
Abstract
Heat acclimation (HA) offers functional neuroprotection in mice after traumatic brain injury (TBI). This study further characterizes endogenous neuroprotection acquired by HA (34±1°C, 30 d) after TBI. We establish here the ability of HA to induce sustained functional benefits and to reduce activation of apoptotic pathways. Neurobehavioral recovery, assessed by the Neurological Severity Score, was greater in HA mice up to 8 days after injury as compared with normothermic controls (P<0.05) and lesion volume was also smaller in the HA group (P<0.05). Reduced apoptotic cell death in HA mice was confirmed using caspase-3 activity measurements and immunohistochemistry. To investigate the underlying molecular pathways, expression levels of intrinsic apoptotic pathway-related proteins were examined. HA mice displayed higher mitochondrial levels of antiapoptotic Bcl-xL, accompanied by lower proapoptotic Bad levels and decreased cytochrome c release, suggesting a higher apoptotic threshold. Taken together with our previous reports, indicating increased Akt phosphorylation and antioxidative capacity, alongside with reduced tumor necrosis α levels after TBI in HA animals, the current results support the involvement of an antiapoptotic effect in HA-induced neuroprotection. Current results warrant further study as TBI-induced apoptosis may persist over weeks after injury, possibly providing a target for belated therapeutic intervention.
Introduction
Traumatic brain injury (TBI), a main cause of chronic disability and death, occurs when the brain is damaged by a mechanical impact. Traditionally, trauma-induced deleterious processes have been divided into two phases, namely primary damage caused directly by the impact and a secondary damage phase in which a cascade of cellular events is triggered by morphologic, excitotoxic, and metabolic changes (Leker and Shohami, 2002; Zhang et al, 2005). Interestingly, distinct modes of cell death dominate these two phases. In particular, though the main cell-death event observed immediately after impact is necrosis, the second phase of cellular death is mainly because of apoptosis (Yakovlev and Faden, 2001; Zhang et al, 2005). Apoptotic cell death can be triggered by a variety of molecular signals (Eldadah and Faden, 2000). More specifically, apoptosis can be induced either through the mitochondrial (intrinsic) or the receptor-mediated (extrinsic) pathways (Yakovlev and Faden, 2001; Zhang et al, 2005), both leading to activation of cysteine proteases (caspases) ultimately resulting in DNA fragmentation by the executioner caspase-3 (Eldadah and Faden, 2000).
It is now well established that TBI-induced apoptosis has a critical role in brain cell death with neurons being the most vulnerable target (Zhang et al, 2005). Consequently, treatment with caspase inhibitors has been shown to lead to improved histologic and behavioral outcome after TBI (Knoblach et al, 2004). Furthermore, transgenic mice overexpressing Bcl-2, a mitochondrial inhibitor of the intrinsic pathway, were reported to display improved histologic outcome after trauma (Raghupathi et al, 1998; Tehranian et al, 2006) suggesting involvement of the intrinsic pathway in postinjury cell fate. It is noteworthy that TBI-induced apoptosis is activated in both rodents (Graham et al, 2000; Raghupathi et al, 2000) and humans (Raghupathi et al, 2000; Zhang et al, 2005). In earlier studies, using the same model of TBI, Reshef et al (2008) investigated the time course of apoptotic cell death and reported that maximal prevalence of apoptotic cells was observed at 24 h after the insult. At 72 h from the insult, only a few scattered cells could be observed, and at 7 days after TBI, there was no evidence for such cells. Similar temporal profile, showing the peak of apoptotic cell death around 24 h was also reported by Stahel et al (2000).
The Bcl-2 protein super family, consisting of death suppressor and death promoter members, is crucial for facilitating intrinsic apoptosis (Graham et al, 2000; Raghupathi et al, 2000). As a neuron is exposed to stress, Bad, a proapoptotic member of the Bcl-2 family, is dephosphorylated and translocates from the cytosol to the mitochondrial outer membrane, in which it displaces antiapoptotic Bcl-2 and Bcl-xL from binding Bax. Subsequently, Bax, which is also part of the Bcl-2 family, is activated to permeabilize the mitochondrial outer membrane releasing inter-membrane proteins such as cytochrome c into the cytosol. In turn, cytochrome c recruits and activates the initiator caspase-9 that subsequently cleaves and activates the executioner caspase-3. DNA fragmentation and protein cleavage are the final steps of apoptosis (Eldadah and Faden, 2000; Yakovlev and Faden, 2001).
The failure of mitochondrial function is only one of the hallmarks of cellular stress response and oxidative stress in TBI-induced cell damage. It is an important mechanism that precedes the onset of neuronal loss after TBI, and drugs that target mitocohondria, such as cyclosporine A, have been shown to be neuroprotective (Sullivan et al, 1999). It should be noted in this context that oxidative stress occurs within minutes after TBI (Beit-Yannai et al, 1997) and may account for the observation of early (30 mins) mitochondrial dysfunction. Oxidative stress is also shown to be associated with apoptotic cell death through the intrinsic pathway (Chong et al, 2004; Sugawara and Chan, 2003).
Previous studies showed that heat acclimation (HA) renders protection against various types of stress through a cross tolerance mechanism. These have been shown to include cardiac ischemia–reperfusion (Levi et al, 1993), brain hyperoxia (Arieli et al, 2003), and TBI (Beit-Yannai et al, 1997; Shein et al, 2005; Shohami et al, 1994). Using a mouse model of closed head injury (CHI), we have established the neuroprotective effects of HA on TBI outcome (Shein et al, 2008a; Shein et al, 2005). Neuroprotection was earlier shown using a similar trauma model in HA rats (Beit-Yannai et al, 1997; Shohami et al, 1994). This has been suggested to involve the activation of protective pathways shared by acclimation and other stressors (Horowitz and Hari Shanker, 2007). Furthermore, previous studies have indicated that HA induces antiapoptotic pathways in the heart, thus generating an antiapoptotic cardiophenotype, which provides protection against stressors including heat stress and cardiac ischemia-reperfusion insult (Assayag et al, 2007; Horowitz and Hari Shanker, 2007).
In earlier studies on the molecular mechanisms by which HA offers neuroprotection, we have reported that HA induces upregulation of hypoxia-inducible factor-1α, hypoxia-inducible factor-1 transcriptional target erythropoietin, and its receptor EpoR (Shein et al, 2008a; Shein et al, 2005). In addition, Akt phosphorylation that is known to occur downstream to EpoR activation and harbor antiapoptotic effects was increased in HA mice shortly after TBI (Shein et al, 2007a). HA animals were also found to display altered expression dynamics of low molecular weight antioxidants, namely attenuated post-TBI reduction in expression, which would suggest enhanced ability of coping with trauma-induced oxidative stress and consequent cell death.
In light of the crucial role of oxidative stress and p-Akt in determining cell fate, the association between the extent of apoptotic cell death and overall outcome and the antiapoptotic and antioxidant effects of HA observed in the heart and brain, this study was designed to investigate the effect of HA on overall apoptotic cell death after TBI and on factors having an important function in this process.
Materials and methods
Animals and Maintenance
The study was approved by the Institutional Animal Care Committee of the Hebrew University. Male Sabra mice weighting 38 to 53 g were used. Animals were kept under controlled light conditions with 12 h/12 h light/dark cycle. Food and water were provided ad libitum. The animals were divided into two groups: control normothermic, maintained at an ambient temperature of 24 ± 1°C; and heat acclimated, held in a climatic chamber at 34 ± 1°C for 30 days, ensuring the achievement of a stable acclimated state.
Trauma Model
Experimental CHI was induced under isoflurane anesthesia using a modified weight drop device developed in our laboratory (Chen et al, 1996). Briefly, after anesthesia, a midline longitudinal incision was performed, exposing the skull. A Teflon-tipped cone (2 mm diameter) was placed 1 to 2 mm lateral to midline in the midcoronal plane. The head was manually held in place and a 95 g weight was dropped on the cone from a prefixed height, resulting in a focal injury to the left hemisphere. After recovery from anesthesia, the mice were returned to their home cages with postoperative care and free access to food and water. Sham control mice received anesthesia and skin incision only.
Neurobehavioral Evaluation
The functional status of the mice was evaluated according to the Neurological Severity Score (NSS) by an observer unaware of the given treatment. This score is a 10 point scale that assesses the functional neurologic status based on the presence of some reflexes and the ability to perform motor and behavioral tasks such as beam walking, beam balance, and spontaneous locomotion (Beni-Adani et al, 2001). Animals are awarded one point for failure to perform a task, such that scores range from 0 to 10, increasing with the severity of dysfunction. The NSS obtained 1 h after CHI reflects the initial severity of injury. Therefore, the extent of the recovery (ΔNSS) can be calculated as the difference between the NSS at 1 h and at any subsequent time point. A positive ΔNSS indicates recovery, 0 reflects no change, and a negative ΔNSS indicates neurologic deterioration. NSS values were measured at 1 h after injury and at 24h intervals during the first 3 days thereafter. Additional measurements were performed at 5, 8, and 14 days after injury. ΔNSS was calculated for each time point.
Western Immunoblotting
After 30 days of acclimation or normothermic period, mice were subjected to either CHI or sham surgery and killed 6, 12, or 24h later (n =4 to 6/group). After decapitation, brains were rapidly removed and frontal segments from left, injured, hemispheres were separated and frozen at −70°C until analysis. Sample separation was performed as previously described (Shein et al, 2005) with minor modifications. Both mitochondrial and cytosolic extracts were prepared from each sample. Tissue segments were incubated in a buffer containing 10 mmol/L HEPES (pH 7.9), 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 10% glycerol, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, and 10% complete EDTA-free protease inhibitor (Roche Diagnostics, Mannheim, Germany) for 30 mins at 4°C and then homogenized. Homogenates were centrifuged at 5,000g for 10 mins and supernatants (cytosolic extracts including cell organelles) were then centrifuged at 12,000g for 30 mins. Supernatants (cytosolic extracts) were frozen at −70°C whereas pellets were re-suspended in buffer as described above and centrifuged at 12,000g for 10 mins. Pellets (mitochondria-enriched fraction) were re-suspended and kept at −70°C. Protein concentrations in all extracts were determined using the Bradford method (Bio-Rad Labratories, Munich, Germany). Equal protein samples (40 μg for cytosolic and 30 μg for mitochondrial extracts) were separated on 10% SDS–polyacrylamide gels with 4.5% SDS stacking gels and electrotransferred onto nitrocellulose membranes (0.2 μm, Schleicher and Schuell, Dessel, Germany). Mitochondrial and cytosolic blots were probed with a rabbit polyclonal anti-Bcl-xL (1:1000; Cell Signaling Technology Inc., Danvers, MA, USA), anti-Bad (1:1,000; Delta Bio Labs, Gilroy, CA, USA), and anticytochrome c (1:1000; Cell Signaling Technology Inc.). Rabbit polyclonal antibody against β-actin (1:1000; Cell Signaling Technology Inc.) was also used for confirming equal protein loading (Xilouri and Papazafiri, 2006). Appropriate peroxidase-coupled immunoglobulin G (1:5,000; Jackson Immunoresearch, Soham, Cambridgeshire, UK) was used for secondary incubations. Reactive bands were visualized using the enhanced chemiluminescence system (Biological Industries, Beit Haemek, Israel). Optical density of reactive bands was quantified using Tina software (Raytest, Strau-benhardt, Germany) and protein levels were expressed as the optical density of the examined factor relative to β-actin within the same lane. To enable multiple blots comparisons a sample of prefixed volume of whole brain extract was loaded on each blot and was used for normalization among blots.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
Normothermic and HA mice were killed after sham surgery or at 6 or 12 h after injury (n = 4 to 8 per group). Segments of the frontal left cerebral cortex were taken and frozen at −70°C until use. All reagents used for RT-PCR were of molecular grade. Total RNA was isolated using TRI reagent (Molecular Research Center Inc., Cincinnati, OH, USA). RNA samples were confirmed free of DNA contamination by 260/280 nm optical density measurement. One micro-gram of total RNA was reverse transcribed using a first-strand cDNA synthesis kit (Fermentas International Inc., Burlington, ON, Canada). Quantitative real-time polymerase chain reaction was performed using Taqman Gene Expression Assays-on-Demand (Applied Biosystems, Foster City, CA, USA). The amplification reaction was performed in an ABI 7700 sequence detection system (Applied Biosystems). Reaction was performed in a 20 μl reaction volume containing 90 ng of cDNA, 10 μl of Taqman universal PCR mix (Applied Biosystems), and 1 μl of the assay solution containing the specific primers and 6-FAM-labeled probe. The thermal profile for qPCR was 95°C for 10 mins followed by 45 cycles of 95°C for 15 secs and 60°C for 60 secs. Relative quantity values were analyzed using ABI 7700 sequence detection system software (Applied Biosystems) according to the ΔCt method, which reflects the difference in threshold for each target gene relative to that of β-actin.
Caspase-3 Activity Assay
Caspase-3 activity was measured using a commercial fluorometric kit according to the manufacturer's instructions (BioVision Research products, Mountain View, CA, USA). Briefly, HA and normothermic mice were killed 24 h after CHI or sham surgery (n = 5/group). This time point was chosen as maximal apoptosis and was shown earlier in our model 24 h after TBI (Reshef et al, 2008). Frontal cortical segments from left injured hemispheres were quickly removed and homogenized in 200 μl of cell lysis buffer supplied in the commercial kit. Whole cell homo genates were frozen in −70°C for batch analysis procedure; 50 μl of each sample, containing 150 μg protein (determined using the Bradford method) was loaded onto 96-well plates and placed on ice for 10 mins; 50 μl of 2 × reaction buffer containing 10 mmol/L dithiothreitol was added to samples followed by 5 μl of 1 mmole/L DEVD-7-amino-4-trifluoromethyl coumarin (AFC) substrate and incubated at 37°C for 2 h. Cleavage product was detected using a fluorometer (FluoroStar 403, BMG LabTech, Offenburg, Germany) equipped with a 390 nm excitation filter and a 520 nm emission filter. Activity was determined by subtracting the bulk from the samples fluorescence.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling
Normothermic and HA mice (n = 4 to 5/group) were killed 24 h after CHI or sham surgery, a time point for maximal apoptosis in this model. Whole brain was placed in 4% paraformaldehyde in PBS for 5 h at room temperature, followed by three washes in 20% sucrose in PBS for 10 mins each. Brains were then placed overnight at 4°C in 20% sucrose in PBS and frozen at −70°C in tissue-tek OCT. Coronal 10 μm-thick sections were obtained from the injured area (approximately + 0.38 mm from bregma) using a sliding cryostat (Leica 3000, GMI Sections, Ramsey, MN, USA) and kept at −70°C until further processing. Brain sections were washed three times in PBS and permeabilized for 4 mins with permeabilization solution containing 0.3% triton and 0.1% sodium citrate. After three washes in PBS, sections were incubated at 37°C for 1 h in 50 μl of labeling solution containing 5 μl of enzyme solution (Roche Applied Science, Indianapolis, IN, USA). Sham sections were subjected to DNase treatment as positive control. Labeling solution without enzyme was equivalently used as negative control. Sections were washed three times in PBS and cell nuclei were labeled with propidium iodide (PI) for 5 mins at room temperature, followed by a DDW wash. Slides were then mounted and coverslipped. DNA strand brakes were visualized using an Olympus FluoView Olympus IX70 fluorescent microscope equipped with 488 nm (red) and 543 (green) filters and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive cells were quantified using × 50 magnitude. Only cells that were double labeled intensely with PI were counted and their percentage out of all nuclei presented in the field was subsequently calculated. Twenty fields, representing the entire injured cortex were counted in each brain section.
2,3,5-Triphenyl-Tetrazolium Chloride
Normothermic and HA mice were killed 48 h after CHI (n = 6 to 8/group) as to allow for the secondary apoptotic tissue damage that develops at that time frame to be included in the lesion volume. Brains were removed and sliced to 2 mm-thick slices. The slices were placed in a 24-well plate containing 2% 2,3,5-triphenyl-tetrazolium chloride (TTC) in PBS and incubated for 1 h at 37°C, allowing the TTC to stain vital tissue. After incubation, 4% paraformaldehyde in PBS was added to each well and maintained overnight. Paraformaldehyde was washed out with three PBS washes. Sections were visualized and photographed using stereoscope Stemi SV11 (Zeiss, Jena, Germany) equipped with a digital photo-camera (Coolpix 4500, Nikon, Tokyo, Japan). Lesion volume was calculated as the relative unstained (white) area to the uninjured contralateral hemisphere area, as to neutralize the effect of edema formation on the injured hemisphere volume. Calculations were conducted using commercial ImageJ 1.40 software and relative lesion areas from all brain sections were grouped to provide lesion volume, representing the percentage of dead tissue within the injured hemisphere.
Statistical Analysis
For statistical analyses, we used commercially available computer software (SigmaStat 2.03). Normothermic and HA treatments were the independent variables and the outcomes of the TBI parameters were the dependent variables. Significance for NSS experimental series was tested using two-way ANOVA for repeated measures, followed by Tukey post hoc tests. Significance within groups (normothermic or HA) for mRNA levels, protein levels, and caspase-3 activity was calculated using one-way ANOVA, and between groups (normothermic versus HA) two-way ANOVA followed by Tukey post hoc tests. For determining significance for TUNEL labeling and lesion volume (TTC staining) t-test was used. P-values < 0.05 were considered significant for all comparisons. Data are expressed as mean ± s.e.m.
Results
Functional Recovery and Lesion Volume
To initially re-confirm the establishment of HA-induced neuroprotection, NSS and lesion volume evaluations were conducted (Figure 1). Injury severity, estimated by NSS at 1 h after injury, was similar in HA and normothermic groups (NSS scores: 6.65 ± 0.14 versus 7.01 ± 0.17, respectively, P > 0.05, data not shown). Spontaneous functional recovery was noted in both normothermic and HA mice, yet this was faster and greater among HA mice. Thus, HA mice displayed better recovery, reflected by increased ΔNSS as compared with the normothermic group at 24 h and on days 3, 5, and 8 after injury (Figure 1A). Postinjury lesion volume was determined at 48 h after injury using TTC staining. Although no detectable lesion was observed in sham mice from both groups (data not shown), postinjury lesion volume was smaller in the HA group as compared with normothermic control mice (14.82% ± 1.28% versus 23.24% ± 2.78%, P < 0.05), as shown in Figures 1B and 1C.
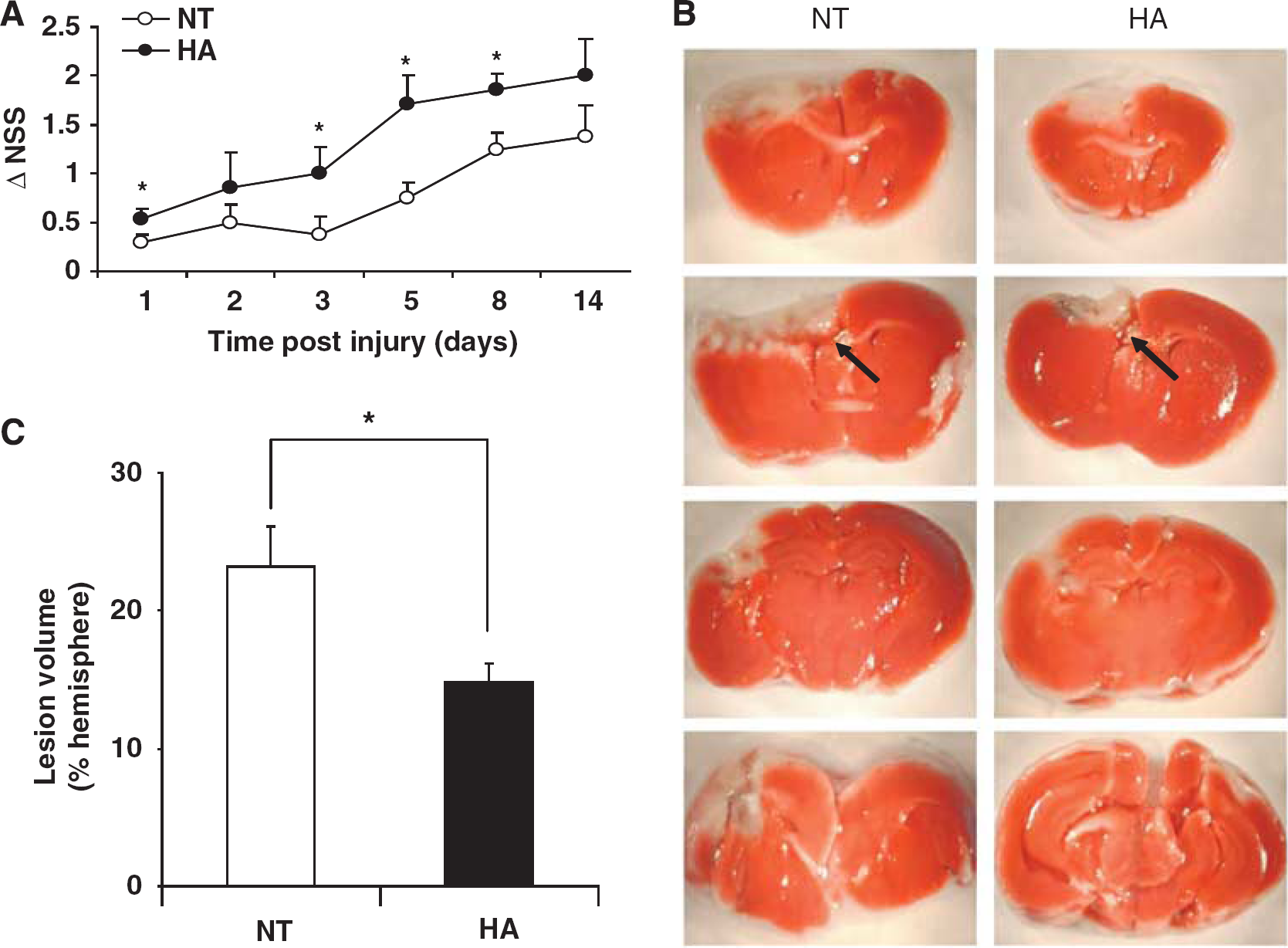
Beneficial effects of heat acclimation (HA) on neurobehavioral recovery and lesion volume after closed head injury (CHI). (
Bad and Bcl-xL Protein Expression
In an effort to study the effect of HA on Bcl-2 proteins, the expression levels of Bad and Bcl-xL, both are key proteins in the intrinsic apoptosis pathway, were measured. Both proteins were detected in the cytosolic and the mitochondrial compartments at 6, 12, and 24 h after CHI, as well as in sham control mice. Figure 2A shows that in the cytosol, Bad levels in HA brains were higher than in normothermic at all time points after trauma, reaching significance at 12 h (1.3 ±0.4 versus 0.27 ±0.12, P < 0.05) and 24 h (1.82 ± 0.30 versus 0.39 ± 0.06, P < 0.01). Although no significant decline in cytosolic Bad levels was observed in the brains of HA mice after CHI, normothermic mice showed a significant decrease in cytosolic levels of this factor at 6 h after injury (1.55 ± 0.56 versus 0.28 ± 0.13, P < 0.05) and at all later time points as compared with the sham state. Cytosolic Bcl-xL levels, shown in Figure 2B, were far higher in sham HA animals than in normothermic controls (1.26 ± 0.10 versus 0.51 ± 0.05, P < 0.001) as well as at 6 h after CHI (1.22 ± 0.18 versus 0.52 ± 0.13, P < 0.01). By 12 h after injury and subsequently at 24h after trauma, there was no difference in Bcl-xL expression between groups, as cytosolic levels observed in HA declined to normothermic values.
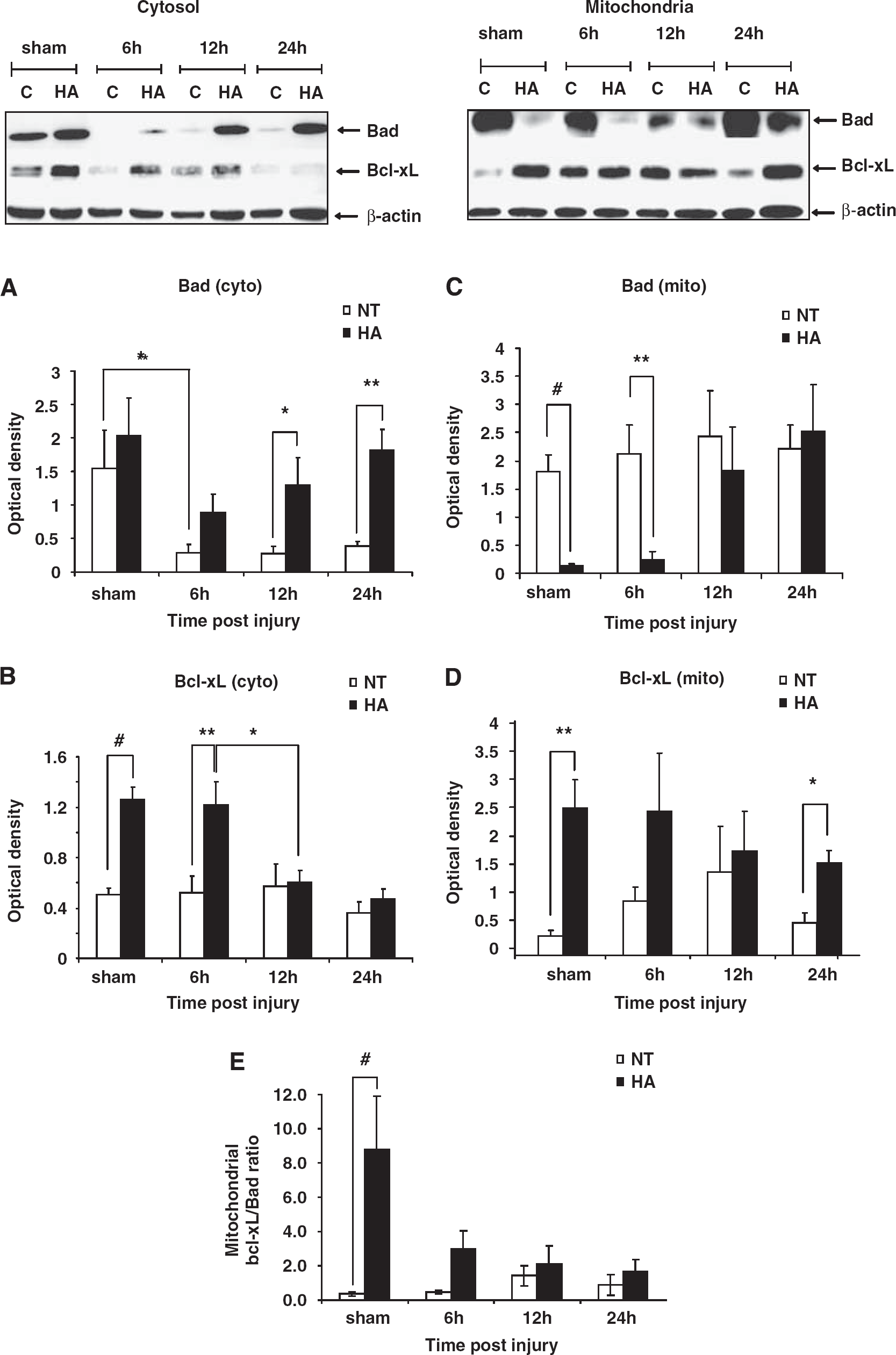
Dynamic changes in Bad and Bcl-xL protein expression in heat acclimated (HA) and normothermic (NT) sham and injured mice. Cytosolic and mitochondrial extracts from injured tissue or sham controls were separated on SDS–PAGE gels and analyzed using Western blotting. Optical density normalized to β-actin is shown. Quantified results are presented for cytosolic (cyto) Bad (
In the mitochondrial compartment, Bad levels were dramatically lower in sham HA animals as compared to normothermic (0.13 ± 0.03 versus 1.81 ± 0.29, P < 0.001). A similar difference was preserved 6 h after CHI as well (0.24 ± 0.15 versus 2.12 ± 0.52, P < 0.01). At 12 to 24h after CHI, no difference was observed between the groups because of a robust increase of mitochondrial Bad in the HA mice (Figure 2C). The Bcl-xL levels tended to be higher in the mitochondrial compartment of HA mice at all examined time points, as shown in Figure 2D, reaching a statistically significant difference from the normothermic group in sham control mice (2.50 ± 0.20 versus 0.32 ± 0.10, P < 0.01) and at 24 h after CHI (1.56 ± 0.22 versus 0.55 ± 0.17, P < 0.05). To express the balance between the antiapoptotic Bcl-xL and proapoptotic Bad within the mitochondria the mitochondrial Bcl-xL/Bad ratio was calculated for each time point, and is depicted in Figure 2E. A significantly higher ratio was evident in HA sham mice, as compared with the normothermic controls. Ratio tended to be higher, yet not significant, in HA mice as well at 6 h after injury, suggesting the presence of a balance favoring anti-apoptosis after acclimation.
Bad and Bcl-xL mRNA Levels
Bcl-xL and Bad mRNA levels were quantified as to determine whether changes in their transcription are associated with the observed differences in protein levels. As shown in Figure 3A, Bcl-xL mRNA levels remained relatively unchanged after acclimation or injury (Figure 3A). In contrast, as shown in Figure 3B, Bad transcript levels were found to be higher in noninjured HA shams (4.96 ± 0.55 versus 1.58 ± 0.20, P < 0.05) and at 6 h after injury (6.52 ± 1.49 versus 2.71 ± 0.74, P < 0.01). Importantly, Bad mRNA levels dramatically decreased between 6 and 12 h after injury in the HA group (6.52 ± 1.49 versus 2.08 ± 0.35 at 6 h and 12 h, P < 0.01) but not in the normothermic group.
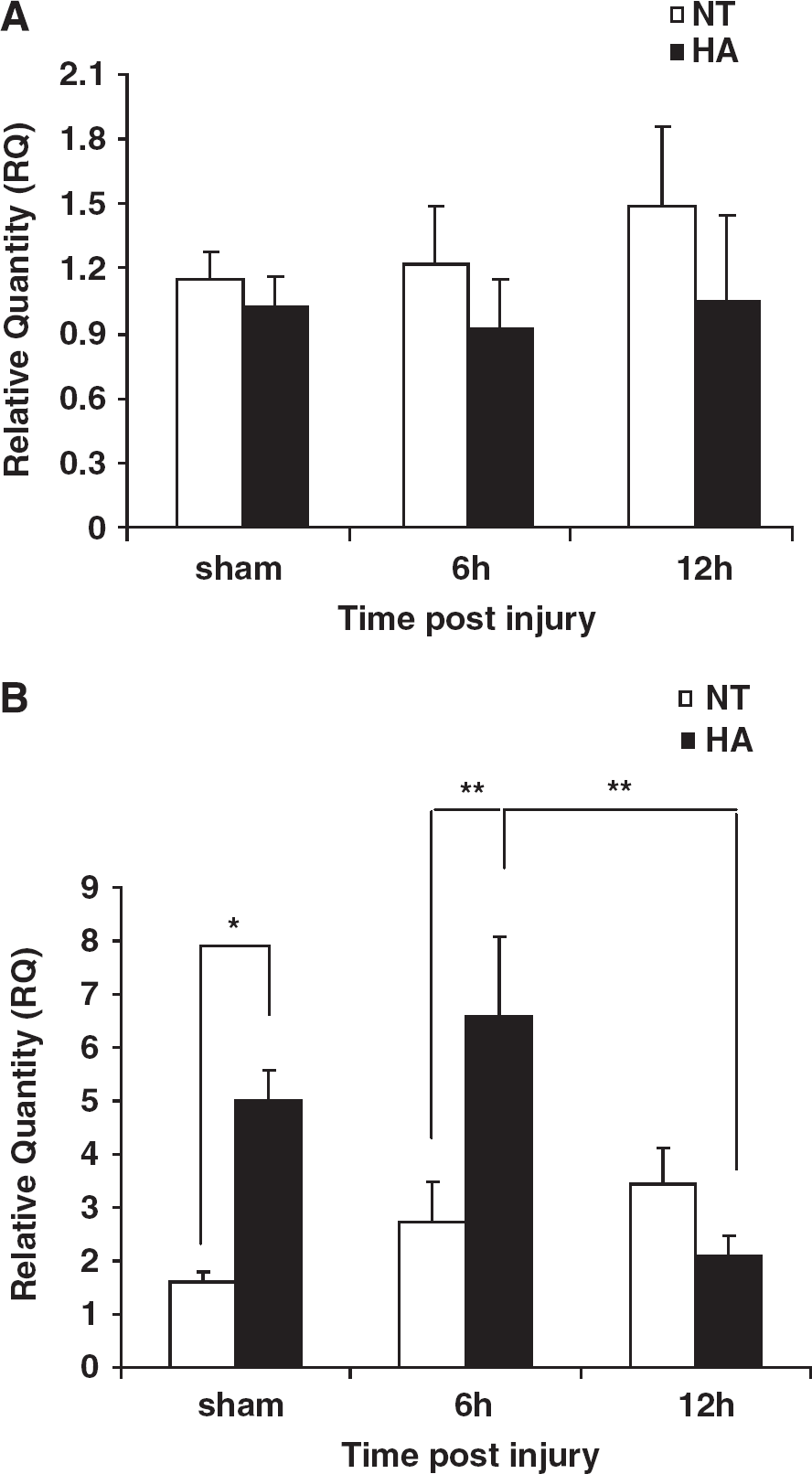
Effect of heat acclimation and closed head injury (CHI) on Bcl-xL and Bad mRNA levels. mRNA in frontal cortical segments from heat acclimated (HA) or normothermic (NT) mice was quantified using real-time polymerase chain reaction. Although Bcl-xL levels remained relatively unchanged (
Cytochrome c Protein Expression
To further study the effect of HA on the intrinsic apoptotic pathway, the levels of cytochrome c protein were measured in cytosolic and mitochondrial extracts prepared from frontal cortical segments of HA and normothermic mice. Cytosolic cytochrome c levels were found to be markedly lower in HA sham mice as compared with normothermic controls, as shown in Figure 4A (0.14 ± 0.03 versus 0.30 ± 0.04, P < 0.001). Similarly, lower cytochrome c levels were found after injury in the cytosol of HA mice at 6 h (0.06 ± 0.010 versus 0.27 ± 0.03, P < 0.001) and 12 h after CHI (0.13 ± 0.02 versus 0.33 ± 0.04, P < 0.001). At 24h after injury, HA mice still tended to have lower levels of cytosolic cytochrome c than normothermic counterparts, however the difference was no longer significant. In the mitochondria (Figure 4B), no difference in cytochrome c levels was detected in sham mice. At all time points after injury, the mitochondrial levels of cytochrome c in HA mice tended to be higher than in normothermic controls, reaching statistical significance between groups at 12 h (1.29 ± 0.12 versus 0.57 ± 0.13, P < 0.001) and 24 h after CHI (1.55 ± 0.25 versus 0.58 ± 0.1, P < 0.001).
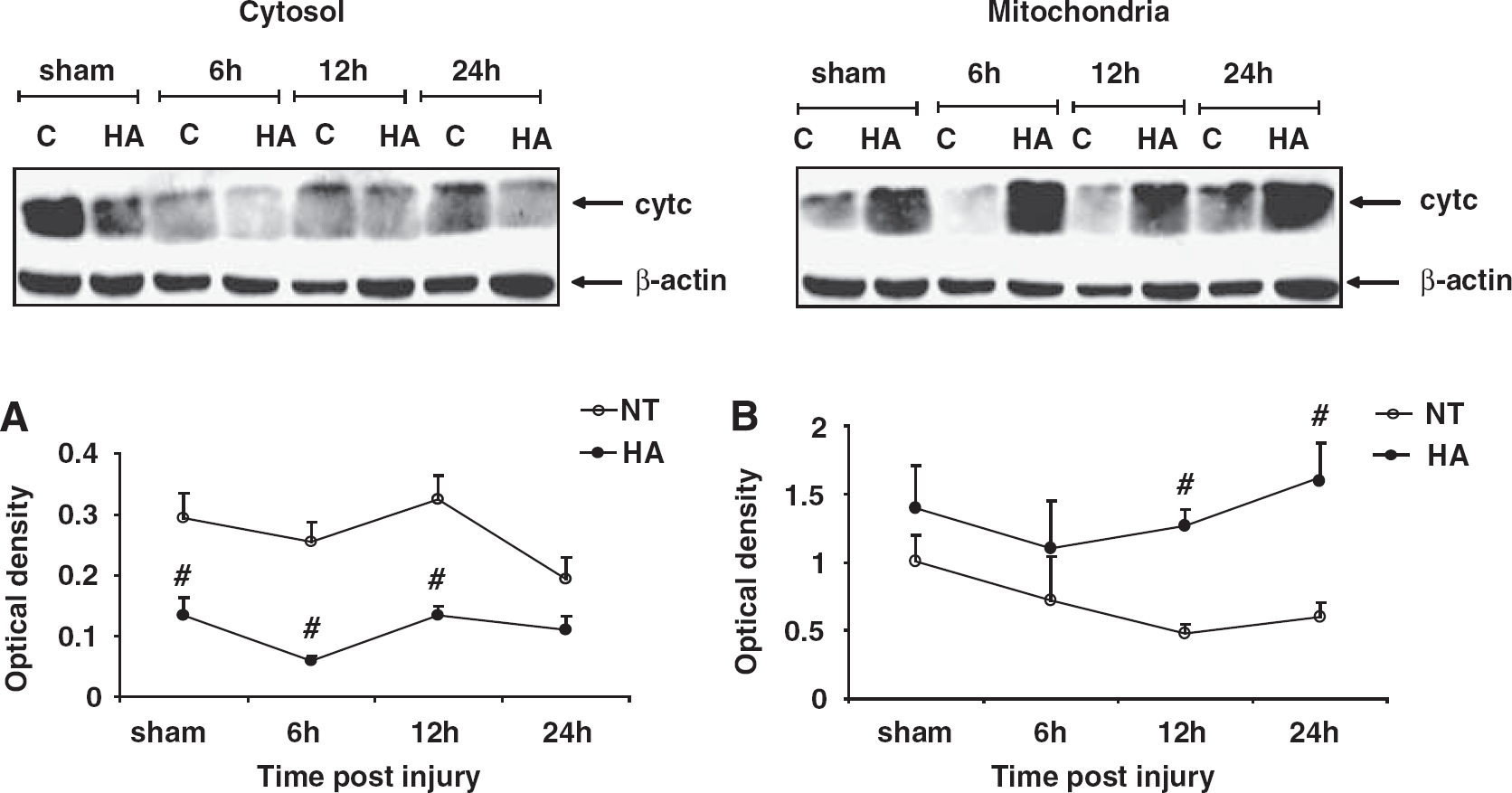
Dynamic changes in cytochrome c protein expression in heat acclimated (HA) and normothermic (NT) sham and injured mice. Cytosolic and mitochondrial extracts from injured tissue or sham controls were separated on SDS–PAGE gels and analyzed using Western blotting. Optical density normalized to β-actin is shown. Quantified results are presented for cytosolic (
Caspase-3 Activity
To determine activation of apoptotic pathways after CHI, we performed a caspase-3 activity assay 24 h after injury. Figure 5 shows that though no significant difference was observed between sham mice of both groups, postinjury caspase-3 enzyme activity in normothermic animals was two-fold higher than in the HA mice after injury (29296.4 ± 2099.3 versus 16392.0 ± 1143.5 fluorescence units, P < 0.001).
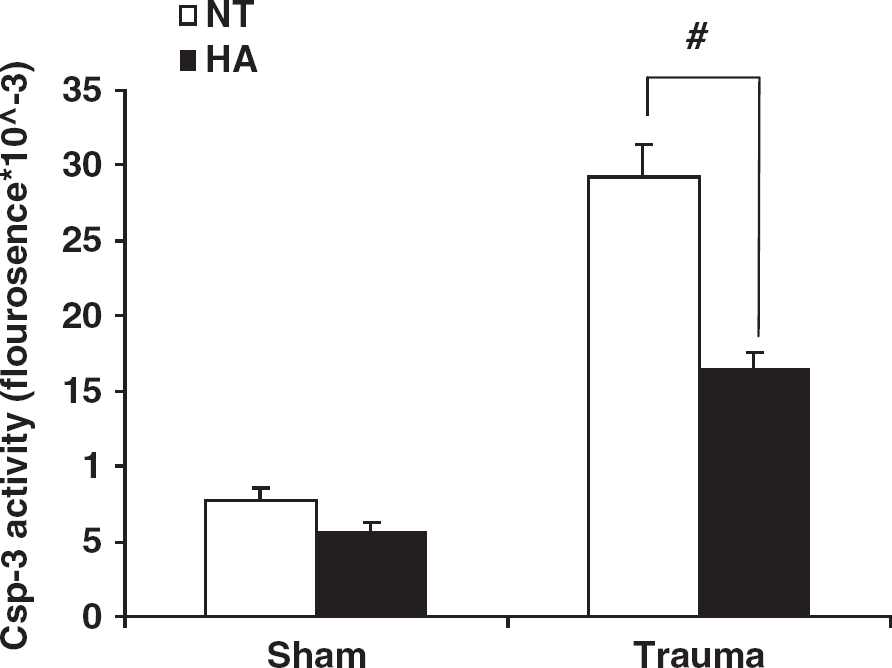
Heat acclimation reduces caspase-3 activity 24 h after injury. Fluorometric caspase-3 (csp-3) activity assay was performed 24 h after injury using whole cell tissue extracts of injured cortex from heat acclimated (HA) and normothermic (NT) mice. (# P < 0.001) as determined by two-way ANOVA followed by Tukey post hoc tests, n = 5 per group.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling
To evaluate overall tissue apoptosis, DNA fragmentation was quantified using TUNEL staining 24 h after injury. Although no detectable fragmentation was observed in sham mice from both groups (data not shown), markedly more cells in close proximity to the injury site showed DNA fragmentation in normothermic mice than in HA mice at 24 h after injury, as shown in Figure 6 (39.4% ± 3.11 versus 24.72% ± 1.89%, P = 0.001). Importantly, no DNA fragmentation was detected in any other region throughout the brain (data not shown).
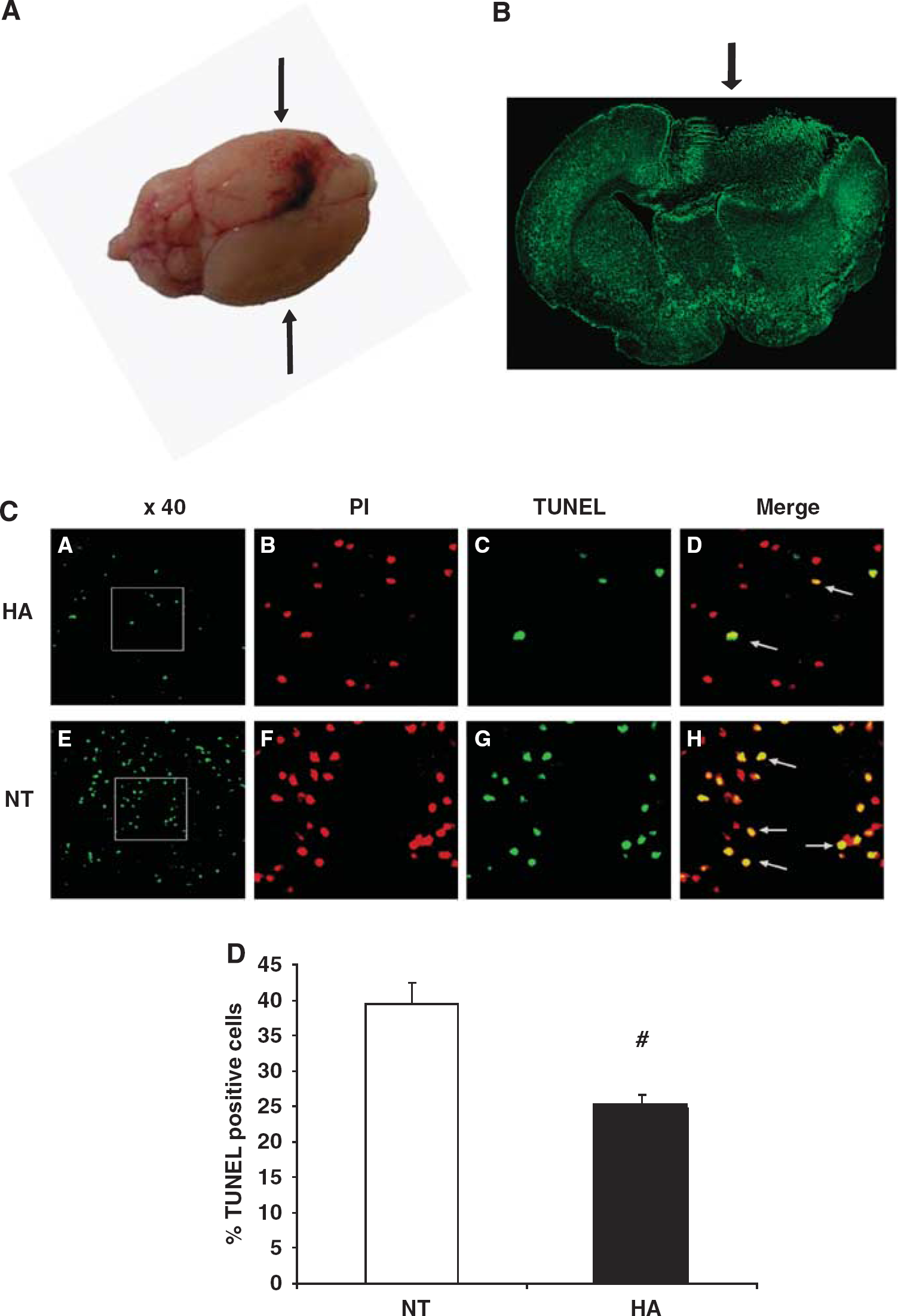
Heat acclimation (HA) reduces apoptotic cell death detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL). (
Discussion
In this study, we show that HA, shown to convey functional neuroprotection after TBI, attenuates postinjury cell death and affects factors associated with the intrinsic pathway of apoptosis induction. Previously, we have shown that HA induces a neuroprotective effect in both rats and mice, reflected by reduced tissue edema formation and improved functional recovery during the initial 24 to 72 h after impact (Shein et al, 2005; Shohami et al, 1994). Our current findings further expand these previous ones, and show that enhanced recovery of function by HA mice is sustained for up to 8 days after injury, suggesting that the protective effect of HA may also affect the belated damage phase after TBI. The observed difference in the time frame of evident functional protection may stem from the fact that the acquired acclimated physiologic phenotype is reversible. In the current set of experiments, we returned the mice to the climatic chamber at 34°C after subjection to CHI, whereas for the previous studies HA animals were kept at room temperature in accordance with reports placing the time frame for deacclimation in rats within several weeks (Arieli et al, 2003; Tetievsky et al, 2008). Given that mice do have a faster metabolic rate than rats (Schmidt-Nielsen, 1964) and in light of the current observations, we suggest that it is likely that the differences between the studies are due to the prevention of deacclimation. Thus, to exert more prolonged neuroprotection, mice should be kept under HA conditions after injury.
Cell death in the injured tissue is a main cause of functional disabilities and has a crucial impact on clinical outcome (Zhang et al, 2005). In the past, we have shown that improved recovery of motor ability and cognitive function in HA mice are accompanied by attenuated tissue damage, namely reduced edema formation and neurodegeneration (Shein et al, 2008a; Shein et al, 2005). Here, we investigated whether HA also leads to reduced overall cell death reflected by smaller lesion volume. Indeed, TTC staining of injured brains 48 h after injury revealed a smaller lesion volume in HA mice as compared with normothermic controls.
TBI not only leads to acute cell death at the core of the injured tissue but also triggers apoptosis, a major component of delayed cell loss. Our results show decreased caspase-3 activity in HA mice along with reduced number of TUNEL-positive cells. Taken together, this indicates that postinjury apoptosis is attenuated in HA mice. Interestingly, caspase-3 activity in HA sham, noninjured animals was also lower than that in normothermic mice, supporting the notion that higher basal apoptotic threshold is acquired by HA.
Multiple pathways trigger apoptosis after TBI (Raghupathi et al, 2000; Zhang et al, 2005). It is a tightly regulated process, with overall outcome determined by complex positive and negative controls. The Bcl-2 super family of proteins is a key regulator of intrinsically induced apoptosis (Gross et al, 1999; Raghupathi et al, 2000; Yakovlev and Faden, 2001). At the end of the acclimation period, HA mice displayed higher basal protein levels, of both Bcl-xL and Bad. Moreover, postinjury dynamic changes in these Bcl-2 family proteins were also different in HA mice from those observed in the normothermic group. Apoptotic suppressor Bcl-xL is abundantly expressed in adult neurons (Miyawaki et al, 2008) and has an essential role in preventing neuronal cell death (González-García et al, 1995; Parsadanian et al, 1998; Raghupathi et al, 2000) Neuroprotective effects of elevated Bcl-xL levels have been shown in transgenic mice overexpressing Bcl-xL. These mice displayed reduced lesion volume and better neurologic outcome after permanent occlusion of the midcerebral artery (Wang et al, 2008; Wiessner et al, 1999). In addition, Bcl-xL has been reported to potently suppress apoptotic cell death in neurons and other cell types in response to various stimuli (González-García et al, 1995; Graham et al, 2000). In HA mice we found increased Bcl-xL protein levels without changes in transcription both before and after injury. Bcl-xL levels were higher in the HA group than in normothermic mice not only in the mitochondria but also in the cytosol, two cellular compartments in which Bcl-xL can exert antiapoptotic effects. Though the classic mitochondrial role of Bcl-xL is to bind Bax and prevent it from promoting cytochrome c release, cytosolic Bcl-xL can directly inhibit caspases and prevent the release of Bad from its cytosolic inhibitor 14-3-3 (Datta et al, 2000). Our observation showing similar constitutive transcription levels of Bcl-xL in HA and normothermic before injury may imply that other modes of regulation of protein levels take place after TBI, independent from basal transcription. As both pro- and antiapoptotic Bcl-2 proteins may be regulated by transcription, phosphorylation, dimerization, translocation, degradation, and cleavage (Gross et al, 1999), it is plausible that increased Bcl-xL levels in HA animals are associated with posttranscriptional modifications, subcellular compartmentalization, or reduced degradation/cleavage rate. This notion is supported by previous reports indicating that injurious stimuli in the brain promote proteolytic cleavage of full length Bcl-xL to create a strong proapoptotic product that acts within the mitochondria (Bonanni et al, 2006; Miyawaki et al, 2008).
Bad protein levels were also measured in the mitochondria and cytosol of sham, noninjured mice and at various time intervals after TBI. Lower levels of mitochondrial, but not cytosolic Bad, were found in HA shams and at 6 h after injury. In addition, a robust decrease in cytosolic Bad was evident in normothermic mice but not in HA counterparts. Transcription levels of Bad were higher in HA sham mice and 6 h after injury, yet only in the HA group mRNA levels were subsequently downregulated.
Augmented Akt phosphorylation after TBI was proposed as a key event in the neuroprotective mechanism offered by HA (Shein et al, 2007a). One way by which phosphorylated Akt induces cell survival is by inhibiting Bad. When phosphorylated by Akt at serine 136, phospho-Bad associates with the cytosolic protein 14-3-3. This prevents mitochondrial translocation of Bad, consequently blocking Bad-mediated displacement of Bcl-xL from association to Bax. Accumulated cytosolic Bad protein, as observed in HA mice after TBI, taken together with preceding increased Akt activation 4 h after TBI (Shein et al, 2007a) in these animals, suggests inhibition of Bad through Akt-mediated phosphorylation and subcellular compartmentalization. However, direct evidence of this mechanism would require evaluating phosphorylated Bad levels within the cytosol. Despite our effort to achieve this in the scope of this study, we were unable to detect phospho-Bad (ser 136) with several commercially available antibodies. Thus, the precise role of Akt in facilitating cytosolic retention of Bad in HA mice remains to be further established and the possible involvement of other regulatory mechanisms in this effect cannot be excluded at this point. These may potentially include interleukin-3, which phosphorylates Bad at serine 112 and serine 136 to inactivate it (Condorelli et al, 2001), Jnk that activates Bad by serine 128 phosphorylation (Kamada et al, 2006), and activated caspase-3, capable of cleaving and activating Bad to mediate both intrinsic and extrinsic apoptotic pathways (Condorelli et al, 2001). In contrast to its effect on Bcl-xL, HA was found to alter both the transcription and protein dynamics of Bad. Enhanced basal transcript levels perse do not necessarily indicate functional consequence in terms of apoptosis, as was confirmed by the lack of TUNEL staining in HA shams. However, the elevated transcription of Bad followed by modification in protein distribution of this factor, suggests that HA affects Bad by more than one mode of regulation, most likely consisting of both transcriptional regulation as well as posttranscriptional modifications. Overall, HA itself increases the mitochondrial Bcl-xL/Bad ratio, changing the preset basal mitochondrial apoptotic threshold. Namely, HA mice may require more robust stimuli than normothermic for initiating apoptosis. Similar results were also found in HA rat heart (Assayag et al, 2007; Horowitz and Hari Shanker, 2007).
Mitochondrial integrity and membrane potential are a result of integrative, proapoptotic, and prosurvival stimuli, mediated by various proteins. The localization and levels of cytochrome c are indicative of mitochondrial integrity. Our results show no changes in cytochrome c levels in the mitochondria of HA mice after injury, whereas these gradually decrease in the normothermic group. The increased release of cytochrome c from the mitochondria to the cytosol, which occurs in normothermic, but not HA mice, supports the notion of lower proapoptotic activity in the HA group. Furthermore, lower cytosolic cytochrome c levels found in sham HA mice reinforce the notion that HA raises the intrinsic apoptotic threshold and attenuates cytochrome c release into the cytosol. However, it should be considered in this context that recent findings indicate that neurons can survive a rise in cytochrome c levels and that increased levels of this factor may not be sufficient to cause cell death (Miyawaki et al, 2008). Furthermore, prosurvival factors may act after cytochrome c is released to inhibit apoptosis, as Akt can directly phosphorylate and inhibit caspase-9 (Fukunaga and Kawano, 2003).
In conclusion, our current findings establish the ability of HA to provide sustained neuroprotection, and to attenuate trauma-induced apoptosis. The effects of HA on factors associated with the intrinsic apoptosis-inducing pathway add to the previous body of evidence pointing to the ability of HA to induce dynamic changes after TBI. Importantly, these dynamic changes have been shown earlier to include augmented expression of hypoxia-inducible factor-1α, erythropoietin and erythropoietin receptor (Shein et al, 2008a; Shein et al, 2005), enhanced Akt phosphorylation (Shein et al, 2007a), and brain-derived neurotrophic factor (Shein et al, 2008b), alongside increased low molecular weight antioxidant levels (Beit-Yannai et al, 1997), augmented preinjury anti-inflammatory interleukin-10, and reduced tumor necrosis factor α expression after injury (Shein et al, 2007b).
As all of the above-mentioned factors are known to directly or indirectly provide protection against apoptotic sentence and in light of the current findings, we suggest that an antiapoptotic mechanism is likely to be involved in HA-induced neuroprotection, with reduced apoptosis in acclimated mice being a consequence of beneficial elements acquired during acclimation and triggered on subsequent injury. Our findings warrant further study of the mechanisms underlying the attenuation of apoptosis by HA and may harbor substantial implications given the long postinjury timeframe at which this process takes place and the potential prolonged therapeutic window for antiapoptotic treatment.
Footnotes
Acknowledgements
This work was supported in part by a grant (# 241/04) from the ISF. ES is a member in the Brettler Center for Research in Molecular Pharmacology and Therapeutics, and the incumbent of the Dr Leon and Mina Deutch Chair in psychopharmacology at the Hebrew University.
The authors declare no conflict of interest.