
Abstract
A novel agent, (R)-(−)-2-propyloctanoic acid (ONO-2506), has a unique property in that it modulates functions of activated cultured astrocytes, including pronounced inhibition of S-100β synthesis. The present study examined whether administration of this agent would mitigate the delayed expansion of infarct volume and the neurologic deficits after permanent middle cerebral artery occlusion (pMCAO) in rats. Daily intravenous administration of ONO-2506 (10 mg/kg) abolished the delayed infarct expansion between 24 and 168 hours after pMCAO, whereas the acute infarct expansion until 24 hours was unaffected. The agent significantly reduced the expression of S-100β and glial fibrillary acidic protein in the activated astrocytes and the number of terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling-positive cells in the periinfarct area. The neurologic deficits were significantly improved, compared with the vehicle-treated groups, as early as 24 hours after the initial administration of ONO-2506. The agent had a wide therapeutic time window of 0 to 48 hours after pMCAO. These results indicate that because of the pharmacologic modulation of astrocytic activation induced by ONO-2506, symptoms can regress whereas delayed expansion of the lesion is arrested. Pharmacologic modulation of astrocytic activation may confer a novel therapeutic strategy against stroke.
Accumulating evidence indicates that the acute increase in infarct volume within the initial 12 to 24 hours after focal cerebral ischemia is generally succeeded by delayed expansion, the duration and extent of which is variable depending on the type of ischemic insult (Baird et al., 1997; Beaulieu et al., 1999; Du et al., 1996; Loihl et al., 1999, Matsui et al., 2002). To explore the underlying pathogenic mechanisms and means of coping with delayed infarct expansion is of utmost importance in view of the disappointing results of clinical trials accumulated over the past decade (De Keyser et al., 1999; Lee et al., 1999).
Using the permanent middle cerebral artery occlusion (pMCAO) model in rats, we showed that there is a significant increase in the infarct volume between 24 and 168 hours after pMCAO (Matsui et al., 2002), which closely resembles the time course of infarct expansion in human stroke as reported by Beaulieu et al. (1999). In our model, delayed infarct expansion has been heralded by astrocytic activation in the periinfarct area at approximately 24 hours, as indicated by the appearance of numerous reactive astrocytes expressing glial fibrillary acidic protein (GFAP) and S-100β. The gradual increase in the infarct volume thereafter is paralleled by an increase in the number of apoptotic cells in the periinfarct area beginning at 24 hours and peaking at 120 hours after pMCAO. Because S-100β is known to induce neuronal cell death through nitric oxide release from astrocytes (Chao et al., 1996; Hu et al., 1996,1997; Murphy, 2000), we hypothesized that an enhanced synthesis of S-100β by the periinfarct-reactive astrocytes may play a role in the occurrence of delayed infarct expansion (Asano et al., 1999; Matsui et al., 2002). The present study attempted to examine this hypothesis from a pharmacologic viewpoint, using a novel agent ONO-2506 [(R)-(−)-2-propyloctanoic acid; Ono Pharmaceutical Co. Ltd., Osaka, Japan].
To find an agent that possesses an inhibitory action on astrocytic synthesis of S-100β, vigorous screening using an in vitro assay system was performed in the Minase Research Institute of ONO Pharmaceutical Co. Ltd. until ONO-2506 was finally discovered. In the preceding experiments, astrocytes or neurons obtained from the cerebral cortex of neonatal Wistar rats were cultured for 14 days in the presence or absence of ONO-2506. Although the agent did not affect the viability or growth of cultured astrocytes or neurons, it dose-dependently decreased the contents of S-100β in cultured astrocytes and nerve growth factor-β (NGF-β) in the culture media as measured by the sandwich enzyme-linked immunosolvent assay method with an ED50 value of approximately 60 μmol/L for each effect (Shinagawa et al., 1999). Using a similar method, the effect of the agent on mRNA expression of various proteins in cultured astrocytes was examined. At a concentration range of 100 to 300 μmol/L, ONO-2506 significantly increased the mRNA expression of glutamate transporters (GLT-1 and GLAST) and GABA receptors (GABAA-R β1, GABAA-R β2, and GABAA-R β3). At a similar concentration range, the agent significantly decreased the mRNA expression of S-100β and NGF-β, and the lipopolysaccharide-stimulated mRNA expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) (Katsumata et al., 1999; Shimoda et al., 1998; Shinagawa et al., 1998, 1999). The mRNA expression of glyceraldehyde-3-phosphate dehydrogenase and GFAP was not affected.
Although the mechanism underlying such a wide spectrum of actions of ONO-2506 in vitro is unclear, cultured astrocytes appear to spontaneously convert to an activated form as judged from a gradual increase in the intracellular levels of S-100β, GFAP, and other proteins (Shimoda et al., 1997; Tateishi et al., 1997). Astrocytes upregulate or downregulate numerous proteins on activation (Eddleston and Mucke, 1993). Therefore, a plausible hypothesis would be that ONO-2506 interferes with the intricate pathways of astrocytic activation upstream to mRNA expression of various proteins. In other words, the agent is considered to act to modulate the gene expression and functional alterations of astrocytes induced by activation (Shimoda et al., 1998; Shinagawa et al., 1998, 1999).
The following data show how such a modulation of astrocytic activation by ONO-2506 affects cell viability. ONO-2506 exerted no significant effects on the viability of otherwise unstimulated neuronal cells cocultured with astrocytes. Glutamate-induced neuronal death was not inhibited by ONO-2506 at doses of 1 to 300 μmol/L when neurons were cultured alone (Shinagawa et al., 1998). However, when neurons were cocultured with astrocytes, a remarkable protective effect of ONO-2506 was observed against glutamate-induced and lipopolysaccharide-induced neuronal death at the concentration range of 30 to 300 μmol/L (Shimoda et al., 1998; Shinagawa et al., 1998, 1999). In addition, ONO-2506 suppressed expression of S-100β mRNA in cultured astrocytes, but did not affect that of GFAP mRNA.
Therefore, we decided to use S-100β and GFAP as markers of astrocytic activation, the time course and to-pography of expression of which have already been delineated in the rat pMCAO model. Furthermore, the effect of S-100β is toxic at least in in vitro, whereas that of GFAP is innocuous or even beneficial (Nawashiro et al., 2000; Xu et al., 1999). Thus, using these two proteins possessing different actions, the present study aimed to obtain insight into the integrated actions of astrocytic activation on evolution of infarct and concomitant neurologic deficits, and to clarify the effect of ONO-2506 on the synthesis of the two proteins, together with their possible roles on stroke evolution and alterations in neurologic deficits.
Routine in vitro pharmacologic evaluations performed in Panlabs, Inc. have disclosed that ONO-2506 does not possess any significant effects on receptors of neurotransmitters, enzymes such as calcineurin, calpain, iNOS, constitutive nitric oxide synthase, protein kinase C, epidermal growth factor, tyrosine kinase, and various ion channels. The in vivo data with various animals obtained in Panlabs, Inc. and the Minase Research Institute disclosed no significant effects of ONO-2506 on the central nervous, cardiopulmonary, renal, or digestive functions, up to the dose of 25 mg/kg (intravenous bolus injection).
Thus, the present study aimed to clarify the effects of ONO-2506 on (1) the infarct volume, (2) astrocytic activation as determined by enhanced production of S-100β and GFAP, (3) the number of terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling (TUNEL)-positive cells (TPCs) in the periinfarct area, and (4) the neurologic deficits in the rat pMCAO model. The obtained data show that ONO-2506 abrogates delayed infarct expansion, suppresses the enhanced synthesis of S-100β and GFAP in the reactive astrocytes, decreases the number of TPCs, and significantly improves the neurologic deficits. These results indicate that pharmacologic modulation of astrocytic activation induced by ONO-2506 leads to the regression of symptoms and the arrest of delayed expansion.
MATERIALS AND METHODS
Surgical procedures and neurologic evaluation
Animal housing and care and the present protocols complied with the Principle of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals (DHHS publication No. [NIH] 85-23, revised 1985), and were approved by the Animal Use Ethical Committee of the Saitama Medical Center/School preceding the experiment. Male Sprague-Dawley rats weighing 280 to 300 g (Japan SLC Co. Ltd., Shizuoka, Japan) were housed in a constant-room-temperature (23°C ± 2°C) environment with a regulated relative humidity (55% ± 10%) and aeration (10 cycles/hour) under an alternating 12-hour cycle of fluorescent light and darkness (lighting, 08:00 to 20:00 hours).
Before the experiment, rats were fasted overnight with free access to water. Rats were anesthetized with halothane (2 to 3% for induction and 1 to 1.5% for maintenance) in a mixture of 70% nitrous oxide and 30% oxygen. The physiologic parameters (arterial Pa
Determination of the optimal dose of ONO-2506: experiment 1
To determine the optimal dose of ONO-2506, 52 rats were subjected to sham (n = 10) or pMCAO (n = 42). Rats subjected to pMCAO were randomly assigned to groups for intravenous administration of saline (n = 10) or ONO-2506 (1 mg/kg daily, n = 12; 3 mg/kg daily, n = 10; 10 mg/kg daily, n = 10). Administration of ONO-2506 was started at 24 hours and repeated every 24 hours after pMCAO until the animals were killed. The neurologic score, Rota-Rod performance, and the infarct volume were evaluated every 24 hours (at 72, 120, and 168 hours) and at 168 hours after pMCAO, respectively.
Effects of ONO-2506 on cerebrospinal fluid and plasma level of S-100β: experiment 2
To examine whether a daily dose of 10 mg/kg ONO-2506 inhibits S-100β synthesis, 30 rats were subjected to pMCAO. The CSF and plasma were obtained from 10 nonoperated rats, which served as the normal control. Intravenous administration of ONO-2506 was started immediately after pMCAO. The CSF and plasma levels of S-100β were determined at 24 hours after pMCAO. This time point was chosen because the preceding study had shown that S-100β present in the CSF at 24 hours is mostly derived from reactive astrocytes in the periinfarct area. Rats subjected to pMCAO were randomly allocated to the vehicle and ONO-2506 groups. At 24 hours after pMCAO, rats were reanesthetized with halothane and approximately 100 μL CSF and 100 μL of plasma were collected for the S-100β assay according to a method described elsewhere (Matsui et al., 2002).
Effects of ONO-2506 on the infarct volume at 72 and 168 hours after permanent middle cerebral artery occlusion: experiment 3
In experiments 3A and 3B, intravenous administration of ONO-2506 was started immediately after pMCAO and repeated every 24 hours until the animals were killed. The effect of ONO-2506 on the infarct volume at 72 or 168 hours after pMCAO was compared between the vehicle-treated and drug-treated groups in parallel experiments as follows. In experiment 3A, rats were randomly allocated to the vehicle (saline) group and drug-treated group; the latter received 10 mg/kg ONO-2506 daily (n = 10 for each group) and were killed 72 hours after pMCAO. In experiment 3B, rats were similarly divided into vehicle (n = 24) and drug-treated groups; the latter received 10 mg/kg ONO-2506 daily (n = 22) and were killed 168 hours after pMCAO. In each experiment, the neurologic score and the Rota-Rod performance were first evaluated at 24 hours after pMCAO and every 24 hours thereafter.
Determination of the therapeutic time window of ONO-2506: experiment 4
A total of 59 rats were subjected either to sham operation (n = 13) or to pMCAO (n = 46). Rats subjected to pMCAO were randomly assigned to receive saline (nontreatment, n = 11) or ONO-2506 (n = 35). The ONO-2506 group was further divided into three subgroups receiving the first intravenous administration of ONO-2506 (10 mg/kg daily) at 24 (n = 12), 48 (n = 12), or 72 hours (n = 11) and every 24 hours after pMCAO until time of death. Until the animals were killed at 168 hours, neurologic scores were evaluated every 24 hours, and the Rota-Rod test was performed at 72, 120, and 168 hours.
Determination of infarct volume
Rats were reanesthetized with halothane and brains were transcardially perfused with 10% neutral-buffered formalin solution for histologic, histochemical, and immunohistochemical studies. At predetermined coronal planes, the fixed brain was divided into two pieces (cerebrum and cerebellum), and the cerebral piece was routinely embedded in paraffin. At 10 different positions, 5-μm thick serial sections were cut from the frontal to the occipital poles at 1-mm intervals. Sections obtained from each site were equally spaced and sequentially labeled as slices 1 to 10 before hematoxylin and eosin- and cresyl violet staining, respectively. The stained section proximal to the posterior surface of each brain slice was examined with a computer-based image analyzer (MCID; Imaging Research, St. Catherine's, Ontario, Canada). The infarct area in each coronal section was determined according to the criteria of Garcia et al. (1993), as described elsewhere (Matsui et al., 2002). To exclude the effect of brain edema, the infarct area was divided with the ratio of the whole area of the affected hemisphere to that of the contralateral hemisphere (Mori et al., 2000). The infarct volume was obtained as the sum of corrected infarct areas of all the 10 slices.
Determination of the number of TUNEL-positive cells
Coronal sections encompassing the anterior commissure obtained in experiments 3A and 3B were used for TUNEL staining as described elsewhere (Matsui et al., 2002). The whole ischemic hemisphere was randomly divided into 130 microscopic fields (x132 magnification) and the number of TPCs in each field was counted. The sum of numbers in all the fields represented the total number of TPCs in the ischemic hemisphere.
Immunohistochemical staining for S-100 and GFAP and the assay of GFAP expression
The section obtained from experiment 3A was used. Double-immunohistochemistry was performed according to the manufacturer's protocol using a Histostain DS Double staining Kit (Zymed Lab. Inc., South San Francisco, CA, U.S.A.) with diaminobenzidine and/or Fast blue as a chromogen. The following antibodies were used: polyclonal antibodies of S-100 (diluted 1:3,000; DAKO, Carpinteria, CA, U.S.A.) or GFAP (diluted 1:500; DAKO). Hematoxylin counterstaining was performed, and controls for the immunostaining specificity were performed with the omission of the primary antibody. To estimate the intensity of GFAP expression in a quantitative fashion, the hemispheric area excluding the infarct area was divided into three areas (area 1, adjacent; area 2, intermediate; area 3, remote), depending on the distance from the infarct border as described in Fig. 7A. Within each area, the percentage of the area reacting positive to GFAP immunoreactivity was measured using an image analyzer (MCID, Imaging Research).
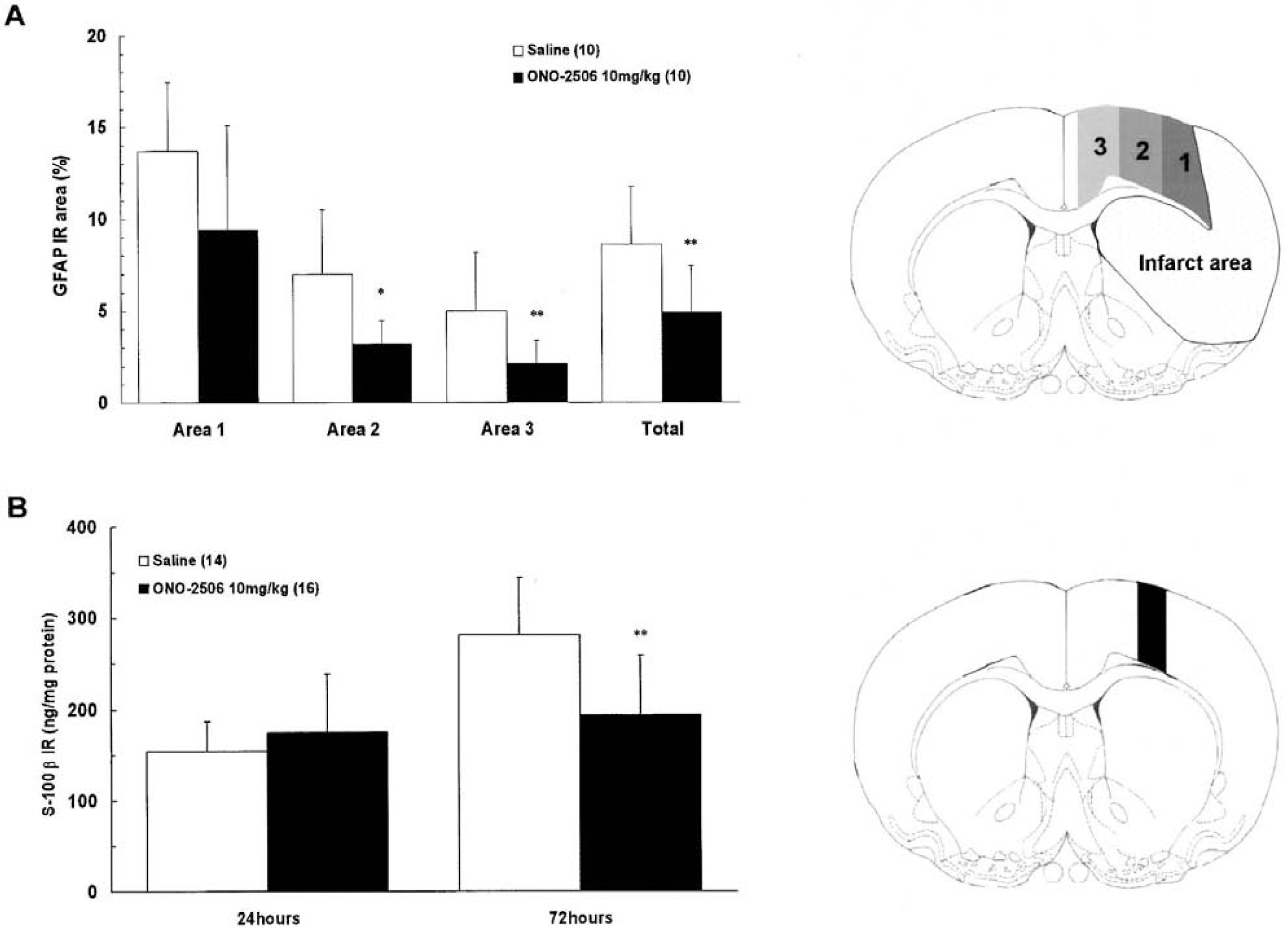
Effects of ONO-2506 on the percentage of glial fibrillary acidic protein (GFAP)-immunoreactive areas
Assay of S-100β levels in brain tissue
A total of 30 rats were subjected to pMCAO and randomly allocated to the vehicle and ONO-2506 groups, each of which was further divided into two groups in which animals were killed either 24 or 72 hours after pMCAO (n = 7, each group). Intravenous administration of 10 mg/kg ONO-2506 daily was started immediately after pMCAO until the animals were killed. Using these rats, the S-100β levels in brain tissues were measured by the following method. At 24 or 72 hours after pMCAO, rats were anesthetized with 35 mg/kg sodium pentobarbital and exposed to microwave irradiation (7.0 to 7.8 kW, 1.3 to 1.7 seconds; Muromachi-Kikai). The coronal brain slice encompassing the anterior commissure (2-mm thick) was dissected and the periinfarct area was collected (Fig. 7B). S-100β was extracted with 0.15% sodium dodecyl sulfate solution containing 2 mmol/L edetic acid and measured by the sandwich enzyme-linked immunosolvent assay method using two different antibodies that detect the β subunit of S-100 protein. Microtiter plates coated with anti-S-100β antibody (diluted 1:1,000, clone: SH-B4, Sigma, St. Louis, MO, U.S.A.) were incubated overnight with standard bovine S-100β protein (0.01–600 ng/mL; Calbiochem-Novabiochem, San Diego, CA, U.S.A.) or the samples at 4°C before the plates were further incubated with anti-S-100 antibody (diluted 1:1,000; DAKO) at 23°C to 25°C for 2 hours. After the plates had been washed, horseradish peroxidase was added, and the plates were incubated for 2 hours at 23°C to 25°C. Horseradish peroxidase substrate was finally added, and the plates were incubated for 5 minutes. The resulting absorbance was measured at an optical density of 412 nm.
Statistical analysis
Data are presented as the mean ± SD. Differences were evaluated by the unpaired t-test, one-way analysis of variance followed by Fisher PLSD, or Kruskal-Wallis test followed by the Mann-Whitney test for statistical significance. P values of less than 0.05 were considered to be significant.
RESULTS
Physiologic parameters
When the brains were isolated, pMCAO at the correct position and the absence of subarachnoid hemorrhage or subdural hematoma were visually confirmed in all rats. Mortalities were not encountered before the end of the experiment. The body temperature of animals remained stable before and after surgery. Representative data regarding the intraoperative alterations in blood gases, blood sugar, and body temperature in animals in experiment 2 are shown in Table 1. In each rat, the neurologic score at 1 hour was 3, which was maintained throughout the observation period until 168 hours. Despite neurologic deficits, all rats consumed food and water normally.
Physiologic values obtained from experiment 2 animals
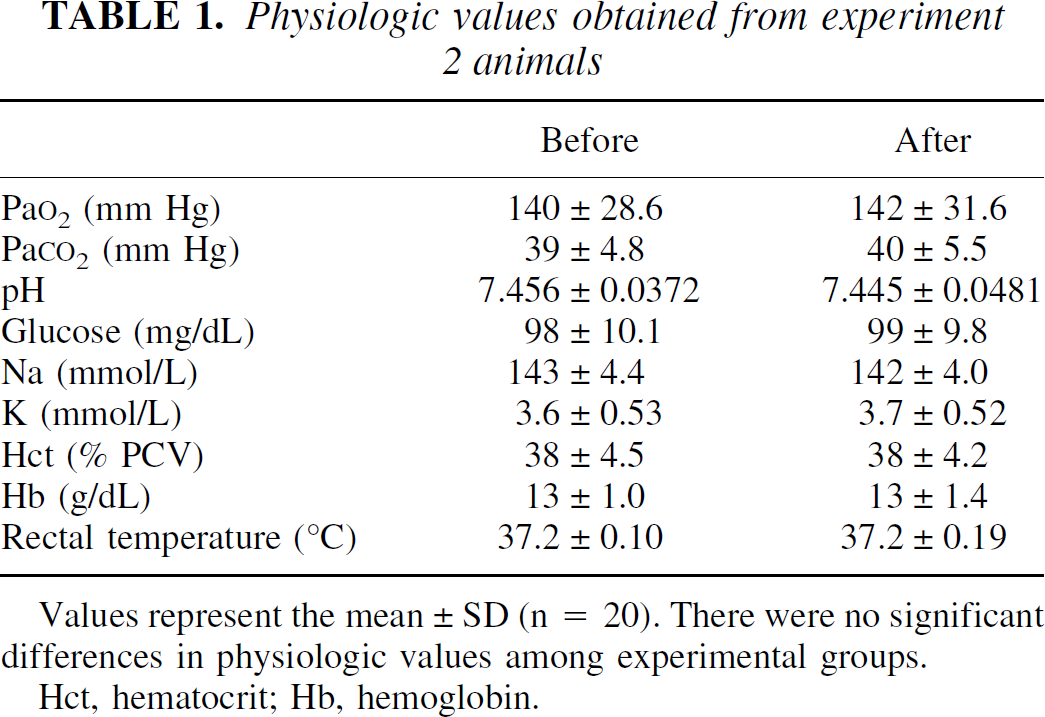
Values represent the mean ± SD (n = 20). There were no significant differences in physiologic values among experimental groups.
Hct, hematocrit; Hb, hemoglobin.
The optimal dose of ONO-2506: experiment 1
Experiment 1 revealed a clear dose dependency regarding the infarct volume at 168 hours and the neurologic score every 24 hours after pMCAO (Figs. 1A and 1B), and regarding Rota-rod performance at 72, 120, and 168 hours after pMCAO (Fig. 1C). Although the results indicated that the optimal dose of ONO-2506 with pMCAO was 10 mg/kg daily (intravenous bolus injection), a significant effect was attained at 3 mg/kg daily. The plasma level of ONO-2506 after intravenous administration of 3 mg/kg was approximately 80 μmol/L, which compares well with the effective concentration range in the preceding in vitro experiment.
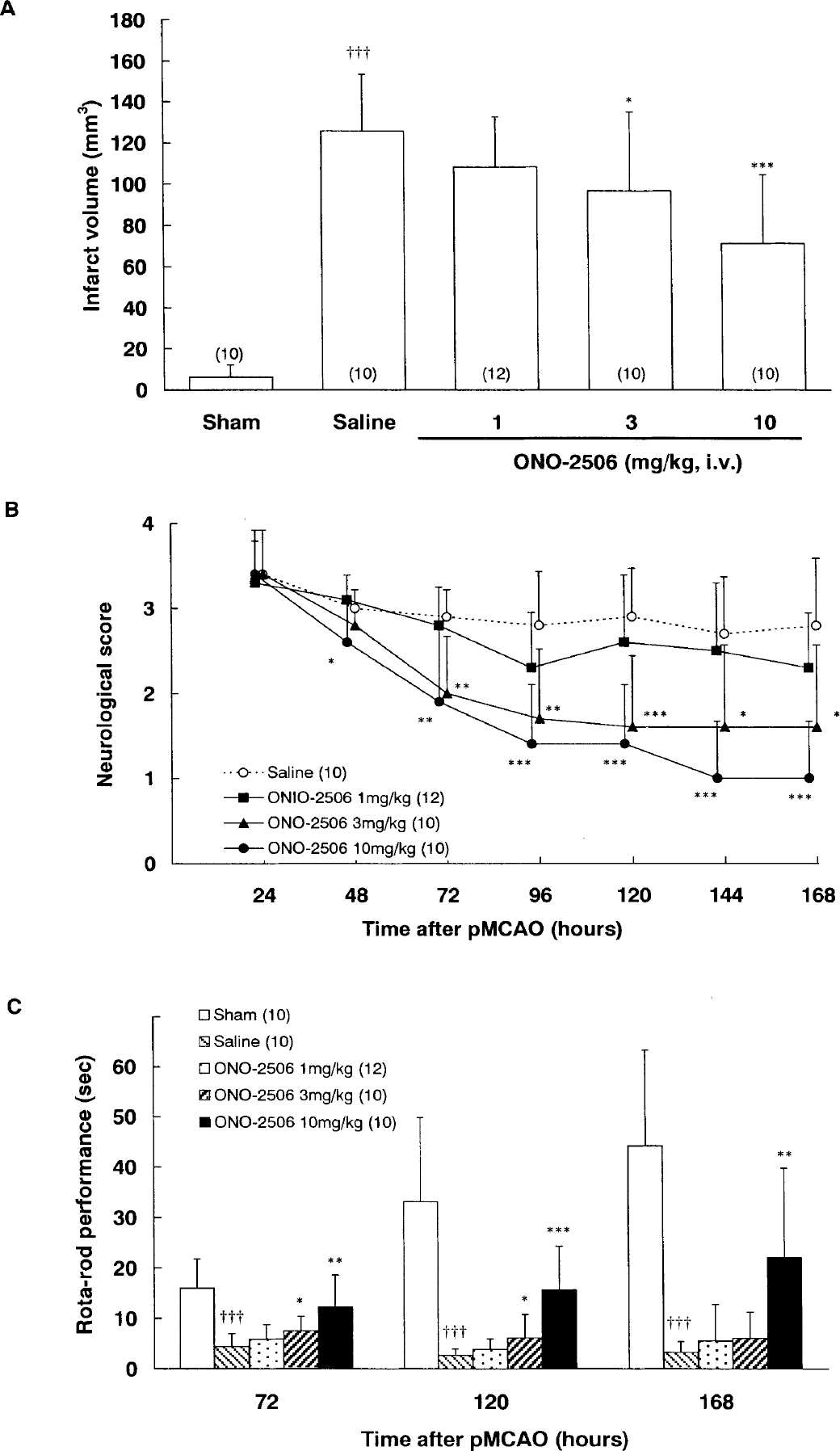
Experiment 1: effects of ONO-2506 on the infarct volume
Effects of ONO-2506 on the cerebrospinal fluid and the plasma level of S-100β at 24 hours after permanent middle cerebral artery occlusion: experiment 2
The CSF and the plasma levels of S-100β were significantly elevated at 24 hours after pMCAO with the following comparisons: CSF (vehicle vs. normal, P < 0.01), plasma (vehicle vs. normal, P < 0.001). Administration of ONO-2506 at a daily dose of 10 mg/kg significantly decreased the CSF level of S-100β without affecting the plasma level at 24 hours after pMCAO (P < 0.01; Figs. 2A and 2B).
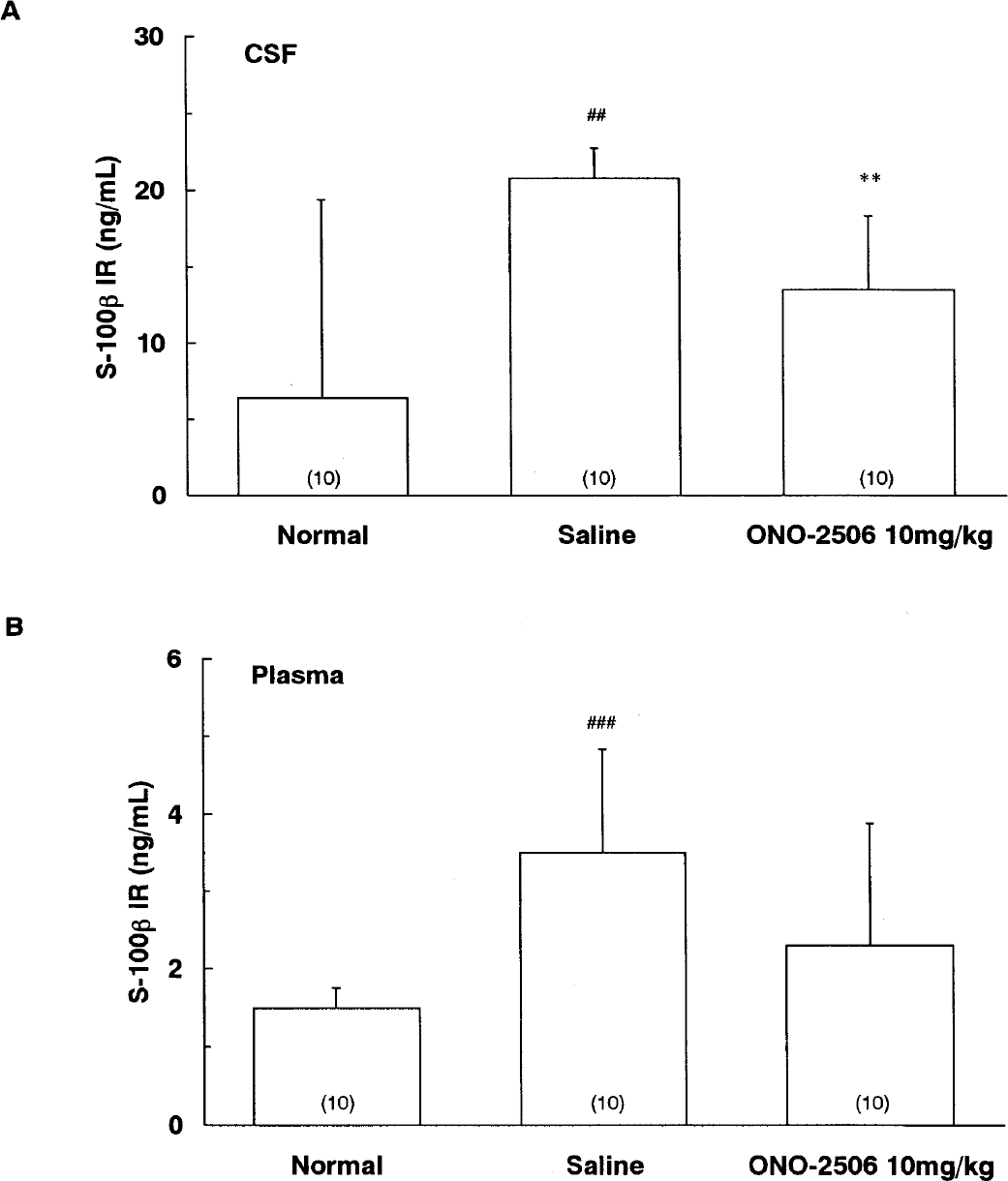
Experiment 2: effects of ONO-2506 on the CSF
Effects of ONO-2506 on infarct volume and neurologic deficits at 72 and 168 hours after permanent middle cerebral artery occlusion: experiment 3
Results of experiments 3A and 3B are illustrated in Figs. 3A to 3D and Figs. 4A to 4D, respectively. At 72 hours after pMCAO, ONO-2506 tended to decrease the infarct volume, but the difference from the saline group did not reach a statistical significance (Figs. 3A and 3B). However, the neurologic score was significantly improved at 72 hours and the Rota-Rod performance was significantly improved at 48 and 72 hours after drug administration (Figs. 3C and 3D). This result is surprising because a significant reduction in the infarct volume had not yet been attained at 72 hours in this experiment. At 168 hours, however, significant differences in both the infarct area and volume were revealed between the vehicle and ONO-2506 (10 mg/kg daily) groups (Figs. 4A and 4B). The neurologic deficits were significantly improved at as early as 24 hours after the initial drug administration, and this improvement persisted throughout the observation period (Fig. 4C). The Rota-Rod performance was significantly improved at 72 hours after the initial drug administration (Fig. 4D). ONO-2506 inhibited delayed infarct expansion without any preferential direction (Fig. 4A) or location (Fig. 4B).
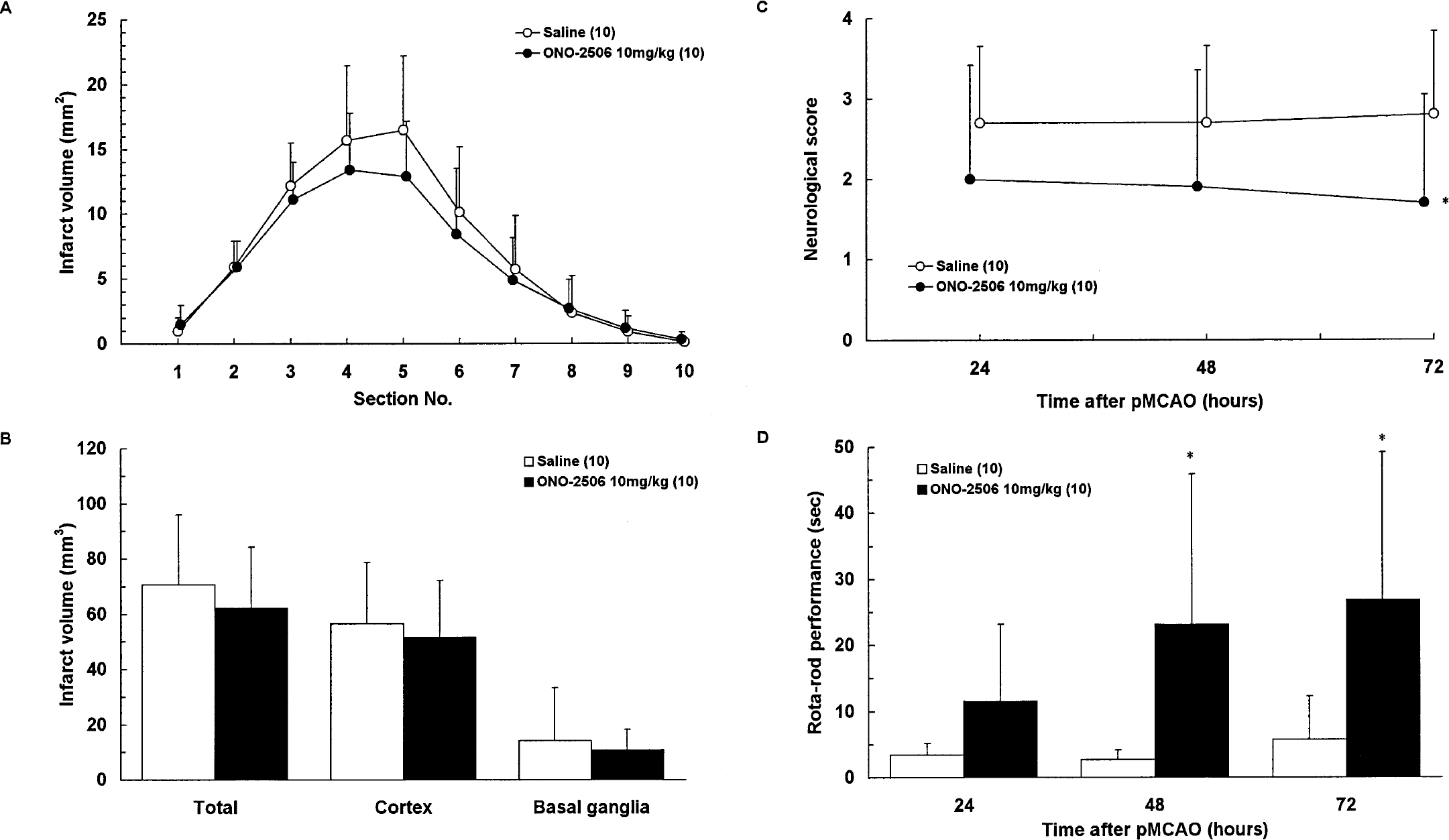
Experiment 3A: effects of ONO-2506 on the infarct area
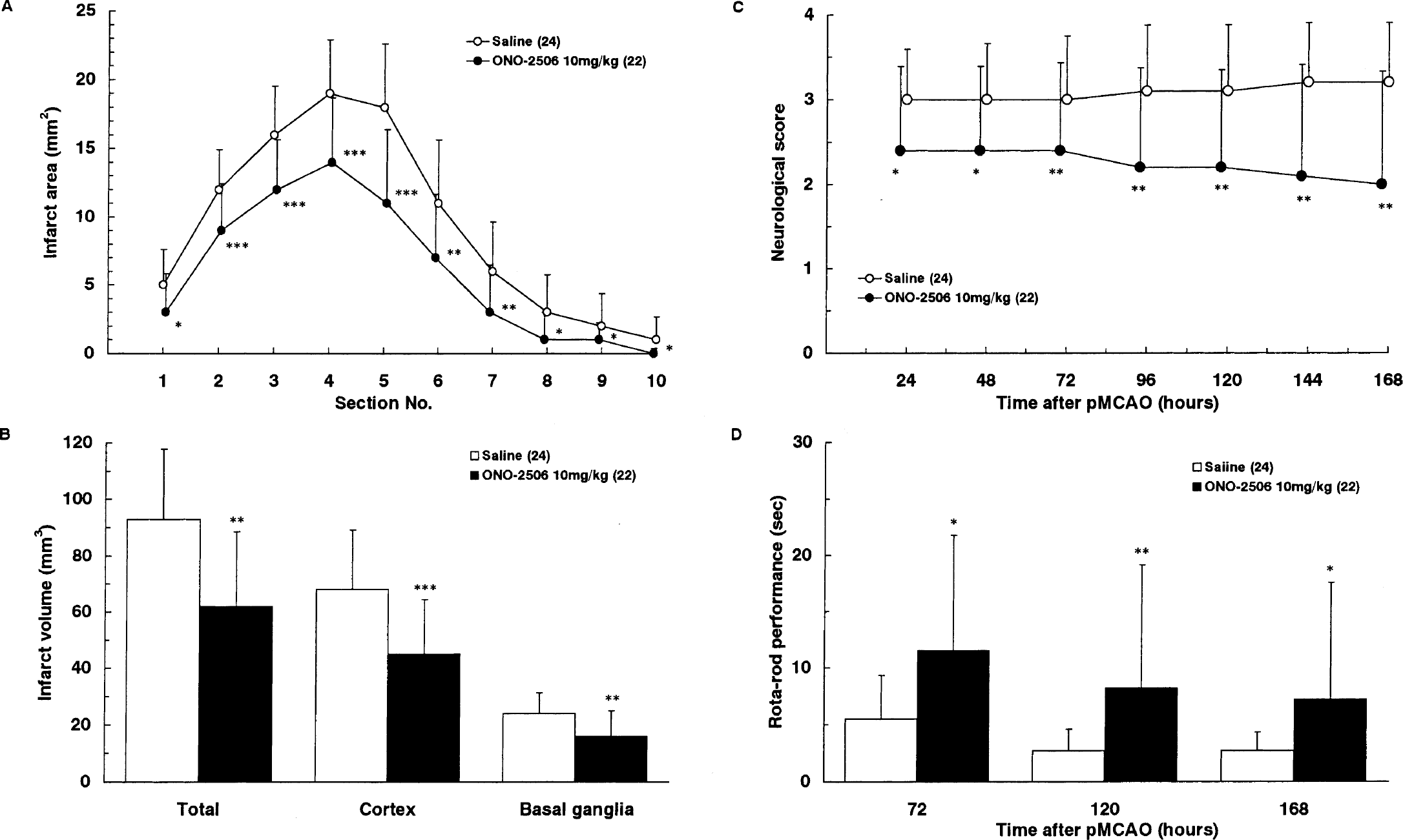
Experiment 3B: effects of ONO-2506 on the infarct area
The therapeutic time window of ONO-2506: experiment 4
Because administration of ONO-2506 (10 mg/kg daily) initiated immediately after pMCAO significantly reduced the infarct volume at 168 hours (Figs. 1 and 4), its therapeutic time window was determined in experiment 4. Administration of ONO-2506 started at 24 hours and 48 hours significantly decreased the infarct volume, though such an outcome was not established with administration initiated at 72 hours (Figs. 5A–5C). Therefore, ONO-2506 displayed a very wide therapeutic time window, ranging from 0 to 48 hours after ischemia (Fig. 5A). The improvement in the neurologic deficits induced by ONO-2506 was also dependent on the timing of its first administration, and a similar therapeutic time window was observed with the Rota-Rod performance (Figs. 5B and 5C). Drug administration started at either 24 or 48 hours induced a significant improvement at 168 hours, whereas the former showed a more striking improvement as early as 72 hours that was significantly different from the vehicle group. Delayed drug administration initiated at as late as 24 to 48 hours after pMCAO induced a swift and marked improvement of neurologic deficits.
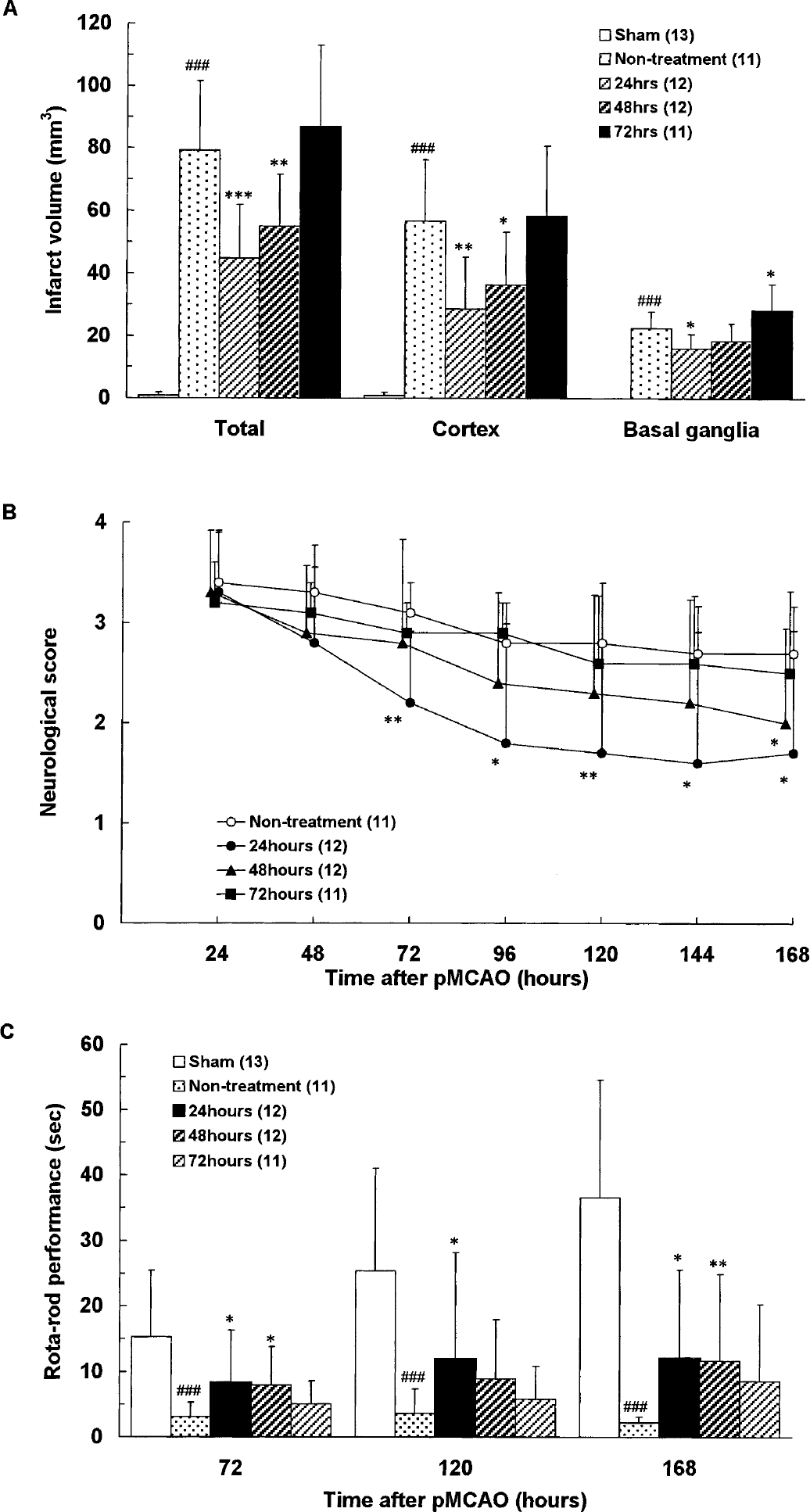
(A-C) Experiment 4: therapeutic time window of ONO-2506. Each value represents the mean + 1 SD. Administration of ONO-2506 was started at 24, 48, and 72 hours and repeated every 24 hours until 144 hours after permanent middle cerebral artery occlusion (pMCAO). For the infarct volume at 168 hours after pMCAO, statistical analysis of differences was performed by comparing with the sham or nontreatment group by one-way analysis of variance followed by Fischer PLSD, where differences of P <0.05 (*), P < 0.01 (**), and P < 0.001 (### or ***) were considered significant. For neurologic scores and Rota-Rod performance, differences between the sham or nontreatment group were compared with the Kruskal-Wallis test followed by the Mann-Whitney test, where differences of P < 0.05 (*), P < 0.01 (**), and P < 0.001 (### or ***) were considered significant.
Effects of ONO-2506 on the number of TUNEL-positive cells
The number of TPCs was 72 (samples obtained in experiment 3A) and 168 (samples obtained in experiment 3B) hours after pMCAO. In the vehicle group, the number of TPCs in the whole ischemic hemisphere was greater at 72 hours than at 168 hours. The number of TPCs in the whole ischemic hemisphere at 72 hours was greatly diminished in the ONO-2506 group as compared with the saline group (Fig. 6).
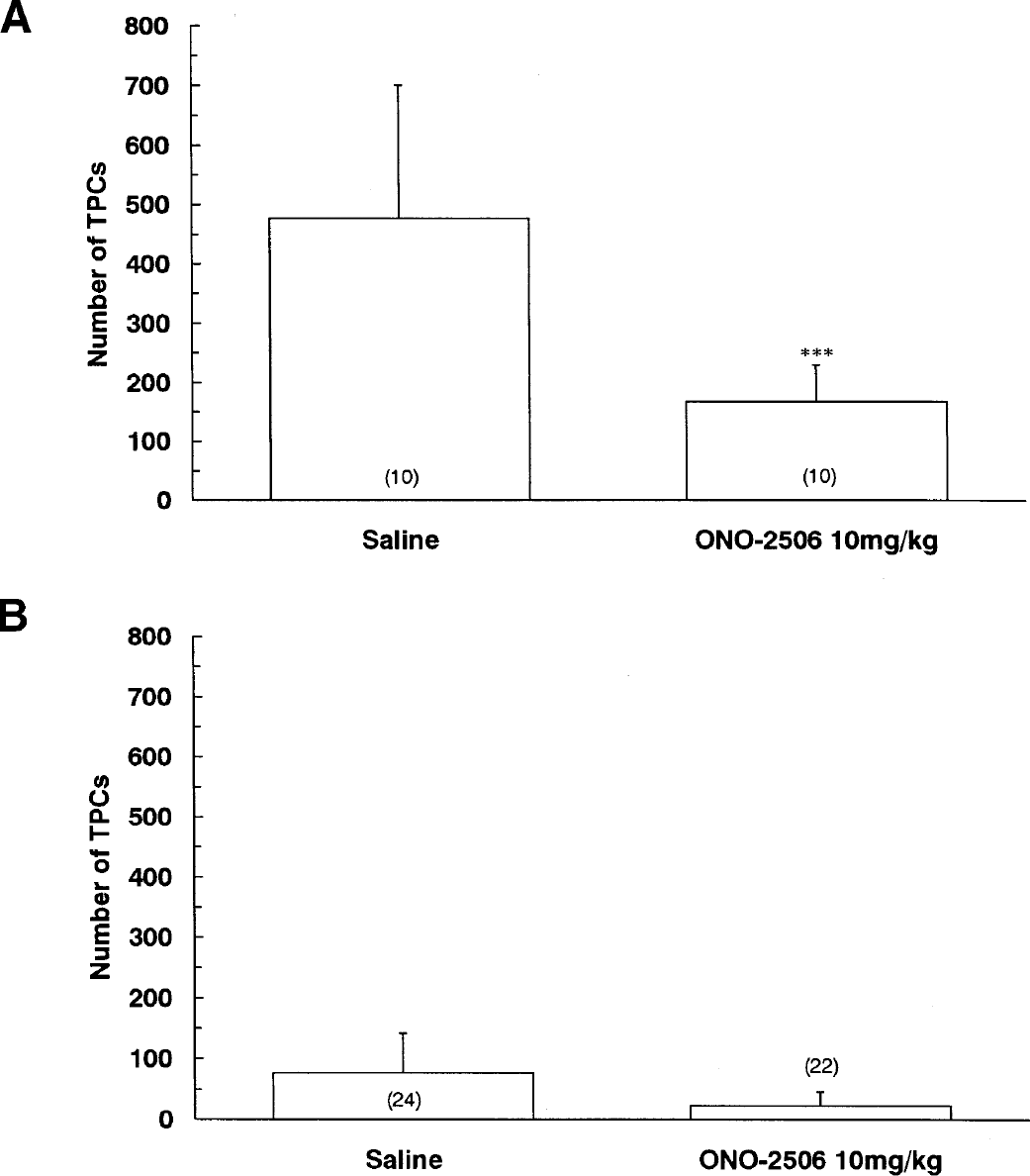
Effect of ONO-2506 on the number of TUNEL-positive cells (TPCs) at 72 and 168 hours after permanent middle cerebral artery occlusion. Samples were obtained from experiments 3A
Effects of ONO-2506 on immunohistochemical staining for GFAP and S-100 and on the tissue level of GFAP and S-100β in the periinfarct area
Astrocytic activation in the vehicle group at 72 hours after pMCAO as evidenced by augmented immunohistochemical staining for GFAP and S-100 was widespread in the whole affected hemisphere, whereas it was most pronounced along the outer boundary of infarct border. The attenuation of the immunohistochemical staining for S-100 in the ONO-2506 group was apparent in the remote and outer boundary. In the ONO-2506 group, the immunoreactivity of immunohistochemical staining for GFAP was decreased in the remote area and the outer boundary of infarct area. These microscopic findings were confirmed by the quantitative determination of GFAP expression in different loci of the periinfarct area (Fig. 7A). Although ONO-2506 did not exhibit the significant reduction of GFAP-immunoreactive areas (%) in the outer boundary (area 1), ONO-2506 reduced GFAP-immunoreactive areas (%) in the remote area (areas 2 and 3). In the vehicle group, the tissue level of S-100β in the periinfarct area was increased as compared with the contralateral region at 24 and 72 hours after pMCAO (Matsui et al., 2002), and was significantly reduced in the ONO-2506 group at 72 hours after pMCAO (Fig. 7B).
DISCUSSION
Our results show that delayed infarct expansion, expression of S-100β in the periinfarct activated astrocytes, the increase in the number of TPCs in the whole ischemic hemisphere, and neurologic deficits were markedly mitigated by administration of ONO-2506. The therapeutic time window of the agent ranged from 0 to 48 hours after pMCAO. From these results, it can be suggested that astrocytic activation, particularly the augmented synthesis of S-100β, plays a pivotal role in the occurrence of delayed infarct expansion and prolonged suppression of neuronal functions in the periinfarct area.
Effect of ONO-2506 on the infarct volume
ONO-2506 significantly reduced the infarct volume at 168 hours but not at 72 hours. During the time interval from 72 to 168 hours, the infarct volume considerably increased in the vehicle group (from approximately 70 to 93 mm), owing to the occurrence of delayed infarct expansion. This increase (33%) approximated well to a finding in our preceding study (from approximately 85 mm3 at 72 hours to 112 mm3 at 168 hours, 32%; Matsui et al., 2002). Because the infarct volume in the ONO-2506 group remained unchanged during the same time interval, the difference in infarct volume between the two groups eventually enlarged at 168 hours to achieve statistical significance, entailing that ONO-2506 abolished delayed infarct expansion from 72 to 168 hours. Therefore, the question is whether ONO-2506 inhibited infarct expansion before the 72-hour post-pMCAO period.
The increase in infarct volume is most rapid during the initial 6 to 12 hours after pMCAO. The time point of 24 hours after pMCAO represents the watershed where the tailing off of acute infarct expansion overlapped with the start of delayed infarct expansion. Because the increase in infarct volume between 24 and 72 hours was approximately 14% in the preceding experiment (approximately 70 to 85 mm3; Matsui et al., 2002), the infarct volume in the vehicle group at 24 hours is calculated as approximately 61 mm3 (70/1.14), which approximated to the infarct volume in the ONO-2506 (10 mg) group at 72 hours (approximately 60 mm). Therefore, it seems reasonable to assume that ONO-2506 inhibited delayed infarct expansion because the effect was initiated approximately 24 hours after pMCAO. Our results show that ONO-2506 administered immediately after pMCAO abolished delayed infarct expansion, whereas the acute infarct expansion before 24 hours was unaffected.
The therapeutic time window of ONO-2506
Regarding the infarct volume at 168 hours, the therapeutic time window of ONO-2506 ranged between 0 and 48 hours. ONO-2506 was administered every 24 hours in this experiment, and the protective effect of the agent administered immediately after pMCAO (92 to 57 mm3, approximately 38%) was actually less pronounced than that administered at 24 hours (79 to 43 mm3, 46%). The above result is in keeping with the recent report that S-100β at nanomolar concentrations exerts a protective action (Ahlemeyer et al., 1999). In fact, the tissue concentrations of S-100β are relatively low during the initial 12 hours after pMCAO (Matsui et al., 2002). The therapeutic time window of the agent encompassed the time interval where the peak tissue level of S-100β was attained (Matsui et al., 2002). This result is consonant with the view that astrocytic activation leading to the augmented synthesis of S-100β plays a cardinal role in the occurrence of delayed infarct expansion.
Expression of S-100β and glial fibrillary acidic protein
Astrocytic activation, as evidenced by an enhanced immunohistochemical staining for either GFAP or S-100β, was most intense along the outer border of infarct, and a similar but less intense change was diffusely present in the ischemic hemisphere, excluding the infarct area. Microscopically, the intensity of this immunohistochemical staining and the morphologic changes, namely hypertrophy characterizing reactive astrocytes (Petito et al., 1990), were considerably attenuated by ONO-2506. This observation is consistent with the result of the assays of the tissue levels of S-100β at 72 hours after pMCAO. Although the tissue level of S-100β was decreased by only 30%, this decrease may be sufficient to prevent the focal rise of extracellular concentrations of S-100β to micromolar levels.
Interestingly, ONO-2506 significantly attenuated GFAP expression in the remote areas according to the quantitative analysis. Expressions of S-100β and GFAP are bound to different intracellular signaling pathways (Ahlemeyer et al., 2000; Barger et al., 1992a, b ; Castets et al., 1997; Eddeleston and Mucke, 1993; Hill et al., 1996; Hinkle et al., 1997; Hu et al., 1996; Krohn et al., 1999), and as described previously, ONO-2506 inhibits the mRNA expression of S-100β, but not of GFAP, in cultured astrocytes. Therefore, the diminished GFAP expression may reflect a generalized inhibition of astrocytic activation induced by ONO-2506 rather than a direct action of the agent on the pathway of GFAP synthesis.
Early improvement of neurologic deficits induced by ONO-2506
ONO-2506 induced a significant neurologic recovery as early as 24 to 48 hours after the first administration. Such an effect was established by the consistent results in four independently performed experiments (experiments 1, 3A, 3B, and 4). Because the improvement in neurologic deficits preceded a significant reduction in the infarct volume, the former cannot be a result of the latter. In this regard, recent reports show that transient MCAO is accompanied by relatively widespread and possibly persisting functional disturbances in the peri-infarct region (Neumann-Haefelin and Witte, 2000; Witte et al., 2000). Thus, the prompt improvement in neurologic deficits observed in the present study should be ascribable to the beneficial actions of ONO-2506 on functional disturbances in the ischemic hemisphere and neural plastic reorganization in both ischemic and nonischemic hemispheres (Yamashita et al., 1996). That functional derangements and delayed infarct expansion were simultaneously mitigated by ONO-2506 suggest that they might stem from a common pathogenic mechanism, namely astrocytic activation. In connection with the relation between the functional derangement in the periinfarct area and the infarct expansion, Dirnagl et al., (1999) stated that symptoms can regress while the lesion actually expands. Results of the present study extend this statement to a view that owing to pharmacologic modulation of astrocytic activation, symptoms can regress while delayed expansion of the lesion is arrested.
The pathomechanism underlying astrocytic activation
Despite a mountain of knowledge acquired through past investigations, the precise mechanism that induces astrocytic activation has not yet been established (Griffith and Sutin, 1996). Nonetheless, periinfarct spreading depression has recently been shown to induce astrocytic activation (Back et al., 1996; Dietrich et al., 2000; Kawahara et al., 1999; Kraig et al., 1991) and expansion of infarct volume during the initial several hours after focal ischemia (Hata et al., 2000; Hossmann, 1994). Furthermore, calcium waves propagating through astrocytic gap junctions participate in the occurrence of periinfarct spreading depression (Nedergaard et al., 1995). Inhibition of periinfarct spreading depression by administration of gap junction blockers leads to a significant reduction in the infarct volume (Rawanduzy et al., 1997; Saito et al., 1997). However, the possibility that peri-infarct spreading depression is directly involved in delayed infarct expansion seems rather unlikely because (1) the occurrence of spreading depression in the otherwise-normal brain does not incur cell damage (Nedergaard and Hansen, 1988), and (2) the occurrence of periinfarct spreading depression has been documented during a narrow time interval of several hours after the onset of ischemia (Hossmann, 1994; Mies et al., 1993; Nedergaard and Hansen, 1993), though it may induce lingering effects on gene expression (Hata et al., 2000; Kawahara et al., 1999). Calcium waves may propagate through astrocytic gap junctions within a wide area of the affected hemisphere, even in the absence of spreading depression-like depolarization (Cotrina et al., 1998; Lin et al., 1998; Pasti et al., 1997). Such a mechanism may explain the wide distribution of activated astrocytes expressing S-100β and GFAP in the affected hemisphere.
ONO-2506 has octanoic acid as its major component. Because octanol is an uncoupler of astrocytic gap junctions (Giaume and McCarthy, 1996; Rawanduzy et al., 1997), it is plausible that ONO-2506 acts as such, and that the effects of ONO-2506 as observed in the present study might be attributable to uncoupling of astrocytic gap junctions. Such a view can explain the beneficial effect of ONO-2506 on neurologic deficits because functional disturbances in the periinfarct area may be causally related to the widespread astrocytic activation (Cotrina et al., 1998; Nedergaard, 1994; Pasti et al., 1997). However, administration of octanol significantly reduced the infarct volume at 24 hours after pMCAO, while inhibiting spreading depression (Rawanduzy et al., 1997). In the present study, by contrast, infarct expansion within 24 hours after pMCAO was unaffected by ONO-2506. Subsequently, whether the major mechanism of action of ONO-2506 is attributable to uncoupling of astrocytic gap junctions remains to be clarified.
The possible interrelation between cytokines, S-100β, and inducible nitric oxide synthase
Because astrocytes upregulate numerous molecules on activation, it remains controversial whether the induced changes are generally beneficial or detrimental (Barone and Feuerstein, 1999; Dietrich et al., 2000; Loihl et al., 1999; Murphy, 2000; Sharp et al., 2000; Simmons and Murphy, 1992; Wang et al., 1997). In this regard, the therapeutic time window of ONO-2506 coincides with the third wave of gene expression after stroke—namely, the expression of numerous cytokines such as IL-1β and tissue necrosis factor α (Barone and Feuerstein, 1999; Gregersen et al., 2000; Mabuchi et al., 2000; Sharp, 2000). Those cytokines exacerbate ischemic brain damage (Barone and Feuerstein, 1999; Gregersen et al., 2000; Mabuchi et al., 2000; Stroemer and Rothwell, 1998), presumably through expression of S-100β and iNOS (Chao et al., 1996; Griffin et al., 1989; Hinkle et al., 1998; Murphy, 2000; Sheng et al., 1996). ONO-2506 and an iNOS-inhibitor aminoguanidine have a similar therapeutic time window (Nagayama et al., 1998), which is consistent with the finding that ONO-2506 significantly inhibited induction of iNOS mRNA in the ischemic brain after transient MCAO in the rat (Shimoda et al., 1998). Moreover, amyloid-β peptide, the expression of which peaks approximately 24 hours after transient global ischemia in rats (Wakita et al., 1992), activates cultured astrocytes (Hu et al., 1998) and stimulates astrocytic S-100β synthesis (unpublished data). S-100β stimulates not only the expression of iNOS (Hu et al, 1997) but also that of other cytokines, chemokines, and COX-2 (unpublished data). In addition, microglial production of TNF-α or IL-1β precedes the astroglial production of S-100β (Barone and Feurerstein, 1999; Giulian, 1993; Gregersen et al., 2000; Loihl et al., 1999; Mabuchi et al., 2000; Sairanen et al., 1997). Hence, a precipitous increase in extracellular levels of S-100β may be attained by a vicious cycle formed by microglial and astroglial activation. Considering the data presented herein, it is tempting to speculate that S-100β plays a key role among inflammatory cytokines produced in the third wave of gene expression.
Because ONO-2506 significantly decreased the tissue level of S-100β, it seems reasonable to surmise that the observed improvement in neurologic deficits, a decrease in the number of TPCs, and abrogation of delayed infarct expansion induced by ONO-2506 are caused by its modulatory effects on reactive astrocytes in the periinfarct area. Obviously, the mechanism of action of ONO-2506 requires further investigation.
In conclusion, our results indicate that ONO-2506 mitigated delayed infarct expansion and neurologic deficits, presumably through modulation of astrocytic activation within the periinfarct area, and the inhibitory action of the agent on astrocytic S-100β synthesis may play a major role. Pharmacologic modulation of reactive astrocytes may confer a novel therapeutic strategy against stroke.