
Abstract
Therapeutic hypothermia (TH) benefits survivors of cardiac arrest and neonatal hypoxic–ischemic injury and may benefit stroke patients. Large TH clinical trials, however, have shown mixed results. Given the substantial pre-clinical literature supporting TH, we explored possible mechanisms for clinical trial variability. Using a standard rodent stroke model (n = 20 per group), we found smaller infarctions after 2 h pre- or post-reperfusion TH compared to 4 h. To explore the mechanism of this discrepancy, we used primary cell cultures of rodent neurons, astrocytes, or endothelial cells subjected to oxygen–glucose deprivation (OGD). Then, cells were randomly assigned to 33℃, 35℃ or 37℃ for varying durations after varying delay times. Both 33 and 35℃ TH effectively preserved all cell types, although 33℃ was superior. Longer cooling durations overcame moderate delays to cooling initiation. In contrast, TH interfered with astrocyte paracrine protection of neurons in a temperature-dependent manner. These findings suggest that longer TH is needed to overcome delays to TH onset, but shorter TH durations may be superior to longer, perhaps due to suppression of astrocytic paracrine support of neurons during injury. We propose a scheme for optimizing TH after cerebral injury to stimulate further studies of cardiac arrest and stroke.
Introduction
Therapeutic hypothermia (TH) is among the most potent neuroprotective therapies studied in experimental cerebral ischemia. Cooling the brain as little as one degree Celsius significantly alters brain responses to ischemia. 1 TH exerts multiple effects at multiple stages of the ischemic cascade.2–5 In addition to reducing temperature-dependent catabolic enzymes, hypothermia inhibits inflammatory responses,2,3,6,7 suppresses free radicals and reactive oxygen species (ROS),8,9 and reduces brain metabolism—consumption of the key substrates oxygen and glucose—thus conserving resources and prolonging penumbral survival.10–14
Translating these robust protective effects of TH to patients has proven difficult. In 2002, two well-recognized studies demonstrated benefit of TH in survivors of cardiac arrest.15,16 Subsequently, the targeted temperature management (TTM) trial suggested that moderate hypothermia to 33℃ may be no better than mild (36℃) hypothermia. 17 In contrast to cardiac arrest studies, trials of TH for stroke revealed highly mixed results.2,4 The ICTuS trials were underpowered to show benefit, although pneumonia appeared to be a significant risk of TH after stroke.18,19 A European trial similar to ICTuS-2 remains ongoing. 20
We sought to understand the sources of variation in trials of TH for global cerebral ischemia and stroke. We found no consensus regarding key treatment parameters, e.g. target depth (33℃ vs. 36℃), treatment duration (12 vs. 24 vs. 48 h), and maximum allowable delay to treatment onset. We hypothesized that variation in the key parameters (depth, duration, delay) could profoundly influence the outcome of TH studies. Furthermore, until recently, studies of cerebral protection addressed the brain as a homogenous unit, with the tacit assumption that all elements responded similarly. Recent delineation of the neurovascular unit (NVU) has drawn attention to the differing roles played by astrocytes, endothelial cells and pericytes in forming the blood–brain barrier (BBB) and protecting brain. 21 We hypothesized that different elements of the NVU respond differently to ischemia and to protection. To model tissue ischemia, oxygen–glucose deprivation (OGD) in brain cell culture has been used extensively. 22 We cultured three elements of the NVU—neurons, astrocytes or endothelial cells—and applied standard OGD of varying intervals. We then attempted to protect the cells from OGD using TH: we compared two target temperature depths against normothermia and varied the delay to and duration of TH.
Materials and methods
Unless otherwise specified, all cell culture reagents were purchased from Life Technologies. Timed pregnant Sprague-Dawley females were purchased from Envigo Laboratories. The Institutional Animal Care and Use Committee (IACUC) at Cedars-Sinai Medical Center approved all animal handling and surgery protocols. The use of laboratory animals in this study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NRC 2011) and local Cedars-Sinai Medical Center IACUC-approved protocols.
Stroke model
All methods used have been published and used extensively in our laboratory.23,24 Male Sprague-Dawley rats weighing 290–310 g (young adult) were purchased from Envigo Laboratories (n = 20/group). All treatments were assigned at random: after each animal was undergoing middle cerebral artery occlusion (MCAo), the surgeon telephoned an off-site assistant who generated a treatment code using a coin flip and a treatment assignment table that varied by odd/even date. All assessments—histological or behavioral—were performed by other investigators unaware of treatment group, as per CAMARADES and ARRIVE guidelines.25,26 Only after all statistics were completed and all data verified was the treatment grouping revealed by the assistant.
We occluded the MCA for 4 h using our method as described previously.23,27 In brief, animals were anesthetized with 4% isoflurane induction and maintained on 1.5–2% isoflurane mixed in oxygen and nitrous oxide (30:70). A midline neck incision was made exposing the left common carotid artery. The external carotid was ligated with 4–0 silk suture and an incision was made in the wall of the external carotid artery close to the bifurcation point of the external and internal carotid arteries. A 4–0 heat-blunted nylon suture (Ethicon) was used for blocking the middle cerebral artery and inserted and advanced 17.5 mm from the bifurcation point into the internal carotid arteries for 4 h. After the occlusion, the nylon suture was removed from the carotid artery to allow reperfusion of blood flow into the MCA. To confirm adequate injury, neurological function was quantified by a blinded rater during reperfusion using a rodent neurological grading system. 28
Physiological monitoring
For physiological monitoring, a Micro-Renathane (#MR3037, Braintree Scientific, MA) tubing was placed in the femoral artery and connected to a pressure transducer (AD Instruments disposable BP transducer, # MLT 0670). We recorded blood pressure and pulse using a digital recorder (Labchart, AD instruments and Power Lab analog/digital converter) and imaged the data using Lab Charts (AD Instruments). About 5 mm of a flexible implantable thermocouple microprobe (AD Instruments MLT 1402 IT-23 connected to a T-Type Pod ML312) was inserted into the temporalis muscle close to the skull and a small drop of surgical glue held the probe in place. We used temporalis muscle temperature as an indicator of brain temperature as we have previously documented temporalis temperature to correlate closely to brain, although 1–1.5℃ lower than brain temperature. 29
Whole animal hypothermia
Our method for rapidly and precisely changing whole body temperature has been described. 24 Briefly, a 4–5 cm long midline incision was made in the abdominal skin and the abdominal contents were carefully removed and quickly covered with warm saline-soaked gauze. A perivascular cooling catheter was looped under the rectum, through the mesentery, and placed on top of the inferior vena cava. Using 4–0 silk suture, the cooling catheter was tacked in place. The ends of the tubes from the cooling circuit were tunneled under the skin to a small exit incision between the scapulae. The external tubing was temporarily packed beneath the intra-scapular skin and the exit incision was closed with one interrupted suture. The abdominal muscle and skin were approximated and closed. The animal was allowed one week to recover before MCAo. To initiate TH, we placed the animal on a homeothermic warming blanket (set to 32.5℃ for the cooled group; 37℃ for the 37℃ group) and wrapped the blanket around the subject to provide improved temperature control. The intra-scapular incision was re-opened and the two ends of the tubing were externalized. The tubing was connected to cold-saline-filled Masterflex tubing (Cole Parmer EW-06424-16). The tubing was wrapped around a donut weight and placed in a 0–5℃ ice water bath and then inserted through the head (Cole Parmer WU-77200-60) of the Masterflex pump (Cole Parmer EW-07528-10).
We recorded the actual time, elapsed time after reperfusion, rectal temperature, temporalis muscle temperature, ice bath temperature, room temperature, blood pressure, pulse and flow rate every 2 min until temperature stabilized; after that, values were recorded at least every 5 min. We could maintain the temporalis temperature precisely at target using the pump flow rate (Supplemental Figure 1). The animal received 10 ml/kg/h of normothermic subcutaneous lactated Ringer's solution during the cooling period every 2 h. At the end of the experiment, the cooling circuit was disconnected from the animal, the microprobe was removed from the temporalis muscle, femoral catheters removed, and the skin incisions were closed with Prolene. Animals recovered from anesthesia on warming pads and when stable were returned to the vivarium for 24 h.
Lesion imaging
At sacrifice, the animals were perfused transcardially with heparinized (1 U/ml) 0.9% saline. The brains were quickly removed and carefully sliced into 2 mm thick blocks. The blocks were placed into a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) in saline at 37℃ for 30 min. After complete development of the red color, sections were incubated in 4% PFA to stop the reaction. Each block was imaged on a flatbed scanner. Using Image Pro Plus (Media Cybernetics, Rockville MD), the total area of each hemisphere, the red viable area, and the pale, ischemic area was determined using semi-automated threshold analysis. The volume of each structure was computed by multiplying the areal measurements by the thickness of the tissue block (2 mm) and then summed over all the blocks from each brain (Cavalieri method). 30 The percent ischemic tissue was computed as the ratio of the volume of pale brain divided by the volume of total brain. If the brains were edematous, a correction method was used. 31
Neuronal cell culture
Primary striatal neuron cultures were prepared from E17-E20 Sprague-Dawley rats, as previously described. 32 Briefly, the striatum was carefully separated and isolated, cleaned of meninges, and finely minced. The tissue was digested in 0.25% trypsin for 5 min in a 37℃ water bath with occasional gentle shaking. DNAse I was added to the cells at 37℃ for another 5 min. The cells were removed, brought up to 50 mL warmed Dulbecco's Modified Eagle's Medium (DMEM) and centrifuged for 5 min at 1000 × g. The cell pellet was resuspended in pre-warmed complete media (Phenol red free DMEM containing 10% horse serum, 1% Glutamax, 1% pyruvate, 1% penicillin/streptomycin, and 1% B27 supplement) and then filtered through a 70 µM diameter membrane, removed, resuspended, and counted for total cell concentration before plating. Neurons were plated on 96-well plates (2.5–3 × 104 cells/well) coated with poly-D-lysine. After four days, the cells were treated with 5-fluoro-2′-deoxyuridine for 24 h to suppress glial cells. Experiments were performed on confluent cultures after 10–14 days.
Astrocyte cell culture
Primary astrocytes were isolated from P1/P2 Sprague-Dawley rat brain striata as described for neurons. The cells were initially grown in T75 flasks in DMEM (Phenol red free) containing 10% fetal bovine serum, 1% Glutamax, 1% pyruvate, and 1% penicillin/streptomycin and 1% N2 supplement at a cell density of 107 cells per flask. After three days, the cells were shaken at 200 r/min for 10 min to remove the floating microglial cells. Cell were washed with phosphate-buffered saline (PBS), dissociated with trypsin, and plated on 96-well plates at a concentration of 2.5–3 × 104 cells/well. This procedure leads to a cell culture in which at least 95% cells are astrocytes, confirmed with GFAP staining (data not shown).
Rat brain endothelial cells
Primary RBEC were isolated from P1 to P2 Sprague-Dawley rats. Meninges were removed from the forebrain, which was then minced into small pieces in ice cold DMEM solution and further dissociated with trituration using a 1 ml pipette. The tissue suspension was digested in 10 ml of DMEM containing collagenase (10 mg/ml) and DNAse 1 (1 mg/ml) for 1 h at 37℃. An additional 10 ml of DMEM was added to neutralize enzymes and the suspension was centrifuged at 1000 × g for 10 min. The pellet was resuspended in 25 ml of DMEM that included bovine serum albumin (BSA, 20%w/v) and centrifuged at 1000 × g for 20 min at 4℃. The myelin and BSA layer was discarded and the pellet was resuspended in 9 ml of DMEM containing 1 ml of collagenase (1 mg/ml) and 0.1 ml of DNAse I and incubated for 1 h at 37℃. Then, 10 ml of DMEM was added and the suspension was centrifuged at 250 × g for 10 min. The pellet was resuspended in DMEM (phenol red free) containing 10% fetal bovine serum, 1% Glutamax, 1% pyruvate, 1% penicillin/streptomycin, 1% basic fibroblast growth factor, and puromycin (4 μg/ml). Cells were cultured in T25 flasks coated with collagen IV. After two days, the medium was changed to fresh media without puromycin. The cells from 80% confluent cultured flasks were passaged and plated on collagen IV-coated 96-well plates at a concentration of 2.5–3 × 104 per well.
OGD
Glucose and serum-free DMEM medium was bubbled with a gas mixture of 95% N2 with 5% CO2 for 30 min at 37℃ to create “OGD medium”. To initiate substrate deprivation in any of the three cell type cultures, the wells were washed with PBS and covered with 200 μl of OGD medium and immediately transferred to an incubator filled with 95% N2/5% CO2 at 37℃. At the end of OGD, the cells were removed from the incubator, the OGD medium carefully removed, and the wells refilled with the appropriate media for respective cell types. The plates were maintained at normal conditions for up to 24 h.
Cell death assay
Neuronal cell death was quantified by measuring lactate dehydrogenase (LDH) activity in the media (Cytotoxicity Detection KitPLUS (LDH) Roche Applied Science, Germany catalog number -04744934001) as published. 32 To determine a maximum LDH value for each well, after removing the media, the remaining cells in each well were lysed and centrifuged at 15,000 r/min for 10 min to deposit the cell debris. Then 100 μl supernatant from each was transferred into a 96-well plate and 100 μl LDH reaction mixture was added. The plates were covered with aluminum foil and incubated with LDH substrate for 30 min at room temperature. Absorbance was measured using a spectrophotometer at wavelength of 490 nm. The LDH detected after cell lysis was summed with the LDH detected in the supernatant to derive the total LDH present in each well. Then, each measurement was normalized and expressed as a percent total LDH in the well.
Cell viability assay
Cytotoxicity was also quantified by measuring the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to produce a dark blue formazan product. 33 The colorimetric assay correlates with cellular metabolic activity due to function and integrity of mitochondria. 34 At the end of each experiment, the cell culture medium was removed and MTT was added to each culture well at final concentration of 0.5% MTT solution (wt/vol). After incubation for 3 h at 37℃, the medium was replaced with solution containing 0.4N HCI in 99% isopropanol for 1 h. The formation of formazan was measured by recording the absorbance at a wavelength of 570 nm and a reference at 630 nm.
Experimental plan
We randomly assigned stable cultures to OGD for various time intervals (1–6 h for neurons, 1–16 h for astrocytes and 1–10 h for RBEC). The results were used to select one OGD interval for each cell type that would reproducibly kill 80% of the cells. To next determine the effect of hypothermia target temperature, treatment duration, and treatment delay, well plates were randomly assigned to one of the treatment groups which included delay times of 0, 30, 60, or 90 min, and treatment durations of 0, 2, 6 or 24 h (Supplemental Figure 2). Temperatures were assigned at random: 33, 35 or 37℃. Plates were placed into and removed from the incubators at specific times. Incubator temperature was confirmed with both the manufacturer installed thermometer and a standard laboratory thermometer placed in a water-filled beaker. Variation of temperature during incubation never exceeded 0.1℃. All media exchanges were performed with pre-cooled media for the designated temperature to avoid prolonged equilibration times.
Immunocytochemistry
To assess OGD-mediated astrocyte activation and the role of hypothermia, astrocytes cultured in 24-well plates were subjected to OGD for 2 h at 37℃ followed by hypothermia for 2, 6 and 24 h duration at 33 or 35 or 37℃.Astrocytes cultures were fixed with 4% paraformaldehyde and stained for GFAP, Serpin-A3N and LNC2. Briefly, coverslips were incubated in 0.02% Triton X-100 in PBS for 15 min and followed by three subsequent washes with PBS followed by incubation in 5% normal goat serum for 1 h at room temperature and followed by overnight incubation with rabbit anti-GFAP (1:2000), goat anti-Serpin-A3N (1:300), rat anti-LNC2 (1:800). Coverslips were incubated with Alexa 488, Cy3 and 594 conjugated secondary antibodies for 1 h at room temperature. Cover slips were washed and mounted on slides and viewed under Olympus (FV10i) microscope. Each experiment was conducted in triplicate and the observer was blinded to the treatment conditions. Each coverslip was divided into four quadrant and 12–14 images at 10 × resolution were captured from each quadrant. GFAP and Serpin-A3N florescence intensity was quantified using NIH Image J software (version 1.51).
Statistical design
All cell culture experiments were conducted in nine wells for each treatment condition. For the MTT assay, the results from each treatment condition were standardized to the mean optical density from control wells observed for 48 h at 37℃ without any OGD. For the LDH assay, the results for each condition were averaged without normalization to a control group. For each assay, we used one-way ANOVA to compare the effect of target temperatures (33, 35 or 37℃), using a random effects model including delay time and treatment duration. We examined all two-way interactions and one three-way interaction. Only if the main effects were statistically significant, we used post hoc tests to differentiate significant differences among study conditions, using p < 0.05 to test for significance. Based on prior experience with the MCAo model, we estimated a Cohen's d of 1.0 for two temperatures. Assuming Power 0.6, alpha 0.05, and a two-sided t-test, we calculated a needed sample size of 11. We raised this to 20 per group to allow greater power.
Results
Using a standard 4 h MCAo model and TTC lesion imaging 24 h later, we found that 2 h TH at 33℃ was superior to 4 h 33℃ in reducing lesion size (Figure 1(a) and (b)). We could not confirm that pre-reperfusion cooling is superior to post-reperfusion cooling.
35
The overall one-way ANOVA was statistically significant (p < 0.05) but due to known variation in this model, routine post hoc testing did not separate individual group differences. With pair-wise t-tests, we could confirm a statistically significant difference between 2 h vs. 4 h TH. During cooling, we noted a cold-pressor response in some animals. When we summarized mean arterial pressure (MAP) for all treatment groups, we failed to find a consistent pressor response (Figure 1(c)). We found no correlation between MAP and lesion volume. The finding that 2 h was superior to 4 h hypothermia led us to test whether the failure of longer cooling durations could relate to the differential susceptibility of elements in the NVU using monocellular cultures subjected to OGD.
Brief hypothermia reduces infarct volume. Animals received standard 4 h MCAo and random allocation to either normothermia (labelled “37 ℃”), immediate post-reperfusion TH for 2 h (labelled “33 ℃-2Hr”) or 4 h (“33 ℃-4Hr”), or pre-reperfusion 2 h hypothermia (“Pre-33 ℃-2Hr”) beginning 15 min prior to recanalization. All hypothermia-treated animals were titrated to 33 ℃. Lesion volume was estimated from TTC-stained sections 24 h later. (a) Representative serial 2 mm thick sections from each of the four groups show smaller infarct volumes with 2 h post-MCAo cooling and 2 h pre-reperfusion cooling. Post-reperfusion cooling to 33 ℃ for 4 h was no better than normothermia. (b) Corrected infarction volume data for each group are shown with mean±SD. The overall one-way ANOVA shows that there is a statistically significant difference among the groups, (F3,113 = 6.6, p < 0.001). Post hoc testing with Tukey's test failed to differentiate the groups. Pair-wise t-tests showed that 2 h hypothermia (either pre- or post-reperfusion) but not 4 h hypothermia, was significantly protective, compared to normothermia (*p < 0.05, **p < 0.01). (c) Aggregate blood pressure recordings for the groups. The mean arterial pressures (MAP) are shown as mean±SD prior to the MCAo, at the time of reperfusion (R, green arrow), and the time the animals reached target temperature (T, grey arrow). At no time did MAP differ among the groups, confirming that the differences in infarct volumes could not be attributed to cold-induced pressor responses. Using Pearson product moment correlation analysis, there was no relationship between infarct volume and MAP. The 37 ℃ group includes some animals kept at target for 2 h and some for 4 h, randomly divided; the groups were combined for clarity after preliminary t-test showed no differences in infarct volumes.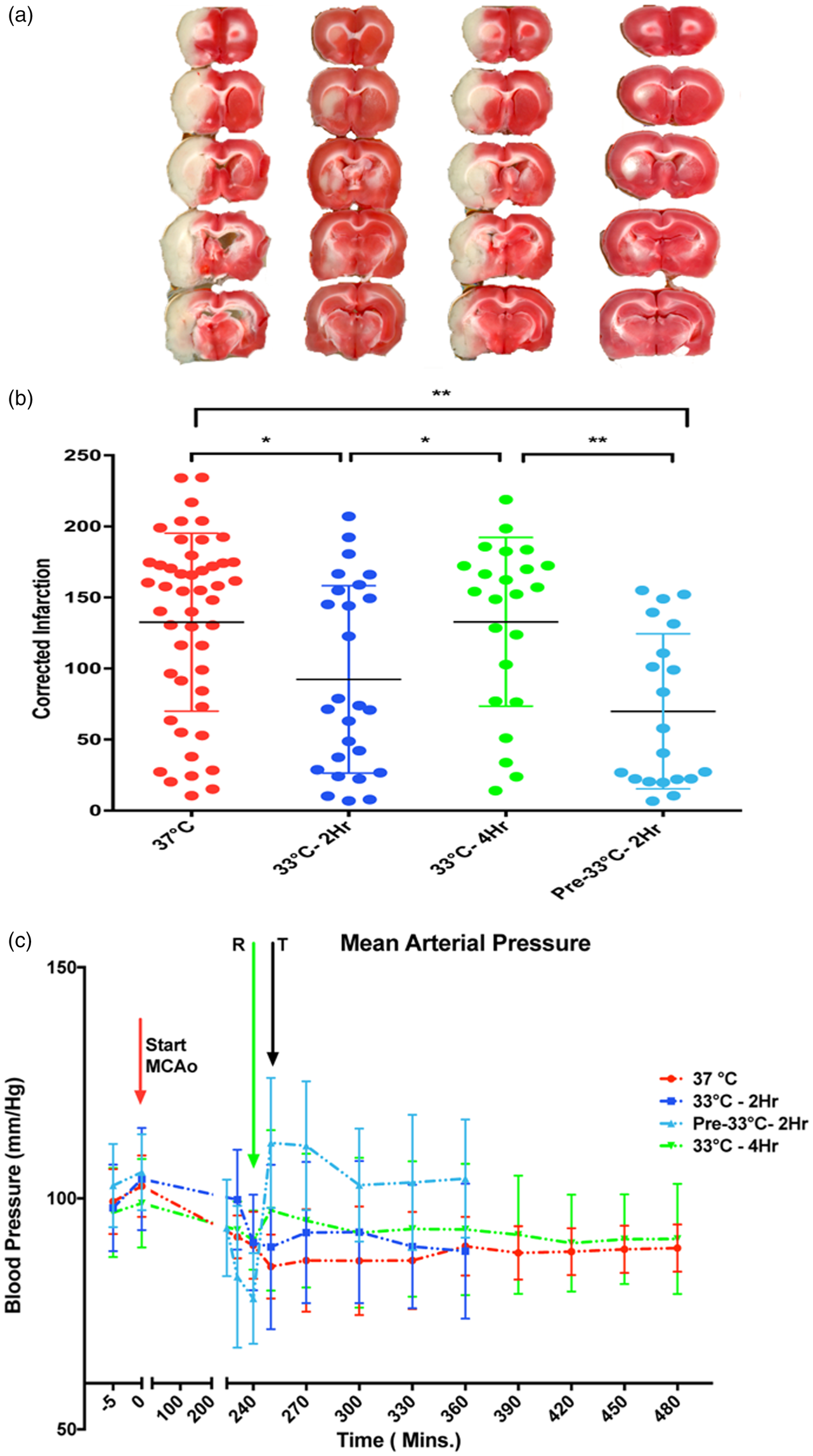
Longer OGD intervals resulted in greater cell death (Figure 2). Significantly, the three elements of the NVU differed considerably in their vulnerability to OGD, with neurons showing the greatest susceptibility and astrocytes the greatest resistance. Both LDH and MTT assays showed comparable results, such that the same interval of OGD that caused 80% cell death in the LDH assay yielded about 20% cell viability in the MTT assay (Figure 2(a) and 2(b)). In all remaining experiments, we used a standard OGD interval to cause 80% cell death: 2 h for neurons, 6 h for endothelial cells, and 10 h for astrocytes.
Effect of oxygen-glucose deprivation on elements of the neurovascular unit. Stable culture plates (neurons: blue lines; astrocytes: green lines; endothelial cells: red lines) were subjected to OGD for randomly assigned intervals (1 to 16 h) and then assayed for cell viability using MTT (Panel a) or for cell death using LDH release (Panel b). All assays were performed by an investigator blinded to the duration of the OGD. Data represent the mean±SD of 9 wells. In Panel (a), the viability assay showed that the three cellular elements of the neurovascular unit respond quite differently to longer durations of OGD, with neurons being the most vulnerable, followed by endothelial cells, followed by astrocytes. In Panel (b), similar vulnerability profile is confirmed using LDH release. The MTT and LDH results at each time point are statistically significantly different from each other in any given cell type (two-way ANOVA, p < 0.0001, Dunnett's post hoc testing p < 0.001). To kill 80% of cells by 24 h (the LD80) required 2 h OGD for neurons, 6 h for endothelial cells, and more than 10 h for astrocytes.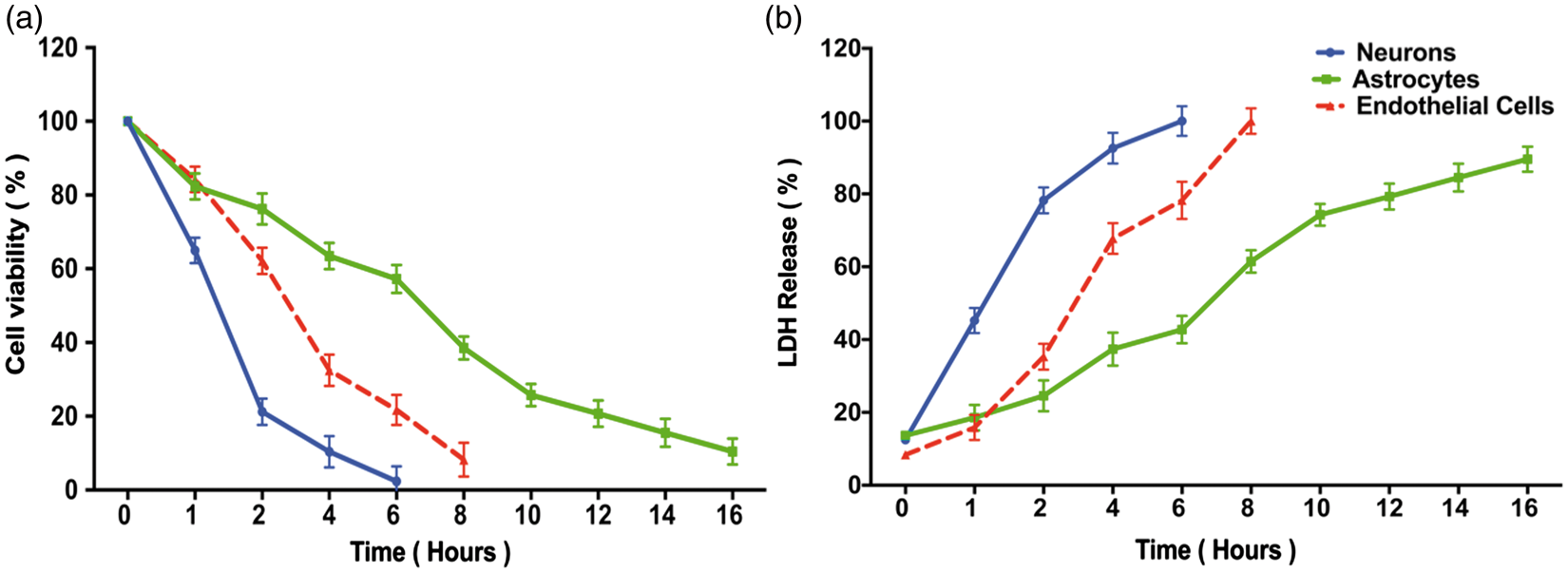
For neuron cultures subjected to 2 h OGD, the cytoprotective effect of TH appeared robust (Figure 3). Deeper hypothermia (33℃) provided significantly superior protection compared to 35℃ regardless of delay intervals and treatment durations, except 2 h cooling after a 90-min delay. Regardless of the treatment duration, the greatest protection was seen with immediate treatment, i.e. delay time of 0 min (Figure 3(d)). For each delay time between 0 and 90 min, however, longer treatment durations were associated with greater viability (Figure 3(a) and (d)) and reduced neuronal death (Figure 3(b)). The three-way interaction among depth, duration, and delay is illustrated in Figure 3(c). The graphical representation illustrates the complementary effect of treatment duration after longer delay times. Also, the superiority of 33℃ vs. 35℃ is clear for all combinations of delay and duration except for 2-h duration after 90-min delay (Bonferroni p < 0.01). Longer duration TH yielded greater cell viability for all delay times (Figure 3(d)).
Effect of hypothermia depth, delay and duration on neurons after OGD. Stable neuron cultures were subjected to 2 h OGD to kill 80% of cells by 24 h reperfusion. At random, culture plates were placed into an incubator at 33, 35, or 37 ºC (normothermia). Randomly, plates were selected for therapeutic hypothermia after delay times of 0, 30, 60 or 90 min, and randomly chosen to receive therapeutic hypothermia for 2, 6 or 24 h duration (Supplemental Figure 2). In Panel (a), the MTT assay, the mean OD for each condition is standardized to control (no OGD). Each bar shows the standardized mean±SD and results were analyzed using one-way ANOVA. When all two-way and one three-way interaction terms were included, all main effects were highly statistically significant: for temperature (F2,233 = 57.72, p < 0.001), for delay time (F3,233 = 269.25, p < 0.001), and for duration of TH (F3,233 = 248.19, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 9.79, p < 0.001), for depth×duration (F2,233 = 12.08, p < 0.001), and for delay×duration (F6,233 = 12.15, p < 0.001). The three-way interaction term depth×delay×duration was also highly significant (F6,233 = 269.25, p < 0.001). Post hoc testing was restricted to selected comparisons of interest, given that all interaction terms were significant. For example, for each combination of delay–duration, the cultures treated at 33 ºC showed higher survival than 35 ºC (ANOVA followed by Dunnet's test, p < 0.01), except for 90-min delay and 2-h duration, and both these were not different from normothermia, suggesting that 2-h therapeutic hypothermia is too short to overcome the deleterious effect of waiting 90 min to initiate treatment. On the other hand, after a 90-min delay, both 6 h and 24 h therapeutic hypothermia at either 33 ºC or 35 ºC were individually superior to normothermia (Dunnet's, p < 0.01) and 33 ºC was superior to 35 ºC (p < 0.01). In Panel (b), LDH release is shown and the control and normothermia wells were treated as in Panel (a). Each bar represents the mean±SD for each condition. As in Panel (a), all main effects were highly statistically significant: for temperature (F2,233 = 520.51, p < 0.001), for delay (F3,233 = 247.223, p < 0.001), and for duration (F3,233 = 221.05, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 27.06, p < 0.001), for depth×duration (F2,233 = 14.18, p < 0.001), and for delay×duration (F6,233 = 10.07, p < 0.001). The three-way interaction term depth×delay×duration was also highly significant (F6,233 = 13.15, p < 0.001). Post hoc testing was restricted to selected comparisons of interest, given that all interaction terms were significant. For example, individual comparisons of 30 min vs. 60 min delay times showed no significant differences (Dunnet's, p < 0.01) regardless of treatment duration. After 90-min delay, there was no beneficial effect of 2 h treatment with either temperature depth, confirming that more than 2 h therapeutic hypothermia is needed to make up for the 90-min delay to treatment. Panel (c) presents a surface contour map of the relationship among target temperature depth, treatment delay, and treatment duration on neuron viability. The treatment effect size is computed from the difference between the standardized mean MTT optical density for treated plates against those of the normothermia groups. For each combination of delay/duration, the difference in cell viability was computed for temperature depth vs. normothermia (computed in Supplemental Table). The differences for the target temperature depths 33 and 35 ºC (y-axis) are plotted for each combination of treatment delay (x-axis) vs. treatment duration (z-axis). The treatment effect size is greater for 33 ºC than for 35 ºC at all combinations except 90 min/2 h. These data illustrate that for essentially all conditions, deeper therapeutic hypothermia provides greater neuroprotection. In Panel (d), data from Panel (a) are re-plotted to illustrate that longer duration overcomes delay to TH. For each delay time (0 to 90 min) longer durations (6 or 24 h) provide better (greater) neuronal cell viability.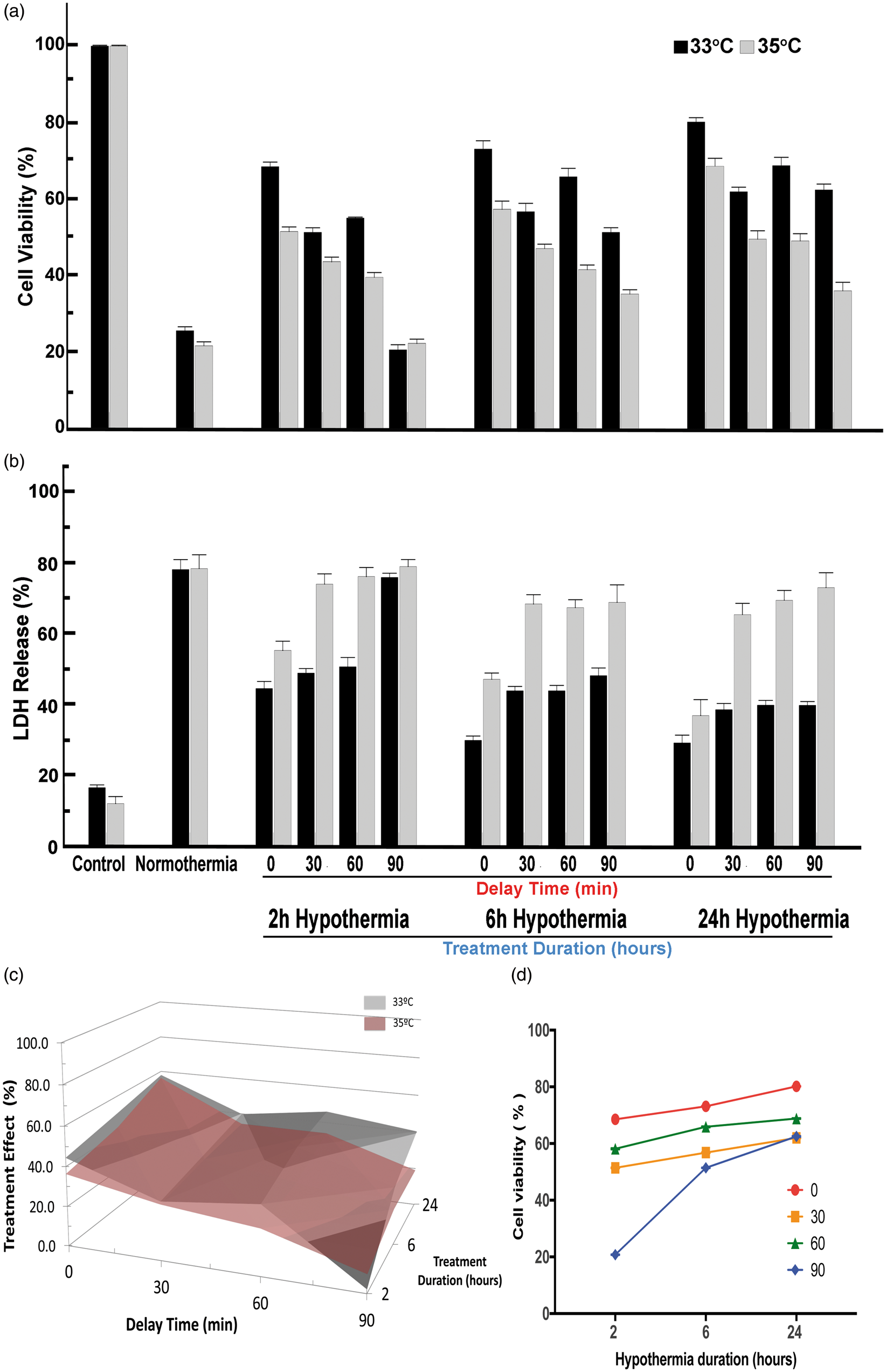
In astrocytes subjected to 10 h OGD, TH to 33℃ was protective over 2, 6, or 24 h treatment duration (Figure 4(a) to (d)). TH at 35℃, however, protected astrocytes only if continued for 6 or 24 h, suggesting the existence of a delay–duration relationship at least for the 35℃ target depth. Deeper TH to 33℃ vs. 35℃ over all delay and duration times proved superior in astrocytes (Figure 4(a) to (d)). Looking at a temperature of 33℃ (Figure 4(d)), longer durations tended to provide greater protection (main effect p < 0.001), but this was not the case for 35℃. In endothelial cells (Figure 4(e) to (g)), we found a more complex result. For cell viability (Figure 4(e)), the overall main effect for treatment depth was highly significant, as were the main effects for delay and for duration. Both two-way and the three-way interaction terms were also highly significant again providing support for a depth–delay–duration effect. However, durations of 6 and 24 h showed metabolic activity (MTT) greater than baseline (Figure 4(e)), indicating endothelial cell proliferation in response to injury;
33
these two durations differed from 2 h, but were not different from each other (p < 0.01 after Dunnett's procedure). The cell proliferation effect was not seen in the cell death assay (Figure 4(f)). The main effects for temperature and duration were all highly statistically significant and the main effect for delay was less so. These data (Figure 4(g)) confirm in endothelial cells that deeper hypothermia is superior, and also confirm that a treatment delay can be overcome with deeper hypothermia for longer duration (Figure 4(h)) although the response is complex.
Effect of hypothermia depth, delay and duration on astrocytes and endothelial cells after OGD. As for neurons in Figure 3, stable astrocyte cultures were randomly assigned to different delays and durations. All treatments were conducted randomly as in Figure 3. In Panel (a), showing MTT results for astrocytes, all main effects were highly statistically significant: for temperature (F2,233 = 161.50, p < 0.001), for delay (F3,233 = 10.30, p < 0.001), and for duration (F3,233 = 39.77, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 29.60, p < 0.001), for depth×duration (F2,233 = 9.96, p < 0.001). However, unlike the case for neurons, the two-way interaction for delay×duration was not significant (F6,233 = 1.65), nor was the three-way interaction term depth×delay×duration significant (F6,233 = 1.40). Post hoc testing confirmed that longer durations provided greater protection, regardless of delay times (Dunnet's test, p < 0.05). For many but not all comparisons, longer delay times were associated with worse outcomes. In Panel (b), the release of LDH from astrocytes after OGD was analyzed as in Figure 3(b). All main effects were highly statistically significant: for temperature (F2,233 = 439.07, p < 0.001), for delay (F3,233 = 62.29, p < 0.001), and for duration (F3,233 = 265.66, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 204.30, p < 0.001), for depth×duration (F2,233 = 20.01, p < 0.001), and for delay×duration (F6,233 = 10.47, p < 0.001). The three-way interaction term depth×delay×duration was also highly significant (F6,233 = 13.28, p < 0.001). Selected individual comparisons (Dunnet's test, p < 0.05) confirmed that longer durations reduced cell death, but the deleterious effect of delay was less obvious. In Panel (c), the three-way interaction among hypothermia depth, delay and duration on astrocytes after OGD is illustrated with surface contour maps. The data from Panel (a) were re-plotted in Panel (d) to highlight the fact that increasing TH duration overcomes the deleterious effect of increasing delay time. The effect of TH on endothelial cell cultures during OGD, as measured with the MTT assay, is shown in Panel (e). In comparison to neurons and astrocytes, the results are quite different: hypothermia caused a protective proliferation response, as evidenced by in viability measures greater than control cultures. All main effects were highly statistically significant: for temperature (F2,233 = 61.63, p < 0.001), for delay (F3,233 = 18.83, p < 0.001), and for duration (F3,233 = 227.96, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 4.02 p < 0.01), for depth×duration (F2,233 = 14.71, p < 0.001), and for delay×duration (F6,233 = 11.91, p<0.001). The three-way interaction term depth×delay×duration was also highly significant (F6,233 = 5.85, p < 0.001). Post hoc testing revealed some differences, compared to neurons and astrocytes. While increasing delay time did reduce viability for the 2 h treatment duration, this effect was not seen for 24-h treatment (Dunnet's test, p < 0.05), mainly because all groups did very well with prolonged therapeutic hypothermia. Comparing 24 h against 6 h duration—for all delay times and both temperatures—there was no statistically significant difference. However, for the 35 ºC treatment only, 90-min delay reduced viability in the 6 h, but not in the 24-h treatment duration, consistent with a delay–duration effect. Although the main effect for temperature was highly significant, individual comparisons revealed a mixed picture, with 35 ºC generally allowing greater proliferation than 33 ºC. In Panel (f), the LDH data analysis was conducted as in Figure 3(b). All main effects were highly statistically significant: for temperature (F2,233 = 61.63, p < 0.001), for delay (F3,233 = 18.82, p < 0.001), and for duration (F3,233 = 277.96, p < 0.001). Significant two-way interaction terms were seen for depth×delay (F3,233 = 4.024, p < 0.01), for depth×duration (F2,233 = 14.71, p < 0.001), and for delay×duration (F6,233 = 11.91, p < 0.001). The three-way interaction term depth×delay×duration was also highly significant (F6,233 = 5.85, p < 0.001) and is illustrated in Panel (g) as a surface contour map. Selected individual comparisons (Dunnet's test, p < 0.05) confirmed that 33 ºC significantly prevented cell death, compared to 35 ºC, over all durations and delay times. Generally, longer delay times resulted in greater cell death, although the powerful protective effect of 24 h hypothermia obscured the delay effect, confirming in endothelial cells a delay–duration relationship, as illustrated in Panel (h).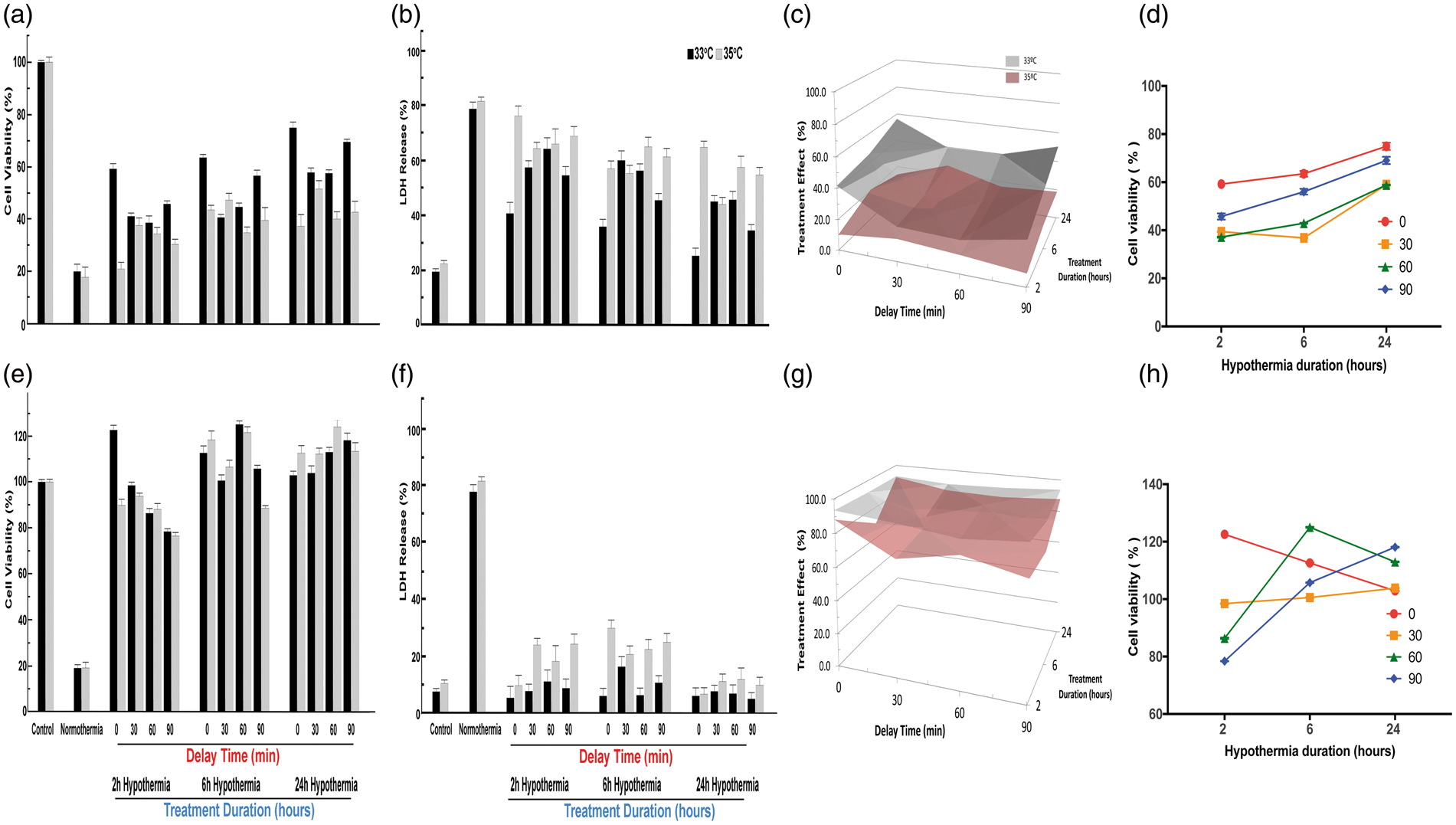
Despite using different OGD intervals to obtain 80% cell death in each cell type, the treatment effect with TH compared to normothermia was several folds greater than the delta between 33℃ and 35℃ (Supplementary Table). For neuronal cultures, the treatment effect size was 37% when we compared 33℃ vs. normothermia, effect size was 30% for 35℃ vs. normothermia, and was 8% for 33℃ vs. 35℃ (Supplementary Table). In other words, the effect size for either target depth temperature greatly exceeded the effect size difference between the two TH targets. Similarly, treatment effect sizes for astrocytes and endothelial cells indicated that the differences between the two TH target depth temperatures were considerably less than for either depth vs. normothermia.
After observing the considerable difference between neuron and astrocyte vulnerability to OGD, we asked whether short duration TH could impact astrocytes negatively. Recent data suggest astrocytes can respond to stress with a toxic or protective phenotype during activation.36,37 Astrocyte-derived paracrine protection of neurons includes a panoply of responses.
38
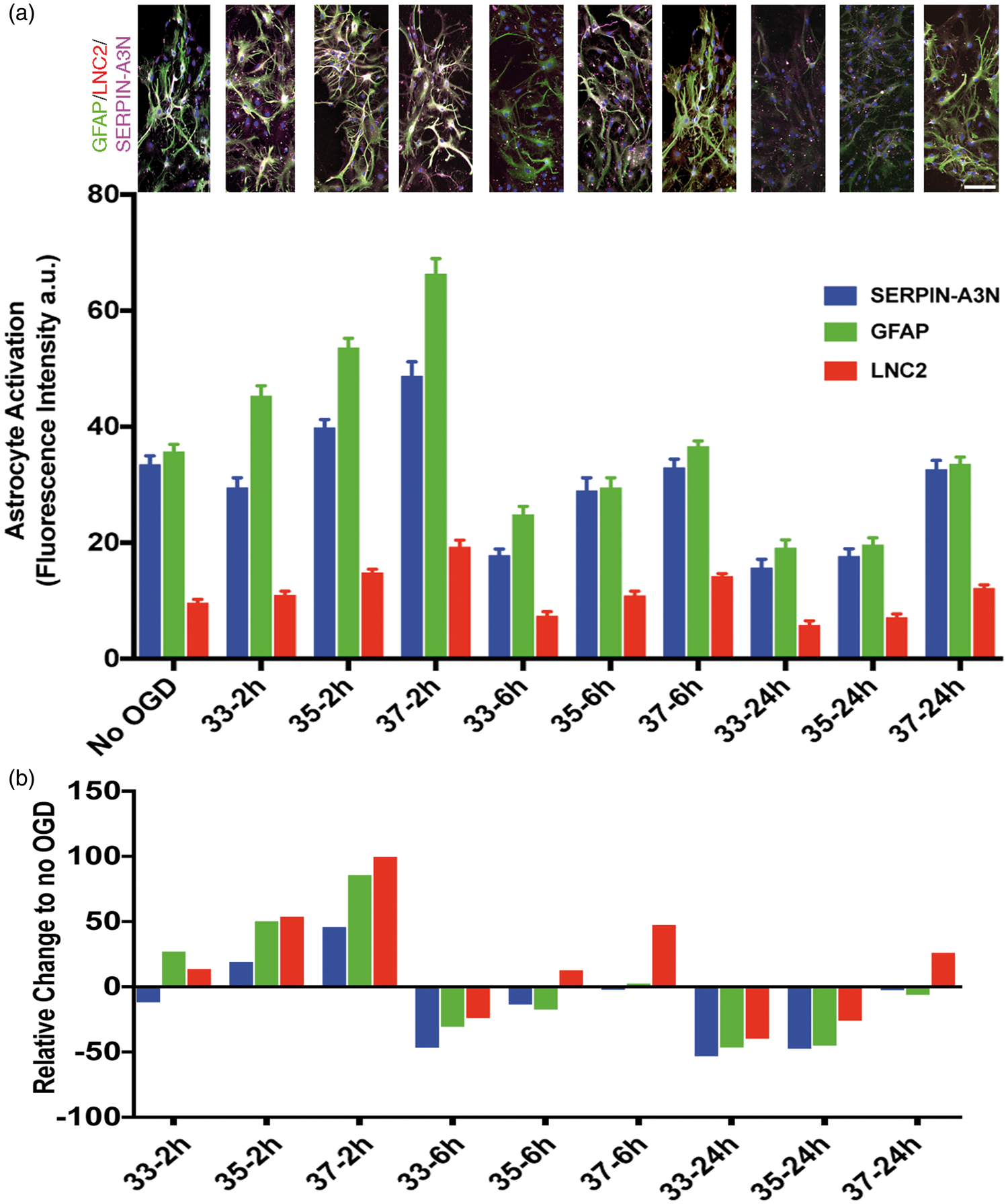
We assessed astrocyte activation using three immunohistochemical markers: GFAP, Serpin-A3N, and LCN2. The first two upregulate in response to injury and have been associated with the so-called protective astrocyte activation phenotype.36,39 The third marker, LCN2, has been associated with the ‘toxic’ astrocyte activation marker.36,40 We first confirmed the known protective effect of astrocyte-conditioned media on neuronal survival during ischemia. Conditioned media was prepared from astrocyte cultures subjected to OGD (range 30–240 min) (Figure 5(a)). When applied to neuron cultures undergoing 2 h OGD, the astrocyte-conditioned media prepared at 37℃ proffered neuronal protection (Figure 5(b)). The neuroprotective effect of conditioned astrocyte media was abrogated by TH in a temperature-dependent manner: preparing astrocyte-conditioned media at 33℃ during OGD completely eliminated the protective property, while 35℃ incubation partially blocked the protective property (Figure 5(b)). As little as 30 min TH during preparation of the conditioned astrocyte media seemed to interfere with the neuroprotective effect. To assess the mechanism of the temperature-dependency, we assessed astrocyte activation at 33, 35, and 37℃ during OGD (Figure 6). Again, in a temperature-dependent manner, TH either totally (33℃) or partially (35℃) blocked astrocyte expression of markers associated with the protective phenotype, GFAP and Serpin-A3N, with minimal effect on LCN2. These data suggest that TH, while cytoprotective under certain conditions, can impede some neuroprotective mechanisms in the NVU, such as astrocyte protective effects on neurons.
Hypothermia reduces astrocyte protective effect on neurons subject to OGD. Conditioned astrocyte media was produced by subjecting stable astrocyte cultures to varying durations OGD (selected at random) and then harvesting the supernatant. Panel (a) shows the experimental setup. At random, during OGD, the astrocytes were held at 33, 35 or 37 ºC for durations ranging from 30 to 240 min. The conditioned astrocyte media was added to neuronal cultures that were then subject to 2 h OGD at 37 ºC. After OGD, the neuron cultures were restored to normal oxygen and normal glucose-containing media; neuron survival was assayed with the MTT assay 22 h later. Panel (b): Using the MTT assay, neuron survival after OGD is shown. After standardizing all OD values to the No-OGD group, the data confirm the results in Figure 1 that 2 h OGD killed 80% of the neurons. Astrocyte-conditioned media (ACM) harvested after 2 h OGD at 37 ºC (labeled “37 – 120 min”) and placed on neuron cultures significantly protected neurons during subsequent neuronal OGD, with about 90% neuron survival. ACM prepared while hypothermia was applied to the astrocyte cultures during OGD suppressed this neuronal protective effect. ACM prepared during hypothermia to 33 ºC for 30, 60, 90 or 120 min (labeled “33 – 30 Min”, “33 – 60 Min”, “33 – 90 Min”, and “33 – 120 Min”) failed to protect neurons during neuronal OGD. Mild hypothermia at 35 ℃ for 120 or 240 min (labeled “35 – 120 Min” and 35 – 240 Min ”) blocked only about half of the ACM protective effect. These data suggest that hypothermia interferes with astrocyte protection of neurons in a temperature-dependent manner. Astrocyte activation after hypothermia was measured using markers for GFAP, LNC2, and Serpin-A3N. Panel (a) shows representative pictomicrographs illustrating astrocyte activation using GFAP (green), LNC2 (red) and SERPIN-A3N (magenta) markers. Increasing durations of hypothermia suppressed astrocyte expression of GFAP and Serpin-A3N, two markers that are associated with the protective astrocyte phenotype. The marker LNC2, which is associated with the toxic astrocyte activation phenotype, was not suppressed by 6 or 2 h TH. The suppression was dose-dependent, in that 33 ºC suppressed more than 35 ºC which suppressed more than 37 ºC. Scale bar = 30 μm. Panel (b) shows the data normalized to cultures maintained at normal oxygen and glucose (No OGD) in order to emphasize the relative change.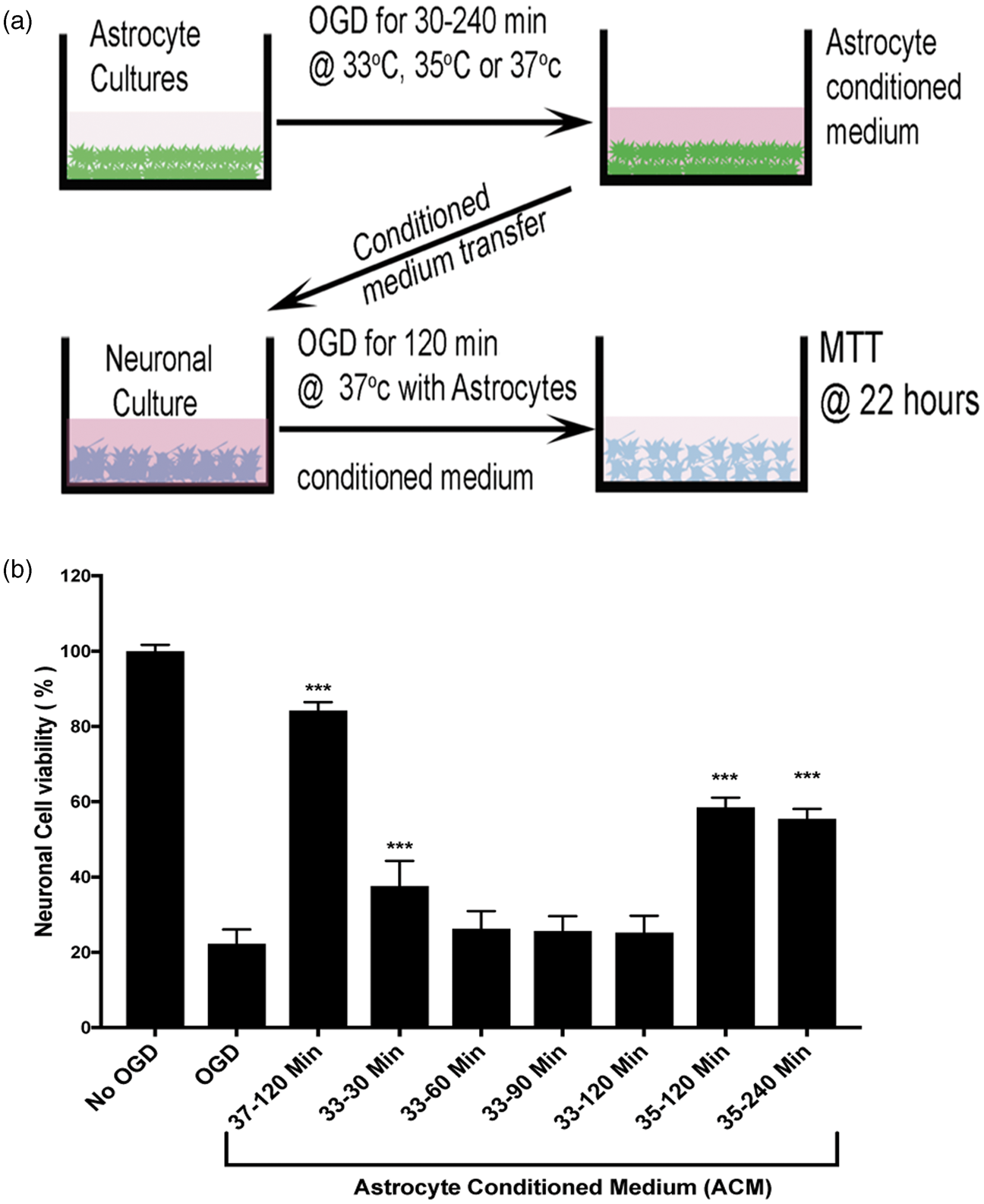
Discussion
TH has failed to translate into unequivocal clinical success, despite an extensive and robust pre-clinical literature. We asked whether this clinical translational failure could result in part from differential effects of TH on the individual elements of the NVU. We observed that short (2 h) was superior to longer duration (4 h) TH using a standard 4 h MCAo and commencing TH immediately upon reperfusion (Figure 1). Investigating the individual elements of the NVU, we found considerable differences in the ischemia tolerance of neurons, astrocytes and endothelial cells (Figure 2). We predicted a relationship between the delay time to TH onset and the required TH duration needed to protect cells, based on seminal work by Corbett and Colbourne in whole animal stroke models.41,42 Using monocellular cultures, we found a remarkably consistent relationship between TH delay and TH duration (Figures 3 and 4): the longer the delay to TH onset, the longer the TH duration needed to salvage neurons and astrocytes; this finding recapitulates prior results in stroke models.43–45 We next asked whether TH interfered with the known paracrine protective effect of astrocytes on neurons.32,38,40,46,47 We found that TH abrogated the neuroprotective effect of OGD astrocyte-conditioned media (Figure 5(a) and (b)) and we related this to TH inhibiting astrocyte activation (Figure 6). These data strongly suggest that one reason for the clinical translational failure of TH could be the interference with astrocyte paracrine neuroprotective effects.
The effect of TH on astrocytes has rarely been studied, but recently, increasing attention in the neuroscience community has focused on the protective role of the astrocytes.38,48,49 We postulate that standard cooling/warming schedules may have failed in human clinical trials due to unappreciated aspects of cerebral pathophysiology, notably the interaction between neurons and astrocytes and maintenance of homeostasis (energy metabolism, neurotransmitter release and signal transmission) during physiological and pathological events in the brain. Considerable data have emerged to show that different elements of the NVU behave quite differently during injury: we and others have shown that astrocytes tolerate substrate deprivation (removal of oxygen and glucose) longer than neurons. 50 Protective effects of astrocytes have been documented by many other investigations,22,32,40,48,51–56 but the mechanism is not yet fully established. We showed that astrocyte-mediated protection of neurons can be blocked by incubating the astrocytes at 33℃ for prolonged periods, whereas incubation at 35℃ does not alter the protective astrocyte paracrine effect as much (Figure 5). Thus, in the human stroke or cardiac arrest patient, single-target hypothermia for a fixed duration could inhibit neuroprotective activity in the NVU. The net effect, despite robust hypothermic protection of neurons, could be unfavorable, due to inhibited NVU-mediated protective responses. If this finding is replicated by others, the implications for clinical application of TH are profound.
Another critical finding here is the superiority of a deeper target temperature across all cell types in the NVU (Figures 3 and 4) Considerable evidence supports the conclusion that deeper TH provides superior protection. A meta-analysis of animal stroke studies suggested superiority of deeper target temperatures.4,5 In global cerebral ischemia, e.g. after sudden cardiac arrest, less data are available, and graded TH seldom has been studied. 17 Our data confirm, across a range of conditions, that 33℃ provided superior protection compared to 35℃. The neurons are the most critical element of the NVU, but they are least resistant to injury. Our data suggest—strongly—that neuronal protection increases with deeper TH (Figure 3). Our finding that deeper levels of TH provided significantly greater cytoprotection of astrocytes (Figure 4(a) to (d)) are somewhat unexpected and quite remarkable, as the astrocytes are traditionally viewed as robust glial cells that resist injury. Failure to protect astrocytes during reperfusion might be fatal to dependent neurons, and our data suggest that 33℃ should be preferred over 35℃ to protect astrocytes. Another novel and highly clinically impactful finding is that for astrocytes, the interaction between treatment delay and duration was not as obvious as for neurons (Figure 4(c)). The explanation for this is not clear but could be an artifact arising from the very long OGD time (10 h) needed to reduce cell viability to 20%. On the other hand, there may be critical differences between astrocyte and neuronal responses to ischemia and TH that require further study.
Endothelial cells responded to OGD and TH in this study quite differently than the neurons and the astrocytes. The brain is protected from circulating factors—toxins, immune cells, antibodies—by the BBB that consists of the endothelial cells, pericytes, astrocytic foot processes, and a basement membrane.49,57 Mild to moderate reperfusion injury in the brain can lead to a number of phenomena, most importantly the swelling of endothelial cells and pericytes that can occlude (“choke”) the distal microcirculatory bed. 58 After severe reperfusion injury, the BBB fails when endothelial cells separate, or porate, or give way to uncontrolled transcytosis. 59 Many survivors of sudden cardiac arrest develop uncontrolled cerebral edema 24 to 48 h after return of spontaneous circulation (ROSC), likely due to gross failure of the BBB. Our data suggest that deeper TH provides superior cytoprotection to the endothelial cells across all delay times and duration times (Figure 4(g)). We did observe a confounding effect of TH in the metabolism assay (Figure 4(e)) consistent with a proliferative response. Although we did not study the pericytes, it is generally accepted that BBB failure depends mainly on endothelial cells.49,57,60–62 Therefore, our data imply that during the reperfusion period after ROSC, deeper hypothermia should be preferred over shallower levels.
A novel and potentially critical finding here is that all cell types in the NVU exhibit the same depth–delay–duration relationship, but on very different time scales. As shown for neurons, delaying treatment for 90 min after restoration of substrate eliminated benefit unless TH was continued for at least 6 h (Figure 3). In astrocytes, 2-h treatment at 33℃ provided some benefit, but at 35℃ only 6 or 24 h provided benefit. As noted, each cell type in the NVU provides different functions, but all three are necessary for recovery of brain function. For endothelial cells, delay time and duration of TH seem less interconnected, although at the longest delay time, 90 min, it did appear that longer durations (24 h) of TH overcame the deleterious effect of the delay (Figure 4(e)). Future clinical trials should incorporate some recognition of the delay–duration relationship and consider titrating the duration of TH based on the delay time to ROSC. The elements of the NVU, however, differ significantly in their resistance to ischemia. If TH is applied to typical survivors of out-of-hospital sudden cardiac arrest, then ROSC will occur within 20 to 40 min of ischemia onset. 17 In this brief time-frame, while it is likely neurons will have suffered some injury, the extent of injury suffered by the astrocytes or endothelial cells will be unknown. Future studies should examine the effects of short duration ischemia on the protective responses of astrocytes, and the barrier function of endothelial cells.
Our data provide a comprehensive validation of the depth–delay–duration relationship at the cellular level: colder is superior and the longer the delay to restoration of substrate, the longer duration of TH required to provide benefit.41,43,63 Seminal publications from Corbett and Colbourne et al. and subsequent studies all show that the duration of TH should be adjusted based on the delay time from injury to therapy initiation, but this is never done in clinical practice.41–43,45,64,65 Our data show that all brain cell types exhibit the same delay–duration relationship: the longer the delay to treatment, the longer duration cooling is needed to show benefit. We showed that cooling to a target-depth of 33℃ was superior to 35℃. In contrast, in all clinical trials, TH has been administered at one target temperature (e.g. 33℃) for a fixed duration (e.g. 24 h), followed by a gradual “ramp” re-warm to normothermia. In several clinical trials published to date (stroke, head trauma), TH has failed to show benefits commensurate with the benefit predicted from animal studies, perhaps due to failure to optimize the administration of TH so as to preserve the protective NVU effect on neurons.
Given the need to titrate duration/depth based on delay and given the differential effects of TH on different cell types, we propose that TH should be carefully optimized. We propose an alternative, highly novel approach to TH, illustrated in Figure 7. The principle assumptions of the method are:
To optimally protect ischemic brain, the duration of hypothermia should be a function of the delay time to restoration of blood flow (ROSC after cardiac arrest or recanalization after stroke). The duration of TH should also be a function of the delay to TH initiation. Time spent at the deepest target temperature should be limited to the briefest possible.
Proposed method for optimized therapeutic hypothermia. The critical time epochs driving TH include the time from onset of ischemia to the time of reperfusion (recanalization after stroke or ROSC after cardiac arrest), labelled T0; and the time from reperfusion to the onset of TH, labelled TR. We propose dividing TH into segments, starting with T1, the initial, deepest target temperature. After an optimum duration of TH at the first target temperature, at time C1, the target temperature changes, perhaps to an intermediate temperature target, or perhaps to normothermia, for another epoch labeled T2. Another change could occur at C2, followed by a third TH epoch, T3. In selected patients who develop cerebral edema, related to endothelial barrier disruption, at time C3 another target temperature change occurs and cooling for cerebral edema begins in epoch T4. Finally, at C4, final warming to normothermia occurs; this could be a ramp rewarm if indicated. The dashed red lines represent the actual transitions that may require time to move from one core target temperature to another.
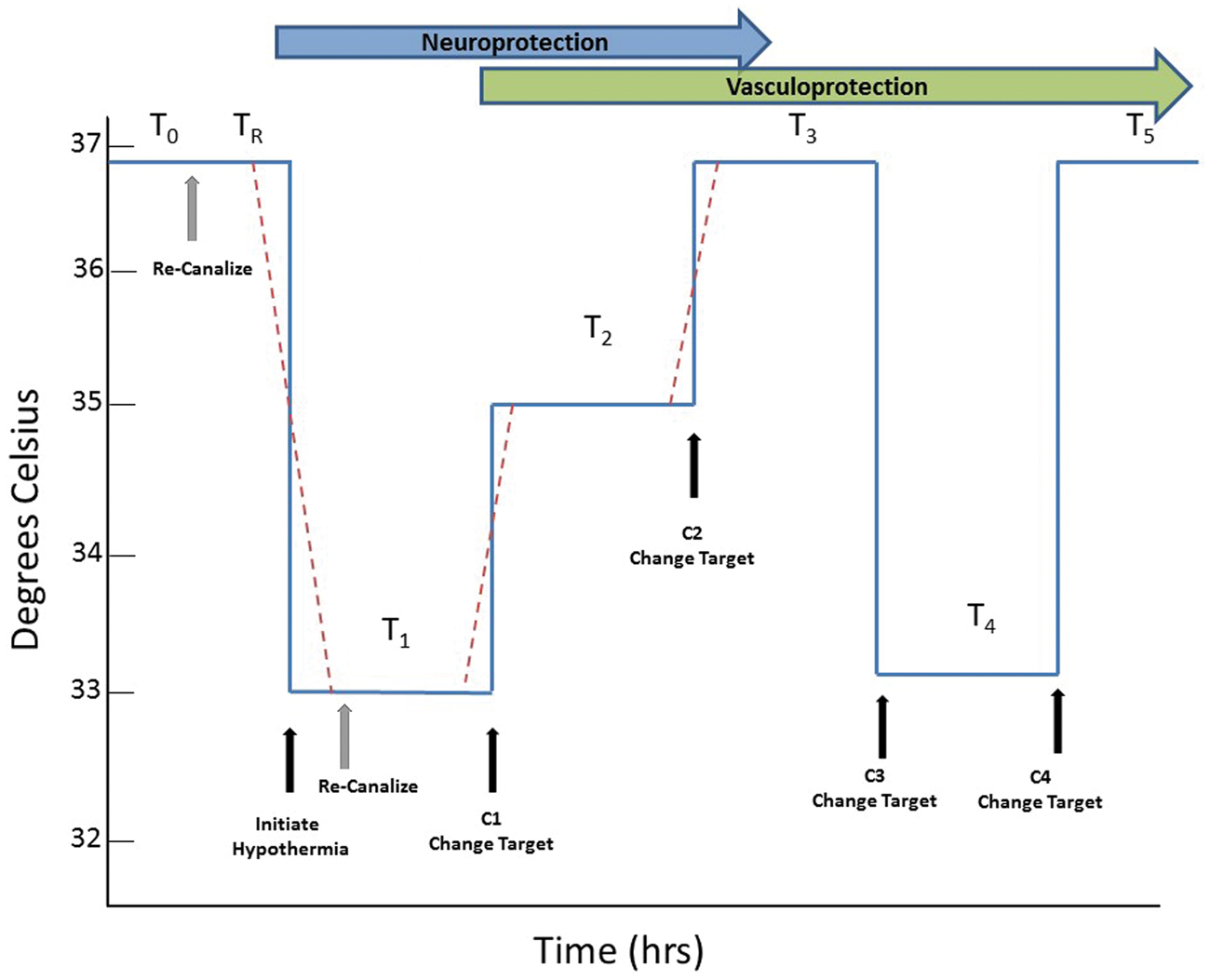
The total duration of TH should be calculated based on the duration of ischemia and delay time to TH onset. Time spent at the deepest temperature should be limited to avoid collateral complications of TH (pneumonia, other infections) but also to avoid inhibiting NVU protective effects. Future studies should seek ways to calculate needed TH duration as a function of delay time, perhaps using a formula to estimate TH duration, or preferably, biomarkers can be found to signal the appropriate transitions to the next temperatures (Figure 7).
There are several limitations of this study. We studied elements of the NVU as monocellular cultures, which eliminate cell–cell interactions during injury. Many of the findings reported here could appear differently when NVU elements are cultured together. In both the MCAo model and in cell culture, there are no co-morbid conditions as found in elderly stroke victims, such as diabetes or the long-term consequences of hypertension. These co-morbid conditions could significantly impact the results. We followed RIGOR and ARRIVE and other standards for quality laboratory modeling of stroke, but our results should be replicated in other laboratories. Our data do not address the effects of re-warming and it is well known that rapid rewarming can lead to rebound cerebral edema.66,67 Truly optimized TH will require considerable further work to determine how to re-warm patients. Given the data presented here, we speculate that a rapid, deep TH period targeting neurons could be followed by an interval return to normothermia to spare astrocytes, followed in selected cases by prolonged TH to control late-developing edema (Figure 7).
In conclusion, after observing short (2 h) duration pre- or immediate post-reperfusion TH to be superior to longer (4 h) duration TH, we showed differential vulnerability of neurons, astrocytes and endothelial cells to OGD in monocellular culture. During OGD, deeper (33℃) TH was superior to mild (35℃) TH, regardless of the delay time to TH or duration of TH. Longer delays to TH onset required longer durations of TH to achieve cytoprotection. In a temperature-dependent manner, TH interferes with paracrine neuroprotective effects of astrocytes during OGD. These data suggest a novel, optimized approach to TH that may be tested in future experiments.
Supplemental Material
Supplemental material for Differential effects of hypothermia on neurovascular unit determine protective or toxic results: Toward optimized therapeutic hypothermia
Supplemental Material for Differential effects of hypothermia on neurovascular unit determine protective or toxic results: Toward optimized therapeutic hypothermia by Patrick D Lyden, Jessica Lamb, Shweta Kothari, Shahed Toossi, Paul Boitano and Padmesh S Rajput in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NINDS R01 NS075930, the Lippman Family Foundation, and the Louis and Carmen Warschaw Chair in Neurology Research.
Acknowledgments
The authors acknowledge the technical assistance of Jilin Bai, MD, PhD. Preliminary results were presented at the American Heart Association Scientific Sessions, November 17, 2014, Chicago.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
All authors contributed to the drafting of the manuscript and approve the version submitted. The experiments were conceived and designed by PL and PR, data were acquired and analyzed by JL, SK, PB, and ST. Funding was arranged by PL. Statistical analysis was performed by PL and PR.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.