
Abstract
An astrocytic protein S-100β enhances the expression of inducible nitric oxide synthase in cultured astrocytes at micromolar concentrations, leading to nitric oxide-mediated death of cocultured neurons. The present study examined whether S-100β production by reactive astrocytes accumulating within the periinfarct area was related to delayed expansion of infarct volume after permanent middle cerebral artery occlusion in the rat. After rapid increases during the initial 24 hours, the increase of infarct volume then decelerated while maintaining the increasing tendency until 168 hours in this model, attaining a significant difference compared with that at 24 hours. In the periinfarct area, the number of reactive astrocytes expressing both S-100 and glial fibrillary acidic protein, the tissue level of S-100β as measured by the sandwich enzyme-linked immunosolvent assay method using anti-S-100β monoclonal antibody, and the number of terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling-positive cells were significantly increased preceding the delayed expansion of infarct volume. The CSF concentration of S-100β showed a biphasic increase, presumably reflecting the immediate release from astrocytes within the ischemic core and the subsequent production in reactive astrocytes within the periinfarct area. These results show for the first time that the enhanced synthesis of S-100β by reactive astrocytes participates in the inflammatory responses within the periinfarct area, which may be related to the occurrence of delayed infarct expansion as a major component of the cytokine network.
It is well known that the rapid increase in infarct volume during the initial 24 hours after focal ischemia is accompanied by apoptotic cell death in the periinfarct area (Charriaut-Marlangue et al., 1996; Li et al., 1995, 1998), the cause of which has been attributed to events such as spreading depression-like depolarization (Hossmann, 1994) and microcirculatory derangement (Garcia et al., 1994a; Zhang et al., 1994). However, the expansion of infarct volume continues after 24 hours (Baird et al., 1997; Beaulieu et al., 1999; Garcia et al., 1993,1995; Li et al., 1998; Nagayama et al., 1998). The pathomechanism underlying such a delayed infarct expansion has largely remained unclear, though leukocyte infiltration (Garcia et al., 1994b; Iadecola et al., 1995b) and expression of inflammatory genes (Barone et al., 1999) may be involved. In the present study, we attempted to correlate the occurrence of delayed infarct expansion and the enhanced production of S-100β in reactive astrocytes within the periinfarct area, using the permanent middle cerebral artery occlusion (pMCAO) model in the rat.
Astrocytic activation after cerebral ischemia has been well documented (Kraig et al., 1995; Petito, 1990). Although astrocytosis is associated with neuronal necrosis after global ischemia (Deleo et al., 1987; Petito et al., 1990; Rischke, 1991), significant increases in glial fibrillary acidic protein (GFAP) staining are present in regions exhibiting intact neurons after transient focal ischemia (Chen et al., 1993). It is an important finding that increased expression of astrocytic GFAP has been noted at the interface between the lesion and the surrounding brain tissue during 6 to 48 hours after pMCAO in rats (Garcia et al., 1993). Astrocytic activation leads to expression of a large number of molecules besides GFAP (Eddleston and Mucke, 1993). Among them, cytokines such as tissue necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) have been the target of intense research (Barone et al., 1999; Davies et al., 1999; Sheng et al., 1996). However, there is paucity of published studies concerning the possible pathogenic role of astrocytic S-100β.
S-100 is a 20-kd Ca2+-binding protein composed of a and β subunits (S-100α and S-100β). S-100β is primarily expressed by astrocytes in the brain (Allore et al, 1990; Hu et al, 1996) and may play a dual role in the regulation of cell functions, being beneficial to cells at low doses and detrimental at high doses (Barger and Van Eldik, 1992; Hu et al, 1996). In a variety of pathologic conditions, such as ischemic brain damage (Aurell et al., 1991; Kim et al., 1996), cerebrospinal trauma (Hill et al., 1996; Hinkle et al., 1997), Down syndrome, and Alzheimer disease (Griffin et al., 1989; Sheng et al., 1996), accumulation of the protein in and around the lesion has been reported. In human stroke, a significant correlation between the plasma concentration of S-100 protein and the volume of cerebral infarct has been reported (Aurell et al., 1991; Büttner et al., 1997; Missler et al., 1997; Persson et al., 1987). Such an increased S-100β expression after acute glial activation has been deemed as one mechanism for the repair of injured neurons (Allore et al., 1990; Barger et al., 1995; Giulian D, 1993; Hu et al., 1997). Indeed, a low concentration (1 to 10 ng/mL) of S-100β protects cultured neurons from glutamate-induced and staurosporine-induced damage (Ahlemeyer et al., 2000), but a high concentration of the protein induces expression of inducible nitric oxide synthase (iNOS) in cultured astrocytes and subsequent production of nitric oxide leads to the death of astrocytes and cocultured neurons (Chao et al., 1996; Hu et al., 1996, 1997; Murphy, 2000). Although induction of iNOS in astrocytes has been well documented previously by exposure of cells in vitro to lipopolysaccharide or combinations of cytokines, such as IL-1β, interferon γ (IFγ), or TNF-α, these findings show that S-100β is a potent inducer of astrocytic iNOS activity. Hence, the role of astrocytic activation appears to be double edged: it may ameliorate or aggravate ischemic brain damage, depending on the magnitude of S-100β synthesis.
The expression of iNOS in the pMCAO model in rats reportedly starts at 12 hours and peaks at 2 days, which accounts for the delayed progression of ischemic brain damage (Iadecola et al., 1995a, b ; 1997; Nagayama et al., 1998). Because the timing of iNOS expression overlaps the period of GFAP expression as revealed in a similar stroke model (Garcia et al., 1993), it seems plausible that the enhancement in the astrocytic synthesis of S-100β leads to the expression of iNOS in the ischemic brain. In this regard, Loihl et al. (1999) recently reported that NOS-2 was expressed in cells infiltrating the infarct, whereas, at a later time, there was also expression in astrocytes around the infarct. This line of thinking impelled us to investigate the possible role of astrocytic synthesis of S-100β in the occurrence of delayed infarct expansion.
In the present study, we undertook time-coursed analysis on the alterations in (1) the infarct volume, (2) the number of terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling (TUNEL)-positive cells (TPCs) in the peri-infarct area, (3) the immunohistochemical staining for S-100 and GFAP in the periinfarct area, and (4) the CSF and tissue levels of S-100β after pMCAO in rats. The present study showed the temporal and topographic correlations between the enhanced syntheses of S-100β in the periinfarct reactive astrocytes and the occurrence of delayed infarct expansion. These results suggest that the enhanced synthesis of S-100β by reactive astrocytes participate in the inflammatory responses within the periinfarct area, and may be related to the occurrence of delayed infarct expansion and prolonged neurologic deficits.
MATERIALS AND METHODS
Surgical procedures
Animal housing and care and the present protocols complied with the Principle of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals (DHHS publication No. [NIH] 85-23, revised 1985) and were previously approved by the Animal Use Ethical Committee of the Saitama Medical Center/School. Male Sprague-Dawley rats (weighting 280 to 300 g; Japan SLC Co. Ltd., Shizuoka, Japan) were housed in a constant-room-temperature (23°C ± 2°C) environment with a relative humidity of 55% ± 10% with aeration (10 times/hour) under an alternating 12-hour cycle of fluorescent light and darkness (light: 08:00 to 20:00 hours). The animals were fasted overnight but were given free access to water before surgical procedures. They were anesthetized with halothane (2 to 3% for induction and 1 to 1.5% for maintenance) in a mixture of 70% nitrous oxide and 30% oxygen. The physiologic parameters (arterial Pa
Experiment 1: determination of infarct volume
A total of 54 rats were randomly allotted for death at 1, 3, 6, 9, 14, 24, 48, 72, 120, or 168 hours after pMCAO. The infarct volume at 336 hours after pMCAO was later examined using a separate group of rats (n = 8). According to the schedule, rats were reanesthetized with halothane and approximately 100 μL CSF was collected by the cisternal puncture technique for the assay of S-100β in CSF. The brain was transcardially perfused with 10% neutral-buffered formalin solution for histologic, histochemical, and immunohistochemical studies. At predetermined coronal planes, the fixed brain was divided into two pieces (cerebrum and cerebellum) and the cerebral piece was routinely embedded in paraffin. At 10 different positions, serial sections (5-μm thick) were cut at a site from the frontal to the occipital poles at 1-mm intervals. Sections obtained from each site were equally spaced and sequentially labeled as slices 1 to 10 before hematoxylin and eosin- and cresyl violet staining, respectively. The stained section close to the posterior surface of each brain slice was examined for the infarct volume using a computer-based image analyzer (MCID; Imaging Research, St. Catherine's, Ontario, Canada).
Histologic features used to identify the lesion, including vacuolation (sponginess) of the neuropil, diffuse pallor of the eosinophilic background, and alterations in shape and stainability of both neuronal perikarya and astrocytic nuclei, were performed according to methods proposed by Garcia et al. (1993). To exclude the effect of brain edema, the infarct area was corrected by dividing with the ratio of the whole area of affected hemisphere to that of the contralateral hemisphere (Mori et al., 1999). The infarct volume was obtained as the sum of corrected infarct areas in all 10 slices.
TUNEL
Separate sections obtained from the coronal brain slice at the level of the anterior commissure were used for TUNEL staining using the In Situ Cell Death Detection Kit (Boehringer Mannheim, Mannheim, Germany) (Ben-Sasson et al., 1995). Fluorescein-labeled DNA strand breaks were incubated with horseradish-conjugated antifluorescein antibody and visualized with diaminobenzidine. For negative controls, terminal deoxynucleotidyl transferase was omitted. In each rat, the whole ischemic hemisphere was divided into 130 microscopic fields (x132 magnification) and the number of TPCs in the ischemic hemisphere excluding the infarct area was counted.
Immunohistochemistry for S-100 and glial fibrillary acidic protein
The serial sections obtained from experiment 1 encompassing the anterior commissure were used. Double immunohistochemistry was performed according to the manufacturer's protocols using a Histostain DS Doublestaining Kit (Zymed Lab. Inc., South San Francisco, CA, U.S.A.) with diaminobenzidine and/or fast blue as a chromogen. The following antibodies were used: polyclonal antibodies of S-100 (diluted 1:3,000; DAKO, Carpinteria, CA, U.S.A.) and GFAP (diluted 1:500; DAKO). Counterstaining was performed with hematoxylin. Controls for the specificity of immunostaining included omission of a primary antibody.
Experiment 2: the assay for S-100β in the brain tissue and cerebrospinal fluid
The CSF samples were collected from the cisterna magna using a 26-G needle and general anesthesia. For the assay of S-100β in the brain tissue, rats were anesthetized with 35 mg/kg pentobarbital according to the time schedule, and exposed to microwaves (7.0 to 7.8 kW, 1.3 to 1.7 seconds; Muromachi-Kikai, Tokyo, Japan). From the isolated brain, the coronal brain slice encompassing the anterior commissure (2-mm thick) was dissected and divided into four predetermined subregions. Subregions 1, 2, 3, and 4 roughly corresponded to the remote cortical area, periinfarct area, infarct area, and the basal ganglia, respectively. The tissue sample was homogenized in 0.15% sodium dodecyl sulfate solution containing 2 μmol/L edetic acid and incubated overnight at 4°C. The homogenate was centrifuged at 100,000 g for 12 minutes and the supernatant was collected. The concentrations of S-100β in CSF and brain were determined according to the sandwich enzyme-linked immunosolvent assay method using anti-S-100β monoclonal antibody (diluted 1:1,000; Sigma, Clone: SH-B4, St. Louis, MO, U.S.A.) as a primary antibody and anti-S-100 polyclonal antibody (DAKO) as a secondary antibody (Missler and Wiesmann, 1995). Bovine S-100 (Calbiochem-Novabiochem, San Diego, CA, U.S.A.) was used as the standard. In brief, microtiter plates coated with 100 μL primary antibody at a dilution ratio of 1:1,000 with 0.1 mol/L bicarbonate buffer (pH 9.6) were incubated overnight at 4°C. After washing with 0.1 mol/L phosphate-buffered saline containing 0.1% Tween-20 and 2 mmol/L CaCl2 [TPBS (+)], the plates were incubated with the blocking reagent (Boehringer Mannheim) for blocking nonspecific binding before the plates were incubated overnight with 50 μL of 4 % bovine serum albumin solution (containing 10 mmol/L CaCl2 and 0.1 % Tween-20) and 50 μL of sample or standard at 4°C. Sample solutions were discarded by decantation, and the plates were washed with TPBS (+). Secondary antibody (100 μL) at a dilution ratio of 1:1,000 with 0.1 mol/L phosphate-buffered saline containing 2% bovine serum albumin and 2 μmol/L CaCl2 was added to the plate and further incubated at 23°C for 2 hours. After washing the plates with TPBS (+), the plate was incubated with horseradish peroxidase-conjugated antirabbit immunoglobulin G (Bio-Rad, Hercules, CA, U.S.A.) at 23°C for 2 hours. Finally, the plates were visualized by adding 100 μL peroxidase substrate kit (Bio-Rad) and the reaction was terminated with 100 μL 2% oxalic acid. The resulting absorbance was measured with the microplate spectrophotometer (SPECTRA-maxTM 250; Molecular Devices Co., Sunnyvale, CA, U.S.A.).
Statistical analysis
Data are presented as mean ± SD. The paired t-test and the one-way analysis of variance followed by the Dunnett test were used for statistical evaluation. A P value less than 0.05 was considered to indicate a statistically significant difference.
RESULTS
Occlusion of the MCA at the correct position and the absence of subarachnoid hemorrhage or subdural hematoma were confirmed in all rats when the brains were removed. There was no death before the end of experiment. The body temperature of animals remained stable before and after surgery. In all rats, the neurologic score at 1 hour was 3, which was maintained throughout the observation period of 168 hours. Despite neurologic deficits, all rats consumed food and water normally.
Alterations in the infarct volume
In the ischemic hemisphere, slight and patchy pallor in the caudoputamen and the cortex was discernible in the hematoxylin and eosin-stained specimen as early as 3 hours after pMCAO. Within the area of pallor, ischemic changes such as shrinkage, scalloping, and swelling of the neuronal perikarya were apparent by 12 hours (Garcia et al., 1993). This was followed by the appearance of red or ghost neurons that were more prominent in the caudatoputamen than in the cortex. Pannecrosis (i.e., irreversible cell injury involving neurons), glia, and cerebral microvessels became recognizable at 72 to 96 hours and peaked at 144 to 168 hours after pMCAO. Cavitation (area of reabsorption of the necrotic debris by macrophage) was first seen in the preoptic area and caudoputamen 5 to 6 days after pMCAO. Six to 7 days after pMCAO, a small area of cavitation was found at the corticomedullary junction. At 336 hours, cavitation became pronounced and was manifested ubiquitously in the whole area of infarct. The edema index, indicated as a ratio of the left hemisphere to the right, peaked at 48 to 72 hours after pMCAO (data not shown). The corrected total infarct volumes at 1, 3, 6, 9, 14, 24, 48, 72, 120, 168, and 336 hours after pMCAO were 5.7 ± 3.6 mm (n = 6), 4.5 ± 0.8 mm (n = 6), 8.6 ± 1.9 mm (n = 6), 28.8 ± 9.7 mm (n = 6), 55.7 ± 25.5 mm (n = 6), 69.7 ± 35.7 mm (n = 6), 80.7 ± 23.3 mm (n = 6), 84.9 ± 33.7 mm3 (n = 6), 98.8 ± 22.7 mm3 (n = 6), 112.1 ± 22.8 mm3 (n = 6), and 84.5 ± 14.5 mm3 (n = 8), in descending order (Fig. 1). The infarct expansion was most rapid during the first 14 hours, and was decelerated between 14 to 24 hours. From 24 to 168 hours, however, the infarct volume continued to increase gradually and became significant (an increase of approximately 41%, P < 0.01), whereas the edema index decreased by only 2.5% (24 hours, 1.134 ± 0.012; 168 hours, 1.106 ± 0.019). Analysis with the noncorrected infarct volume in terms of the edema index yielded a similar result, showing a significant difference between the volumes at 24 and 168 hours (data not shown). Thus, the increase in infarct volume occurring between 24 and 168 hours is considered to be a phenomenon independent of brain edema or artifact at the time of paraffin-slide preparation, and is hereafter designated as delayed infarct expansion, in contrast to the acute infarct expansion occurring between 0 and 24 hours. These results show that the delayed infarct expansion manifested similar extensions in the cortex and the basal ganglia without any preferential direction.
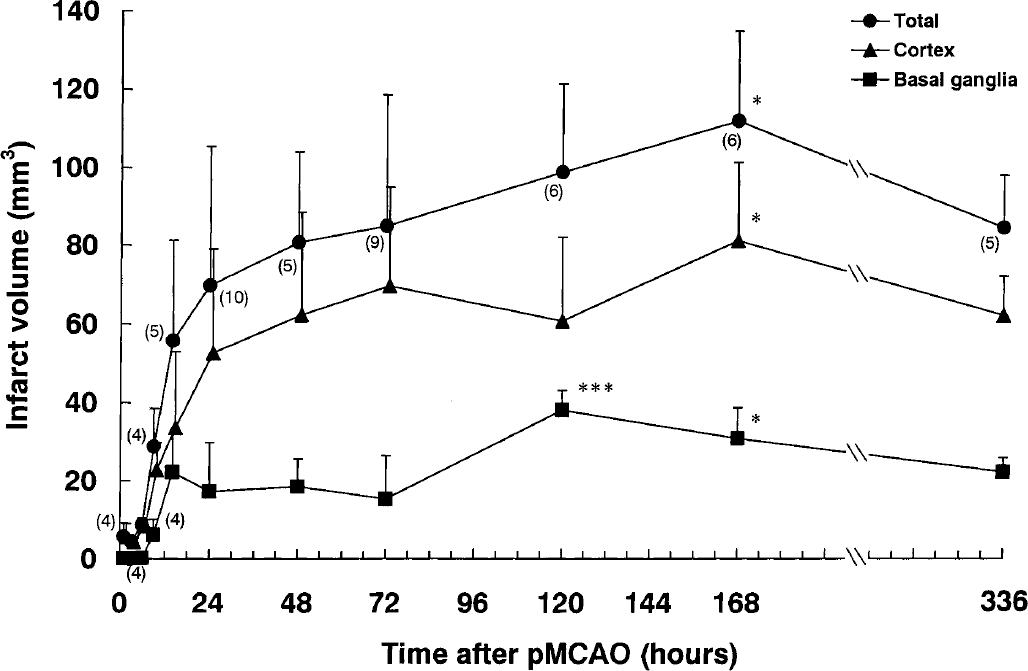
Alterations in the infarct area in the total, cortex, and basal ganglia after permanent middle cerebral artery occlusion (pMCAO). Each value represents the mean + 1 SD. Statistical analysis was performed by comparing the differences with the 24-hours group by one-way analysis of variance followed by the Dunnett test, where differences of P < 0.05 (*) and P < 0.001 (***) were considered significant.
The number of TUNEL-positive cells in the periinfarct area
At every time point examined, the TPCs were preferentially located along the infarct border, and there was an obvious tendency that they were numerically more within the infarct than outside. However, because the purpose of the present study was to examine the outward expansion of infarct, only the number of TPCs outside the infarct border was counted. Although TPCs were meager outside the infarct border by 24 hours, the number increased gradually to peak at 120 hours after pMCAO (P < 0.05 vs. 24 hours) (Figs. 2 and 3).
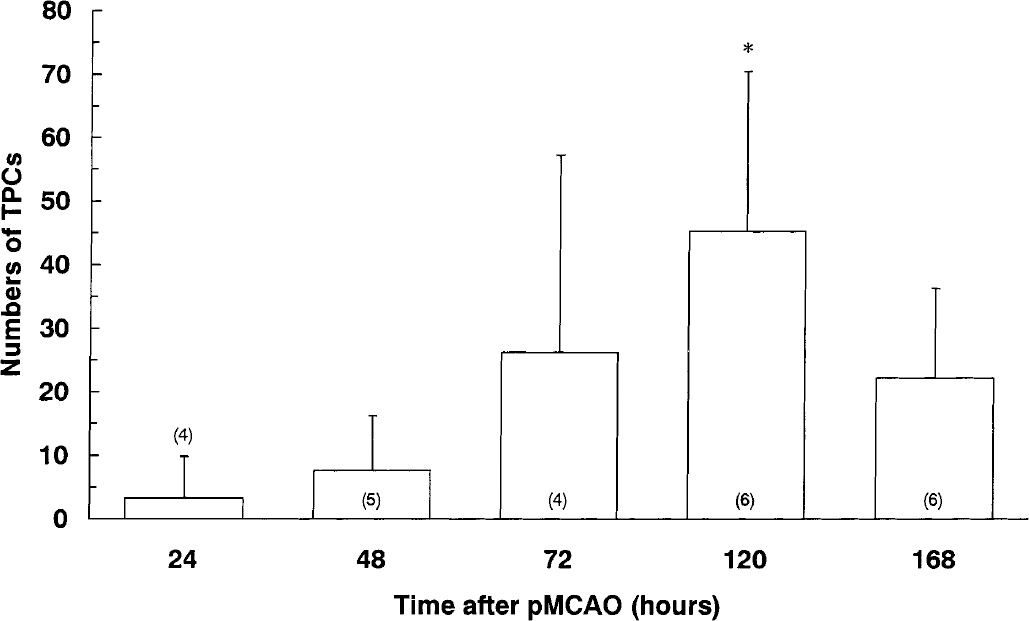
Alterations in the number of TUNEL-positive cells (TPCs) in the periinfarct area after permanent middle cerebral artery occlusion (pMCAO). Each value represents the mean + 1 SD. Statistical analysis was performed by comparing the differences with the 24-hours group by one-way analysis of variance followed by the Dunnett test, where differences of P < 0.05 (*) were considered significant.
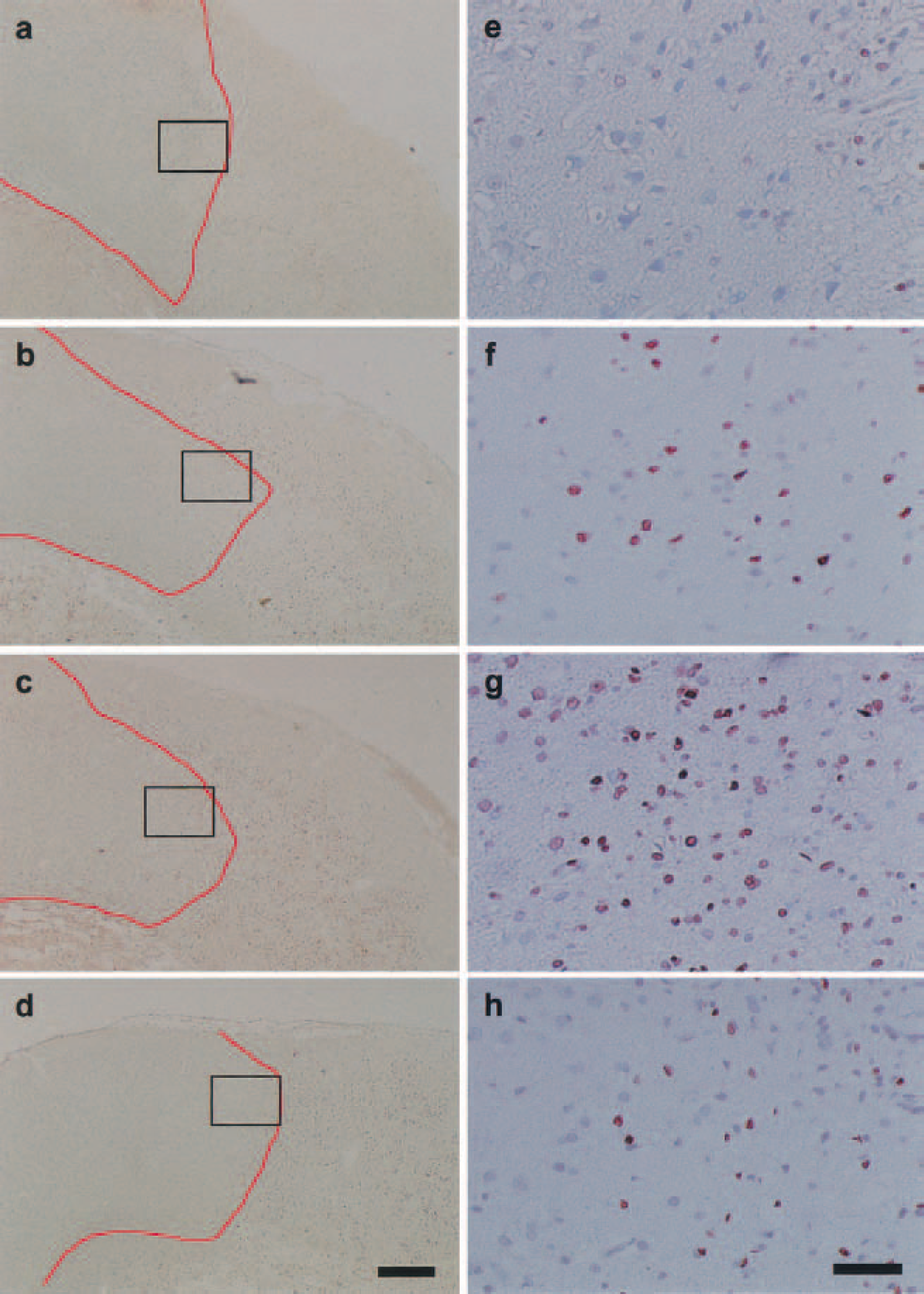
The representative features of TUNEL-positive cells (TPCs) in the periinfarct area after permanent middle cerebral artery occlusion (pMCAO). The photographs
Alterations in the immunohistochemistry of S-100 and glial fibrillary acidic protein
The astrocytic response to ischemia was always most remarkable in the periinfarct area, not within the ischemic focus, thus ac-counting for time-dependent alterations of the immunohistochemical distribution in the periinfarct (Figs. 4 and 5) and alterations in the S-100 immunohistochemistry (Figs. 4A–4H). In the normal control cortex (Fig. 4A), small astrocytes were visible, and a small amount of S-100–positive granules were present in their nuclei. Although cytoplasm was not apparent under the light microscope, numerous minute S-100–positive granules, which were probably within the astrocytic processes, were dispersed between neurons. At 3 hours after pMCAO (Fig. 4B), the ischemic region showed a spongy appearance due to pronounced swelling of the astrocytic cytoplasm and its processes. In this period, S-100–positive material was absent in both the nucleus and cytoplasm. At 24 hours (Fig. 4C), some astrocytic nuclei were strongly positive to S-100, whereas others showed the absence of chromatin. The astrocytic processes, though scanty in number, showed a foamy appearance and were faintly stained for S-100. At 48 hours (Fig. 4D), the stainability for S-100 was further increased in the foamy astrocytic processes. Astrocytic nuclei were filled with hematoxylin, and some reacted positive for S-100. At 72 hours, both the astrocytic nuclei (Fig. 4E) and processes (Fig. 4F) reacted strongly positive for S-100. At 96 hours (Fig. 4G), the astrocytic processes were swollen and strongly positive for S-100; some astrocytes were in the division process. At 168 hours (Fig. 4H), the stainability for S-100 was considerably diminished. Alterations in the GFAP staining are shown in Figs. 5A to 5F. GFAP immunoreactivity in astrocytes as shown in the normal control (Fig. 5A) was unchanged at 24 hours (Fig. 5B) and slightly enhanced at 48 hours, particularly in the swollen astrocytic processes (Fig. 5C). At 72 and 96 hours (Figs. 5D and E), there were numerous reactive astrocytes strongly positive for GFAP. At 168 hours, the stainability for GFAP was still maintained although the number of reactive astrocyte was markedly decreased.
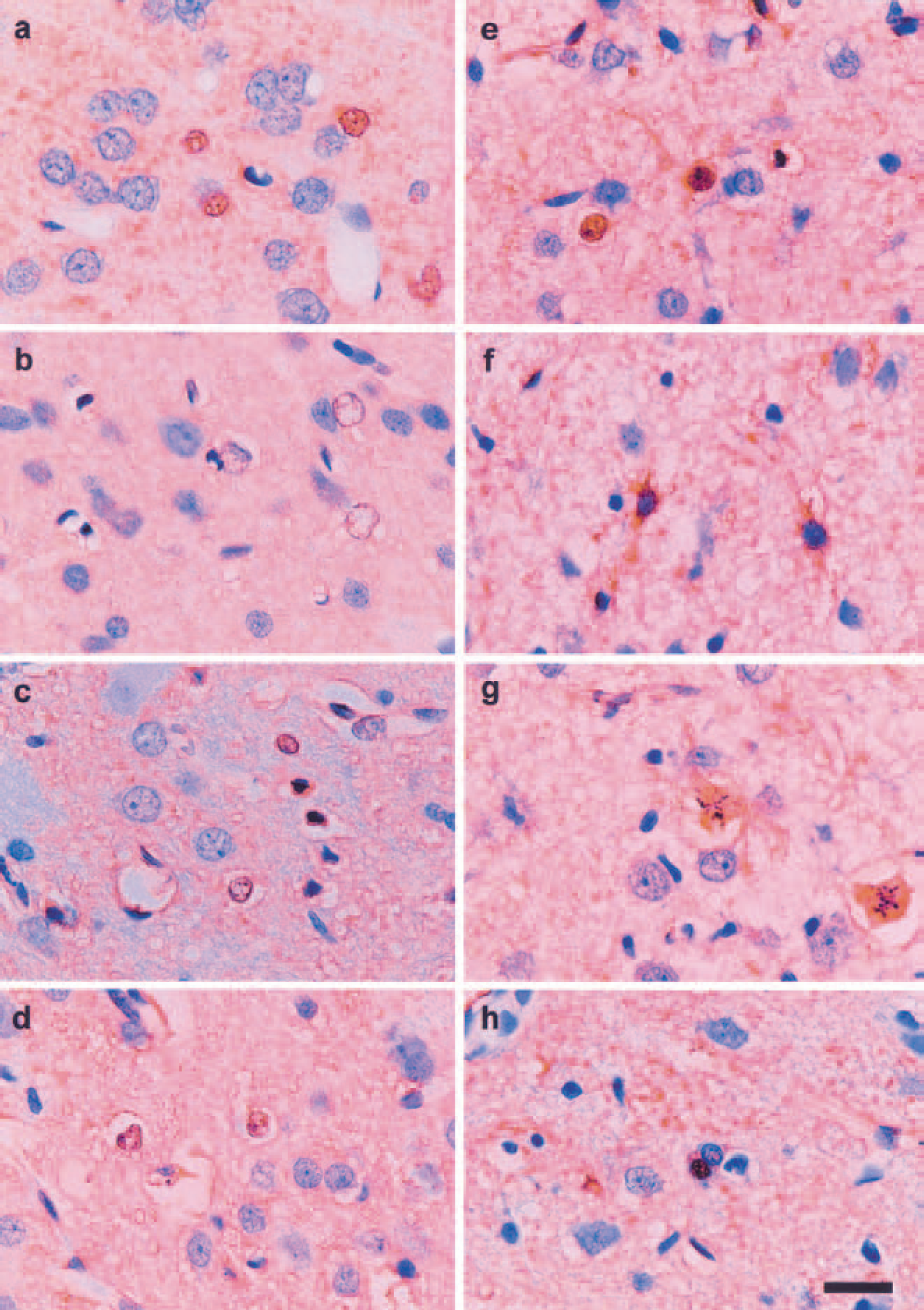
The representative features of S-100–positive astrocytes in the periinfarct area. Each photograph shows the immunohistochemical features of S-100–positive astrocytes in the periinfarct area. In the normal control cortex
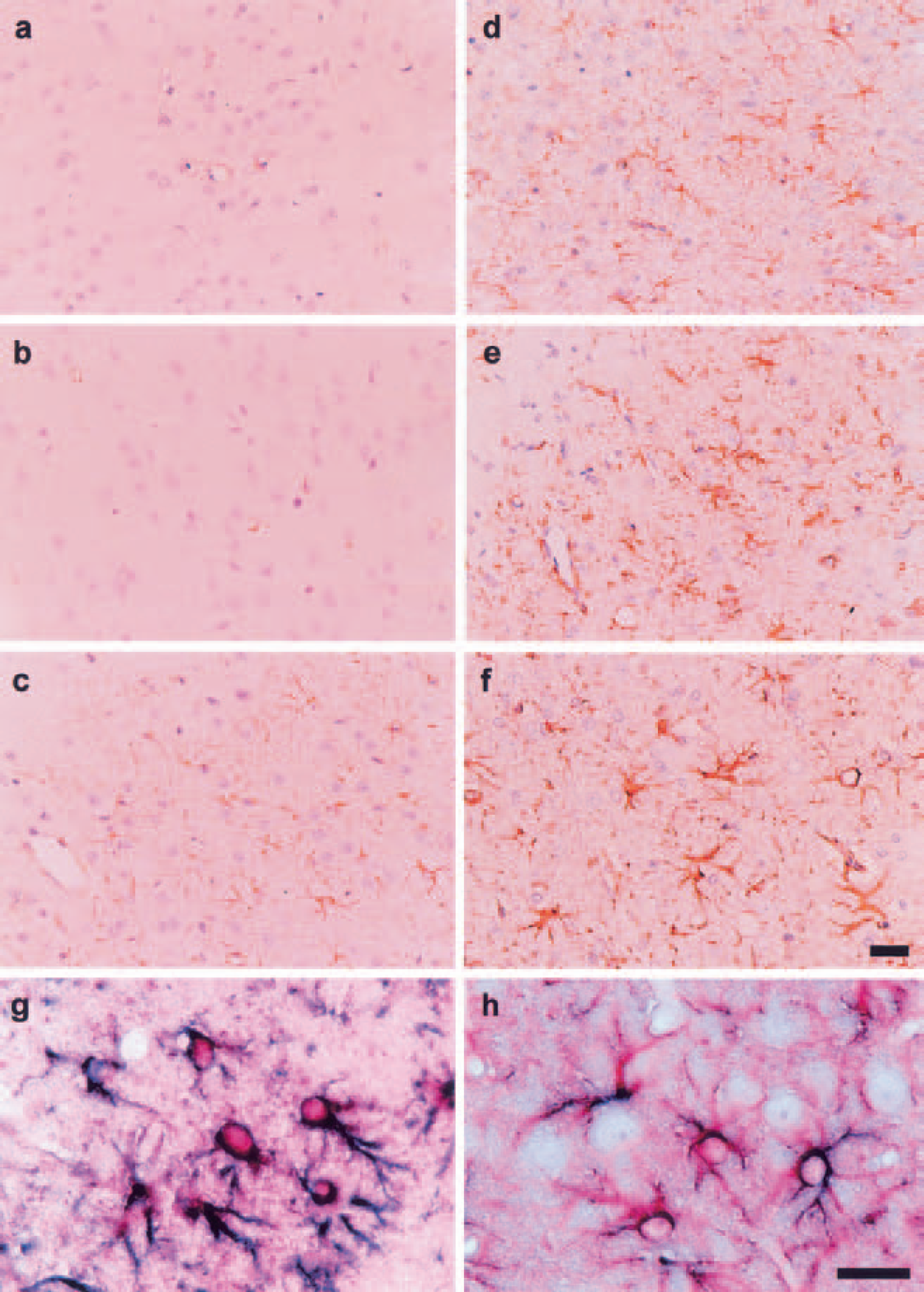
Reactive astrocytes in the periinfarct area. Each photograph shows the immunohistochemical features of glial fibrillary acidic protein (GFAP)-positive astrocytes
These results show that S-100 normally expressed in astrocytes was dispelled from both the periinfarct and infarct areas for a period of nearly 24 hours after ischemia. At approximately 24 hours, the astrocytes in the periinfarct area became strongly positive for S-100, which was maintained during the succeeding several days. At 168 hours, the stainability for S-100 was reduced. The expression of GFAP in the periinfarct area became noticeable only after 24 hours in this model, hence its expression was preceded by that of S-100. Double immunohistochemistry confirmed that S-100 and GFAP were colocalized in the reactive astrocytes after 48 hours (Figs. 5G and 5H).
Time courses of S100β levels in the brain tissue and cerebrospinal fluid
Based on the alterations in the tissue level of S-100β in the four brain regions at 1, 6, 9, 14, 24, 48, 72, 120, and 168 hours after pMCAO (Fig. 6), the S-100β level in the remote area (region 1) tended to be decreased throughout the experiment, whereas that in the infarct area (region 3) immediately decreased after pMCAO and never recovered thereafter. In the periinfarct area (region 2), the S-100β level conspicuously increased at approximately 12 hours and peaked at 24 hours (P < 0.05) before declining gradually. The wide standard deviations of S-100β level in this region are thought to be largely because this region encompassed the infarct area to a variable degree in rats. Consequently, the conspicuous rise of S-100β in the periinfarct area, which was previously disclosed by an immunohistochemical study, tended to be blurred in the net measurement. The same condition applied to the basal ganglia (region 4), which contained both the infarct and the periinfarct areas. Nonetheless, the S-100β level in this region significantly increased during the initial 48 hours only to declined thereafter.
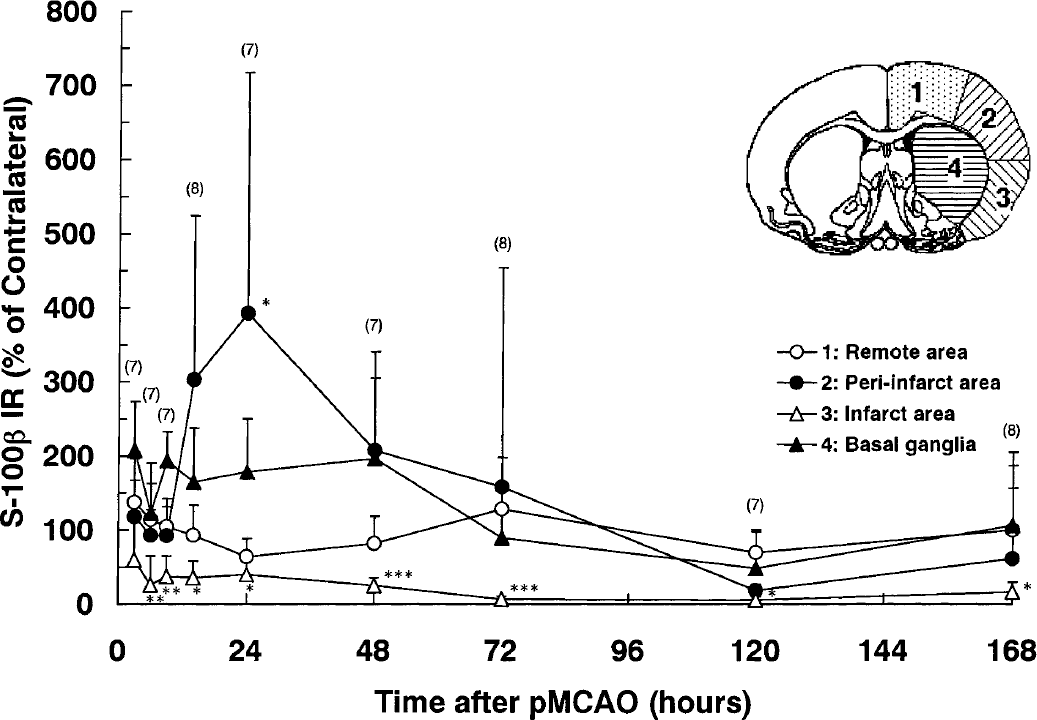
Alterations in the tissue level of S-100β in each brain region after permanent middle cerebral artery occlusion (pMCAO). Each value represents the ratio of the S-100β immunoreactivity (IR) level in one region of the ischemic hemisphere to that of contralateral hemisphere (mean + 1 SD). Statistical analysis where differences with P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were verified by the paired t-test.
The CSF concentration of S-100β (Fig. 7) in the normal control was 2.6 ± 1.0 ng/mL. Immediately after pMCAO, it increased to a small peak at 6 hours (21.0 ± 8.6 ng/mL), tended to decrease for the succeeding 6 hours, and then increased again to form a secondary peak at 14 hours (62.3 ± 36.0 ng/mL). Thereafter, the S-100β level in the CSF gradually decreased, though a relatively high level (> 30 ng/mL) was maintained until 72 hours. Seven days after pMCAO, S-100β eventually recovered to the baseline level.
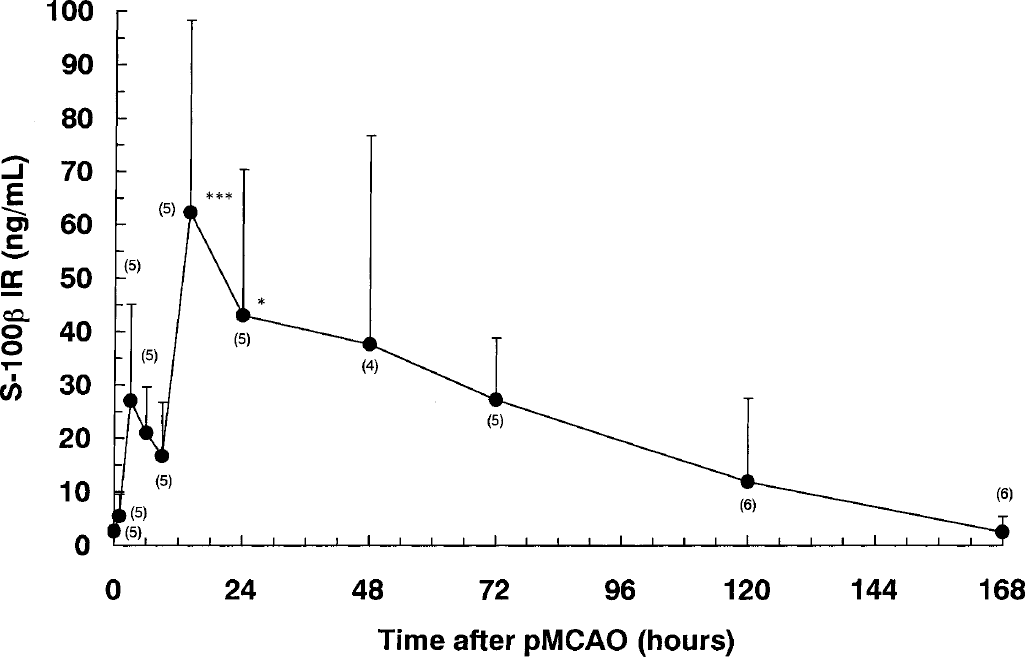
Alterations in the cerebrospinal fluid (CSF) level of S-100β after permanent middle cerebral artery occlusion (pMCAO). Each value represents the mean + 1 SD. The CSF concentration of S-100β immunoreactivity (IR) in the normal control was 2.6 ± 1.0 ng/mL. Statistical analysis was performed by comparing the differences with the normal control group by one-way analysis of variance followed by the Dunnett test, where differences of P < 0.05 (*) and P < 0.001 (***) were considered significant.
DISCUSSION
Delayed infarct expansion
Although studies have shown that the expansion of cerebral infarct after permanent focal ischemia is arrested by 12 to 24 hours (Garcia and Kamijyo, 1974; Kirino et al., 1988; Zhang et al., 1994), recent evidence obtained with animal models and in human stroke patients indicates that the infarct volume continues to increase for a much longer period (Asano et al., 1999; Baird et al., 1997; Beaulieu et al., 1999; Garcia et al., 1993; Iadecola et al., 1997). In the present study, the infarct volume slowly increased from 24 hours until as late as 168 hours, a trend that is strikingly similar to the mode of infarct expansion observed in cases of human stroke (Beaulieu et al., 1999). This discussion is focused on the issues apparently relevant to the pathogenic mechanism underlying delayed infarct expansion.
In a recirculation model in rats, histologic features with hematoxylin and eosin staining at 46 hours of ischemia were divided into three subregions within the damaged tissue: the ischemic core (diffuse pallor of the eosinophilic background), and the inner (obvious vacuolation or sponginess of the neuropil) and outer (between sponginess and entirely intact tissue) boundary zones of the lesions (Li et al., 1998). The inner boundary zone was difficult to delineate in the present study because obvious vacuolation or sponginess of the neuropil was always associated with pallor of the eosinophilic background. Therefore, we adopted the criteria of Garcia et al. (1993) for the delineation of infarct border because such a subtle difference in the histologic features likely stems from the absence of recirculation in our model.
Garcia et al. (1993) used Wistar rats and occluded the MCA by the intraluminal filament technique. Because this model is essentially one of internal carotid artery occlusion, it yielded a much larger infarct than did our model. Although the alterations in total infarct volume were not shown, the infarct area in the section encompassing the anterior commissure was significantly increased during the time interval from 6 to 72 hours. Similarly, our model revealed a statistically significant increase in the infarct volume from 24 to 168 hours. The considerable difference in the time course of delayed infarct expansion between these two models is probably due to the difference in the strain of rats, the method of middle cerebral artery occlusion (MCAO), the method of infarct area/volume measurement, and the severity of ensuing focal cerebral ischemia. Using the filament technique of MCAO leads to a long-term increase in core temperature of approximately 1.5°C (Zhao et al., 1994), which may have accelerated infarct evolution in Garcia's model.
More important, Beaulieu et al. (1999) have recently reported that, in human stroke, the delayed infarct expansion is dependent on whether the area of cerebral ischemia as defined by the initial perfusion-weighted magnetic resonance image is larger than that of the cerebral infarct area as defined by the diffusion-weighted magnetic resonance imaging. This finding is consistent with the view that the existence of wide ischemic penumbra is a prerequisite for the occurrence of delayed infarct expansion (Hossmann, 1994). Because occlusion of the MCA at its origin is bound to a relatively large area of ischemic penumbra, it is not surprising that delayed infarct expansion was most pronounced in our model. Collateral circulation to the periphery of ischemic core is thought to be relatively poor in the MCAO using an intraluminal filament in the internal carotid artery, not to mention further restriction of circulation in the basal ganglia.
TUNEL-positive cells in the periinfarct area
There is now ample evidence that apoptosis contributes significantly to cell death in focal ischemia (Lipton, 1999; MacManus and Linnik, 1997), and that the apoptotic cells present in the periinfarct area (boundary zones) contribute to expansion of the ischemic lesion (Charriaut-Merlangue et al., 1996; Li et al., 1995, 1998; Sharp et al., 2000). Although TUNEL is not selective for apoptosis (Portera-Cailliau et al., 1995), we adopted it in the present study as a convenient method to identify dying cells due to apoptosis within the periinfarct area (Ben-Sasson et al., 1995). In our model, TPCs became apparent in the periinfarct area at 24 hours, and steadily increased in number from 48 to 120 hours, and then declined (Fig. 3). The important fact is that the TPCs in the periinfarct area were increased in number only after 24 hours, being preferentially located adjacent to the infarct border. Therefore, it is certain that some unknown cell-damaging mechanism is in play at the interface between the infarct and the surrounding brain, leading to delayed infarct expansion. In this regard, periinfarct spreading depression plays a cardinal role in acute infarct expansion (Hossmann, 1994). However, the involvement of spreading depression in delayed infarct expansion seems unlikely, because it has been observed in animal stroke models only during the initial several hours.
Alterations in the S-100β levels in the brain tissue and cerebrospinal fluid
Results of immunohistochemical and the chemical assay studies indicate that focal ischemia induces remarkable alterations in the intraparenchymal distribution and the astrocytic synthesis of S-100β. During the initial several hours after the onset of pMCAO, S-100β was virtually absent in either the infarct or the periinfarct areas whereas its level in the CSF was rapidly increased. Together, the aforementioned events indicate that the ischemic insult led to an immediate release of S-100β stored in astrocytes into the extracellular space, inducing a rapid increase in the CSF level of S-100β. After reaching a small peak at 6 hours, the CSF level of S-100β declined once but rapidly increased again, reaching a secondary peak at 14 hours. It seems clear that the secondary increase in CSF S-100β is caused by the elevation of the tissue level in the periinfarct area due to vigorous synthesis of the protein in the reactive astrocytes.
The immunohistochemical findings clearly show that the astrocytic synthesis of S-100 was markedly augmented outside but not inside the infarct border. The existence of such a steep gradient in its concentration across the infarct border hindered exact measurement of the regional concentration of S-100β. In this regard, the peak value of CSF concentration of S-100β was approximately 60 ng/mL (5-nmol concentration), which is well beyond the concentration (1 to 10 ng/mL) at which S-100β exhibited neuroprotective action (Ahlemeyer et al., 2000). Because the concentrations of S-100β should be much higher in the periinfarct area than in the CSF, it is possible that the tissue concentration of S-100β attains micromolar ranges, albeit temporarily, in those loci of the periinfarct area where an intense staining of S-100 was present.
The possible mechanism underlying astrocytic activation after cerebral ischemia
Immunohistochemical and the chemical assay studies consistently show that reactive astrocytes in the periinfarct area started to vigorously synthesize S-100β at 12 hours. The time course of S-100 expression largely overlapped that of GFAP, and the two proteins were colocalized in the reactive astrocytes. S-100β is one of the proteins upregulated on astrocytic activation.
Although astrocytic activation is hallmarked by expression of GFAP, the precise underlying mechanism remains unclear. Besides environmental factors such as pH, K+, and glutamate, participation of spreading depression has hereby been suggested (Dietrich et al., 2000; Hossmann, 1994; Kawahara et al., 1999; Kraig et al., 1995; Kunkler and Kraig, 1997; Yamashita et al, 1996). Activated astrocytes show upregulation of a number of proteins that can stimulate nitric oxide generation, including IF-7, IL-1β, and S-100β (Eddeleston and Mucke, 1993). An interesting phenomenon is that S-100β in turn induces nitric oxide, lactate dehydrogenase release, and cell death without inducing morphologic alterations, events consistent with glial activation (Chao et al., 1996; Hu et al., 1996). This finding contrasts that regarding GFAP, which has recently been shown to be essential for astrocytes acting as a group to physically constrain certain types of inflammatory lesions, such as deposition of substrate-bound β-amyloid protein (Xu et al., 1999). Inasmuch as the precise pathway whereby astrocytic activation leads to an enhanced expression of S-100β remains obscure, there is mounting evidence that IL-1β is involved in the pathogenesis of ischemic brain damage (Barone and Feuerstein, 1999; Stroemer and Rothwell, 1998).
Davies et al. (1999) have shown that IL-1β–reactive putative microglia are present in the ischemic hemisphere as early as 6 hours after pMCAO. At 24 hours, these cells were far more numerous and intensely stained, being distributed along the margins of the infarct and in the adjacent penumbra. Regarding the in vivo action of IL-1β, however, there is some discrepancy in the reported results. Stroemer and Rothwell (1998) have reported that IL-1β alone is not neurotoxic in normal brain, and a major site of action of endogenous IL-1β in the pathology of ischemic brain damage is in the striatum. By contrast, Holmin and Mathiesen (2000) have recently reported that the intracerebral injection of IL-1β induces astroglial activation and apoptotic death of neurons. Apart from the possible toxic effects on neuronal cells, IL-1β bears particular importance in that it regulates S-100β expression in Down syndrome and Alzheimer disease (Griffin et al., 1989) and in cultured astrocytes (Hinkle et al., 1998). Intracerebral injection of IL-1β significantly increases the tissue level of S-100β and two β-amyloid precursor protein isoforms (Sheng et al., 1996). All of these findings raise an intriguing possibility that the first microglial and then astrocytic generation of IL-1β may at least partially participate in the enhanced astrocytic synthesis of S-100β in the periinfarct area.
The relevance of S-100β to delayed infarct expansion
The period when the astrocytic synthesis of S-100β was increased in the periinfarct area coincided with that of the third wave of inflammatory gene expression according to Barone et al. (1999). The appearance of TPCs after 24 hours and the increase in their number in the periinfarct area was most likely caused by the inflammatory reactions provoked by interactions among numerous toxic cytokines, nitric oxide, and reactive oxygen species (Barone et al., 1999; Sharp et al., 2000). Because the increase in the number of TPCs paralleled the delayed infarct expansion, the inflammatory reaction in the periinfarct area is considered the cardinal cause of delayed infarct expansion.
As to the possible role of S-100β in inflammatory response, several preceding reports show a pronounced toxicity of S-100β. In a variety of brain pathology, numerous proteins and/or cytokines act together to induce cell death (Barone and Feuerstein, 1999; Nogawa et al., 1998; Sharp et al., 2000; Xu et al., 1999), and S-100β plays a central role among those deleterious factors in some types of brain pathology (Sheng et al., 1996). In addition, we have recently observed that the protein stimulates the expression of TNF-α, cytokine-induced neutrophil chemoattractants-1, and cyclooxygenase-2 in cultured astrocytes at a concentration range similar to that which induces iNOS. Such an effect of the protein was markedly enhanced by the presence of a small amount of β-amyloid (unpublished data).
In conjunction with available evidence, results of the present study may be interpreted as indicating that the following events are causally related with delayed infarct expansion. Periinfarct spreading depression-like depolarization is generated by the ischemic core and spread to the peripheral zone (Nedergaard and Astrup, 1986). This periinfarct spreading depression, inducing a marked increase in the metabolic rate of the tissue in the face of low oxygen supply, was considered to be responsible not only for the expansion of the infarct but also for the widespread inhibition of protein synthesis that leads to periinfarct disseminated neuronal loss (Hossmann, 1994). However, because spreading depression elicited by mechanical stimulation or high K+ application to the cortical surface does not result in neuronal damage (Back et al., 1996; Nedergaard and Hansen, 1988; Obeidat et al., 2000), there may be additional mechanisms that augment the deleterious effects of periinfarct spreading depression. One such mechanism might be the activation of astrocytes in the periinfarct. Activated astrocytes synthesize a large amount of S-100β, a process augmented by cytokines liberated from activated microglia accumulating in the periinfarct area. In glial and neuronal cells, extracellular S-100β evokes increases in intracellular free calcium concentrations associated with hydrolysis of phosphoinositides, suggesting mobilization of calcium from intracellular stores (Barger and Van Eldik, 1992). Thus, extracellular S-100β, in the presence of other cytokines, would reinforce astrocytic activation, leading to a further increase in astrocytic production of the protein. Such a vicious circle would culminate in a salient rise in the extracellular concentration of S-100β, subsequent expression of iNOS and cyclooxygenase-2 in astrocytes, and eventual death of ambient neurons.
In conclusion, our results support a causal relation between astrocytic activation in the periinfarct area and the occurrence of delayed infarct expansion. Whether S-100β plays a central role among inflammatory cytokines deserves further investigation.