimuli lead to the liberation of Bim and LC8 from the rest of the complex; this released Bim translocates to the mitochondrial membrane, eventually culminating in cell demise (Puthalakath et al., 1999). Therefore, we investigated whether Bim and the dynein motor complex are associated with each other in the brain and, if so, how cerebral ischemia might affect the interaction between them. The intermediate chain is known to serve as a link between LC8 and the heavy chain in the dynein complex (Hirokawa et al., 1998); therefore, if Bim is associated with the dynein motor complex, it should coimmunoprecipitate with the intermediate chain. We first checked the expression of the dynein intermediate chain by Western blot analysis. The expression of dynein intermediate chain declined after ischemia: its expression level began to decrease at 3 hours of MCAO, and became negligible thereafter (Fig. 5A). It is very intriguing that the timing of the decreased expression of the dynein intermediate chain coincided with the increased recruitment of Bim to the mitochondria, as previously demonstrated (Fig. 2). Furthermore, a coimmunoprecipitation assay using the cytoplasmic fraction of brain lysates revealed that BimEL coimmunoprecipitated with the intermediate chain in normal and ischemic brains (Fig. 5B). Moreover, we carried out a coimmunoprecipitation assay using mitochondrial samples. The immunoprecipitates that were recovered using anti-Bim antibody did not exhibit any significant immunoreactivity for the intermediate chain of dynein, which strongly implied that the dynein intermediate chain did not bind to Bim on the mitochondria (Fig. 5C). Collectively, these results raise the possibility that the ischemia-induced loss of the dynein intermediate chain might result in the liberation of Bim from the dynein motor complex, allowing this proapoptotic BH3-only protein to localize to the mitochondria.
DISCUSSION
The present study demonstrated that a BH3-only protein, Bim, localizes to the mitochondria during permanent MCAO. We showed that Bim immunoreactivity is enhanced in both neurons and oligodendrocytes in the ischemic MCA territory. Judging from the temporal relation, this phenomenon is likely to be linked to the release of cytochrome c from the mitochondria into the cytosol and the subsequent activation of caspase-9. Furthermore, we provided evidence that a possible mechanism underlying the increased mitochondrial localization of Bim is the dissociation of this cell death—promoting protein from the dynein motor complex. This is the first in vivo study reporting a pathogenic role for Bim in the diseased brain.
Many biochemical and genetic studies have shown that the Bcl-2 protein family plays critical roles in ischemic brain damage (for review, see Graham et al., 2000). The most convincing examples include the resistance to ischemic neuronal death in Bcl-2–overexpressing mice (Martinou et al., 1994) and the exacerbation of ischemic damage in brains treated with antisense bcl-2 (Chen et al., 2000). The BH3-only proteins are a subfamily of the Bcl-2 proteins named after their characteristic structure, and include mammalian Bad, Bik, Blk, Hrk/DP5, Bid, Bim, Noxa, and C elegans EGL-1 (for review, see Huang and Strasser, 2000). All these proteins, including Bim and Bad, can directly bind to Bcl-2 or Ced-9 and induce the release of cytochrome c into the cytosol, which culminates in cell death (Ottilie et al., 1997; O'Connor et al., 1998; Gross et al., 1999). Although the mechanisms by which they induce cell death have yet to be completely clarified, it is postulated that mammalian BH3-only proteins bind directly to and inhibit the antiapoptotic Bcl-2 and Bcl-XL, which normally suppress proapoptotic Bcl-2 family members, such as Bax and Bak (Huang and Strasser, 2000; Cheng et al., 2001). Consistent with this, recent studies have clearly shown that BH3-only proteins require another proapoptotic Bcl-2 subfamily, Bax or Bak, to execute cell death (Harris and Johnson, 2001; Wei et al., 2001; Zong et al., 2001). Bim, Bad, and Bid cannot induce apoptosis in bax−/− or bak−/− cells in vitro, and their ability to induce apoptosis is restored by delivery of the bax or bak gene (Wei et al., 2001; Zong et al., 2001). The results obtained in the present study are consistent with these in vitro data. Because we demonstrated the coexistence of Bim, Bcl-2, Bax, and Bak and cytochrome c release in the mitochondria at 3 and 6 hours of MCAO, we can deduce that the cytochrome c release was due to the action of Bim in cooperation with Bax and/or Bak, which overrode the opposing action of Bcl-2. We could see no apparent phosphorylation at Ser112 or mitochondrial translocation of Bad, another BH3-only protein, during MCAO. It is demonstrated that Bad is phosphorylated at Ser112 by mitochondria-anchored protein kinase A (Harada et al., 1999). Our observation that there was no Ser112 phosphorylation of Bad supports the view that Bad did not translocate to mitochondria during MCAO. Collectively, it appeared that Bad had no significant role in the ischemia-induced tissue damage in our model.
We did not observe any translocation of Bax from the cytosol to the mitochondria during permanent MCAO. As stated previously, Bax is considered to be an important proapoptotic Bcl-2 family member that directly affects the release of cytochrome c into the cytosol (Antonsson et al., 1997; Jurgensmeyer et al., 1998; Narita et al., 1998). Cao et al. (2001) analyzed the intracellular translocation of Bax after transient MCAO in detail. They observed that the mitochondrial localization of Bax became detectable at 3 hours after 30 minutes of MCAO. This temporal profile suggests that reperfusion injury rather than cerebral ischemia per se caused the Bax translocation in their experimental model. In our study, at 6 hours of MCAO, Bim and Bak were clearly identifiable in the mitochondria, although mitochondrial Bax became negligible at this time. Gillardon et al. (1996) also reported that Bax immunoreactivity decreased in the ischemic core in a permanent MCAO model. Regarding the role of Bax and Bak, it is a matter of controversy whether they have an equivalent role in the BH3-only protein—induced cytochrome c release and cell death. The BimEL-induced apoptosis of cerebellar granule cells is dependent only on Bax (Harris and Johnson, 2001). Therefore, in neuronal cells, Bax may be more important in Bim-induced cytochrome c release than Bak. The small amount of mitochondrial Bax could explain why the cytochrome c release was not remarkable at 6 hours of MCAO. The relatively early release of cytochrome c in our study (around 3 hours after the induction of MCAO) is consistent with the previous studies using rodent permanent MCAO models (Fujimura et al., 1999, 2001; Sasaki et al., 2000).
As for the mechanisms of the increased Bim localization to the mitochondria, semiquantitative RT-PCR showed that Bim mRNA did not increase at 3 hours of MCAO. A recent study showed that a dominant-negative mutant of c-Jun reduced Bim expression, and attenuated the apoptosis of sympathetic neurons after nerve growth factor withdrawal (Whitfield et al., 2001). Another study reported that the induction of Bim in sympathetic neurons after nerve growth factor withdrawal was accompanied by the phosphorylation (activation) of c-Jun (Putcha et al., 2001). These results suggest that a c-Jun—mediated transcriptional mechanism (or mechanisms) is important for Bim induction. However, we did not observe any enhancement of c-Jun phosphorylation by Western blot analysis. Taking these observations together, we conclude that there was no significant transcriptional upregulation of Bim. Another known mechanism that may account for the increased localization of Bim in the mitochondria is the mobilization of Bim from the dynein motor complex. The dynein motor complex is a microtubule-associated structure that is involved in organelle transport and mitosis (for review, see Hirokawa et al., 1998). An in vitro study demonstrated that BimEL and BimL, but not BimS, are bound to the dynein motor complex under normal circumstances (Puthalakath et al., 1999). This is considered to be a defense mechanism that inhibits the inappropriate localization of Bim to the mitochondria in the normal state. Certain death-related stimuli, such as the withdrawal of interleukin-3, result in the liberation of Bim and a dynein light chain, LC8, from the remaining dynein motor complex, allowing Bim to localize to the mitochondria (Puthalakath et al., 1999). We showed that MCAO induced a time-dependent loss of the dynein intermediate chain, a component linking LC8 with the dynein heavy chain. The cytoplasmic dynein may be sensitive to ischemic injury, given that its expression is also greatly attenuated after transient forebrain ischemia (Aoki et al., 1995). Together with our immunoprecipitation assay showing the association of Bim and the dynein intermediate chain, the liberation of Bim from the dynein motor complex is a likely mechanism underlying the increased mitochondrial distribution of Bim during MCAO.
Our immunohistochemical analysis showed the appearance of obvious Bim immunoreactivity in both neurons and oligodendrocytes in the MCA territory at 3 and 6 hours of MCAO. A previous study showed that Bim immunoreactivity is localized to neurons in the nervous system (O'Reilly et al., 2000). Our study is the first to demonstrate the presence of Bim in oligodendrocytes. Because information about the roles of Bcl-2 family members in oligodendrocytes is scarce (Bonneti and Raine, 1997; Kuhlmann et al., 1999; Soane et al., 1999; Tanaka et al., 2001), this finding is of particular relevance. The oligodendrocytes in the same region were dead at 24 hours of MCAO, as witnessed by extensive DNA fragmentation detected by the TUNEL method. We previously reported that hypoxia-and ischemia-induced oligodendroglial death is mediated partly by cytochrome c release and caspase activation (Shibata et al., 2000). Therefore, Bim may be a good candidate molecule that works upstream of cytochrome c release and caspase activation in ischemia-induced oligodendroglial death. Because dynein family proteins are present in oligodendrocytes (Gould et al., 2000) as well as neurons (Hirokawa et al., 1998), the posttranslational control of Bim intracellular distribution by the dynein motor complex may be operative in both these neural cell types. Based on our assumption that the intracellular redistribution rather than de novo synthesis of Bim is the activation mechanism of Bim at 3 to 6 hours of MCAO, the enhanced Bim immunoreactivity in the immunohistochemical analysis should not be attributable to increased protein expression. We speculate that it may reflect the redistribution of Bim to discrete organelles (mitochondria) from the entire cytosol, rendering its immunoreactivity more discernible from the background. Alternatively, binding sites of the polyclonal antibody we used to detect Bim might be physically blocked when Bim is bound to the dynein motor complex.
Caspase-9 is known to be activated downstream of cytochrome c release into the cytosol (Li et al., 1997). In agreement with this, we showed that the peak of caspase-9 activation occurred after cytochrome c release. The timing of caspase-9 activation in our ischemia model was also consistent with that found in a recent study (Benchoua et al., 2001). Caspase-9 resides in both the cytosol and mitochondria (Krajewski et al., 1999; Zhivotovsky et al., 1999). Whether the release of caspase-9 from mitochondria occurs in response to ischemic insult has not been established, although there are a few studies reporting its occurrence (Krajewski et al, 1999; Cao et al., 2001). Because we did not find any significant change in the amount of the mitochondrial caspase-9 pro-form during MCAO, our findings do not support the idea that mitochondrial caspase-9 is activated to bring about cell death, which is in agreement with another recent study (Noshita et al., 2001).
In summary, we clarified the temporal profile of expression and intracellular translocation of Bcl-2 family members during permanent MCAO in a mouse model. We found that Bim translocates to the mitochondria at the early stage of MCAO, and that the mitochondrially localized Bim may drive cytochrome c release followed by caspase-9 activation. Bim was detected in oligodendrocytes as well as neurons in the ischemic brain. These results provide the first convincing evidence that Bim plays a pathogenic role in ischemia-induced brain damage. Based on the growing biochemical and genetic evidence that Bcl-2 is protective against ischemic brain damage, Bim, a Bcl-2–antagonizing protein, may be a potential therapeutic target for ameliorating ischemic brain damage.