
Abstract
Inflammation is an essential component for glial scar formation. However, the upstream mediator(s) that triggers the process has not been identified. Previously, we showed that the expression of CD36, an inflammatory mediator, occurs in a subset of astcotyes in the peri-infarct area where the glial scar forms. This study investigates a role for CD36 in astrocyte activation and glial scar formation in stroke. We observed that the expression of CD36 and glial fibrillary acidic protein (GFAP) coincided in control and injured astrocytes and in the brain. Furthermore, GFAP expression was attenuated in CD36 small interfering RNA transfected astrocytes or in the brain of CD36 knockout (KO) mice, suggesting its involvement in GFAP expression. Using an
Introduction
In response to CNS (central nervous system) injury, astrocytes change their morphology, proliferate, and migrate to the injury sites to form a scar. These reactive astrocytes display hypertrophic morphology and express high levels of glial fibrillary acidic protein (GFAP), a major intermediate filament protein (Pekny and Nilsson, 2005). The formed glial scar isolates and protects the noninjured tissue from injury, inhibits the spread of inflammation, and regulates the extracellular milieu (Bush et al, 1999; Faulkner et al, 2004). However, the scar creates a physical barrier for neurite outgrowth and also produces redox reactants and inflammatory mediators that cause further tissue damage (Askalan et al, 2006; Bush et al, 1999; Silver and Miller, 2004; Sofroniew, 2005). Accordingly, strategies aimed at reducing scar formation have been suggested to overcome the physical barrier to promote axonal growth (Desclaux et al, 2009).
Sterile inflammation, an inflammatory response in the absence of infection, occurs in postischemic tissues (Bamboat et al, 2010a, 2010b). It is a rapid and coordinated process involving an array of cellular and molecular events that leads to glial scar formation. Literature suggests that inflammation is an essential component for the formation of glial scar. Several inflammatory factors including TGF-
CD36, a class B scavenger receptor, has been implicated in pathological conditions associated with inflammation including stroke, atherosclerosis, and Alzheimer's disease (Kim et al, 2008; Febbraio et al, 2000; El Khoury et al, 2003). The receptor is expressed in many different cell types such as microglia, microvascular endothelial cells, monocytes/macrophages, and platelets (Febbraio et al, 2001; Febbraio and Silverstein, 2007). Substantial evidence suggests a role for CD36 in stroke pathology associated with inflammation (Cho et al, 2005; Cho and Kim, 2009). Ischemic stroke produces several CD36 ligands including oxidized/modified low-density lipoprotein and thrombospondins (Kim et al, 2008; Lin et al, 2003; Qin et al, 2011). We previously reported that CD36 contributes to free radical generation in the postischemic brain and that its deficiency ameliorates stroke-induced inflammation and injury (Cho et al, 2005; Cho and Kim, 2009; Kim et al, 2008). In addition to CD36 function in stroke-induced inflammation, its expression also occurred in a subset of GFAP+ astrocytes in the peri-infarct area where the glial scar forms (Cho et al, 2005). The purpose of this study is to address a potential role for CD36 in astrocyte proliferation and glial scar formation in stroke. Using an astrocytic cell line and a mouse model of transient focal cerebral ischemia in wild-type (WT) and CD36 knockout (KO) mice, here we report that CD36 is a novel mediator for astrogliosis and scar formation in stroke.
Materials and methods
Cell Culture
Mouse astrocytic cell line C8-D1A was obtained from ATCC (CRL-2541, American Type Culture Collection, Manassas, VA, USA) and cultured in DMEM (Dulbecco's Modified Eagle's Medium; high glucose, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Mediatech, Manassas, VA, USA), 100 I.U. Penicillin and 100
Animals
The use of animals and performance of procedures were approved by the Institutional Animal Care and Use Committee of Weill Medical College of Cornell University. C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Breeding pairs of each WT and CD36 KO strain (seven times backcrossed with C57BL/6, 99.7% C57BL/6 background) were derived at the time of the heterozygote cross. From these breeding pairs, only male F1 generation were used for the study without further sister–brother mating. Breeding pairs were replaced every 6 to 9 months by another heterozygote cross. This strategy prevented the genetic drifting associated with the propagation of homozygote lines. The procedures for genotyping have been described previously (Febbraio et al, 1999, 2000).
Transient Middle Cerebral Artery Occlusion
The procedure for middle cerebral artery occlusion (MCAO) has been described previously (Cho et al, 2005; Kim et al, 2008; Qin et al, 2011). Briefly, 8- to 10-week-old male mice were anesthetized with a mixture of isoflurane/oxygen/nitrogen and a 6-0 Teflon-coated black monofilament surgical suture (Doccol, Redland, CA, USA) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the circle of Willis to obstruct the origin of the middle cerebral artery. The filament was left in place for either 30 or 45 minutes and then withdrawn. The cerebral blood flow in the center of the ischemic territory was monitored by Laser-Doppler flowmetry (Periflux System 5010; Perimed, Jarfalla, Sweden). Animals exhibiting reduced cerebral blood flow >80% during MCAO and restored cerebral blood flow 80% of baseline by 10 minutes following reperfusion were included in the study. The criteria resulted in reproducible infarcts involving both the cerebral cortex and the striatum.
Tissue Preparation for Infarct Volume and Biochemical Measurement
The brains were excised, frozen, and sectioned using a cryostat. Temporal CD36 and GFAP mRNA changes in the postischemic brain were initially performed to select time points to compare stroke-induced GFAP levels. For this temporal study, we collected an entire hemisphere from C57BL/6 mice. The rest of the samples were serially collected by an unbiased stereological sampling strategy. Infarct typically spans about 6 mm rostrocaudal, roughly from +2.8 to −3.8 mm bregma. To collect tissue to closely reflect the infarct area, the infarct region was cryosectioned for infarct volume measurement and slide immunohistochemistry (20
RNA Interference
C8-D1A cells were transfected with mouse CD36 Stealth RNAi small interfering RNA (siRNA) (Invitrogen) using lipofectamine RNAiMAX (Invitrogen) for 12 hours according to the manufacturer's instructions. In all, 20 nmol/L of siRNA (Oligo ID: MSS202775, MSS202776, and MSS202777, abbreviated to 75/76/77) were added either alone or in combination. For controls, an equivalent amount of nonspecific control siRNA (Stealth RNAi siRNA Negative Control Low GC, Invitrogen) was used. Since siRNA 75 was the most effective, this siRNA (sense, 5′-UAG CUU GGC CAA UAG GAC AAA UUC C-3′; antisense, 5′-GGA AUU UGU CCU AUU GGC CAA GCU A-3′) was used for the further experiments.
Real-Time Reverse Transcription PCR
The levels of mRNA were quantified with real-time quantitative reverse transcription PCR using fluorescent TaqMan technology. Total RNA was reverse transcribed using a QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). PCR primers specific for CD36, GFAP, MCP-1, IL-6, and internal controls 18s rRNA (for the RNA interference experiments) and
Western Blot Analysis
Protein was extracted from cultured astrocytes and brain tissues, and Western blot analysis was performed as described previously (Cho et al, 2005; Kim et al, 2008). Briefly, samples were homogenized in radioimmunoprecipitation assay buffer (for cells, Sigma-Aldrich, St Louis, MO, USA) or CelLytic MT Mammalian Tissue Lysis/Extraction Reagent (for tissues, Sigma-Aldrich) with freshly added protease inhibitor (Roche Diagnostics, Indianapolis, IN, USA). After incubating for 15 minutes on ice, the homogenate was centrifuged at 10,000 r.p.m. for 10 minutes at 4 °C. In all, 20
Cytotoxicity Assay
Cell viability was assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Invitrogen). Briefly, 48 hours after scratch, cells treated with control or CD36 Stealth RNAi siRNA were incubated in the cell culture medium containing MTT for 4 hours at 37 °C. After washing with phosphate-buffered saline, the cells were dissolved in the sodium dodecyl sulfate HCl solution for 16 hours at 37 °C. Absorbance was measured at 570 nm.
Degree of cell death was determined by TdT-mediated dUTP nick end labeling (TUNEL) staining (DeadEnd Colorimetric TUNEL system, Promega, Madison, WI, USA). Briefly, 24 and 48 hours after scratch, cells treated with control or CD36 Stealth RNAi siRNA, were fixed with 4% paraformaldehyde, treated with ethanol/acetic acid and triton, and incubated in an equilibration buffer as described in the kit. The TdT enzyme and nucleotide mix were then added at proportions specified in the kit. Nuclei were counterstained by methyl green.
Assessment of Cellular Proliferation
Immunohistochemistry of Ki-67 was performed 12 and 48 hours after scratch to investigate the effect of CD36 Stealth RNAi siRNA on cellular proliferation. For each culture, six areas along the scratched edge (about 250
Wound Healing Assay
To gauge the degree of wound healing, nonclosed gap area was measured at 0, 24, and 48 hours by using CytoSelect 24-Well Wound Healing Assay Kit (Cell Biolabs, San Diego, CA, USA) and Axiovision software (Zeiss). Briefly, 600
Immunohistochemistry
Following MCAO, the brains were excised, frozen, and sectioned at a thickness of 20
Data Analysis
Gene and protein levels in cultures were normalized by
Results
CD36 and Glial Fibrillary Acidic Protein Expression in Astrocytes Occurs in a Coordinated Manner
CD36 expression in numerous GFAP-positive (GFAP+) cells in peri-infarct area (Cho et al, 2005) led us to investigate a role for astrocytic CD36 in injury-induced scar formation. We first determined CD36 and GFAP expression in astrocyte cultures following scratch-induced injury. CD36 and GFAP mRNA levels were increased similarly in a time-dependent manner in C8-D1A astrocyte cells (Figure 1A). Similarly, the protein levels were significantly increased in C8-D1A cells 48 hours postscratch, reflecting the gene changes (Figure 1B). The results demonstrate that CD36 and GFAP expression occur in a coordinated manner in injured astrocytes.
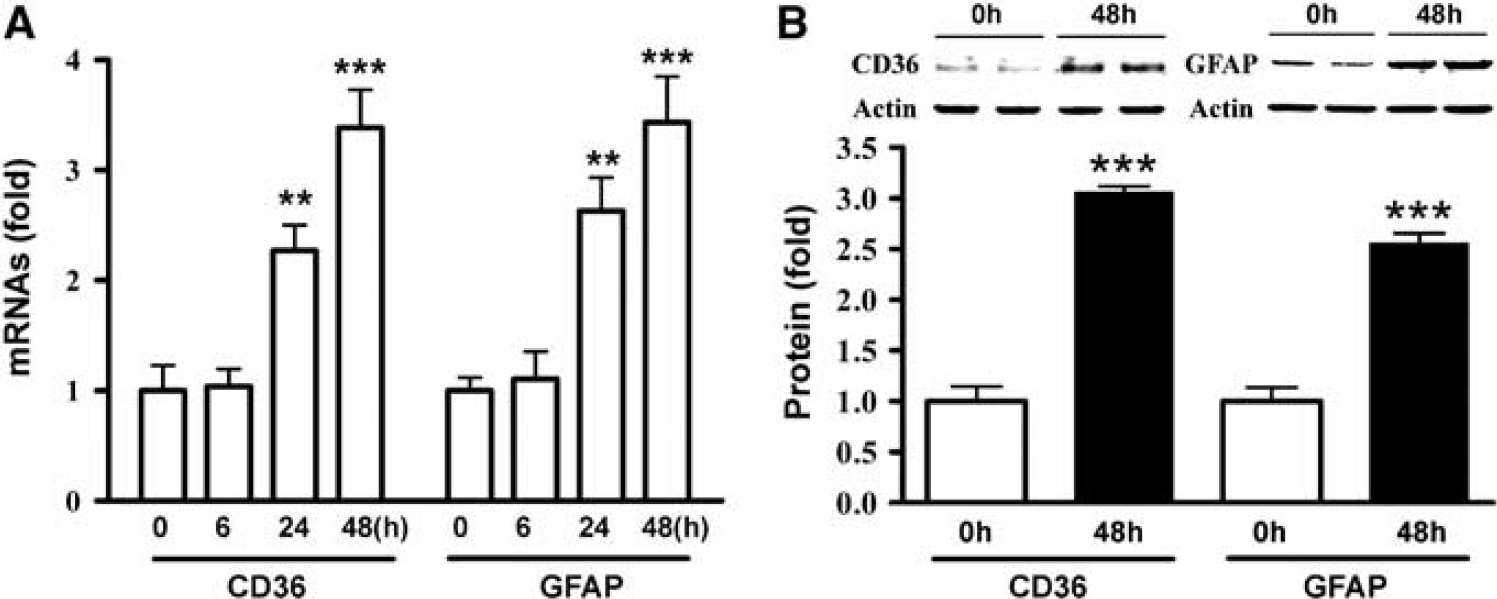
Coordinated expression of CD36 and glial fibrillary acidic protein (GFAP) expression in astrocytes. (
CD36 Absence Reduces Glial Fibrillary Acidic Protein Expression, Delays Wound Healing, and Reduces Inflammatory Factor Expression in Astrocytes
To investigate the requirement of CD36 for GFAP expression, GFAP mRNA levels were determined in CD36-silenced cultures. Transfection of CD36 siRNA 75, 76, and 77, either singly or in combination, significantly attenuated CD36 mRNA levels 48 hours postscratch when compared with their respective control (Figure 2A). There were corresponding reductions in GFAP mRNA levels in CD36 siRNA-treated cultures (Figure 2B), suggesting the necessity of CD36 for GFAP expression.
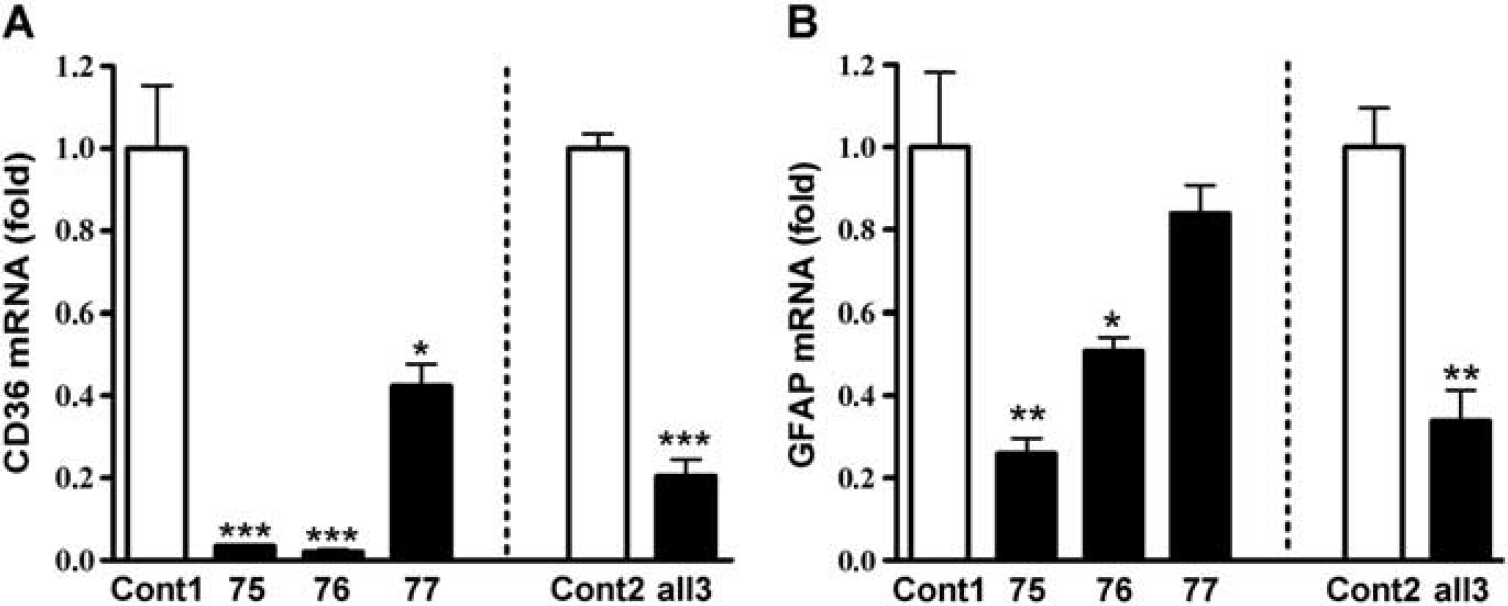
Reduced glial fibrillary acidic protein (GFAP) expression in CD36-silenced astrocytes. Effect of CD36 Stealth RNAi small interfering RNA (siRNA) on fold changes in CD36 (
To address functional significance of CD36 expression, effect of CD36 knockdown on cell viability and also on the extent of wound healing using an
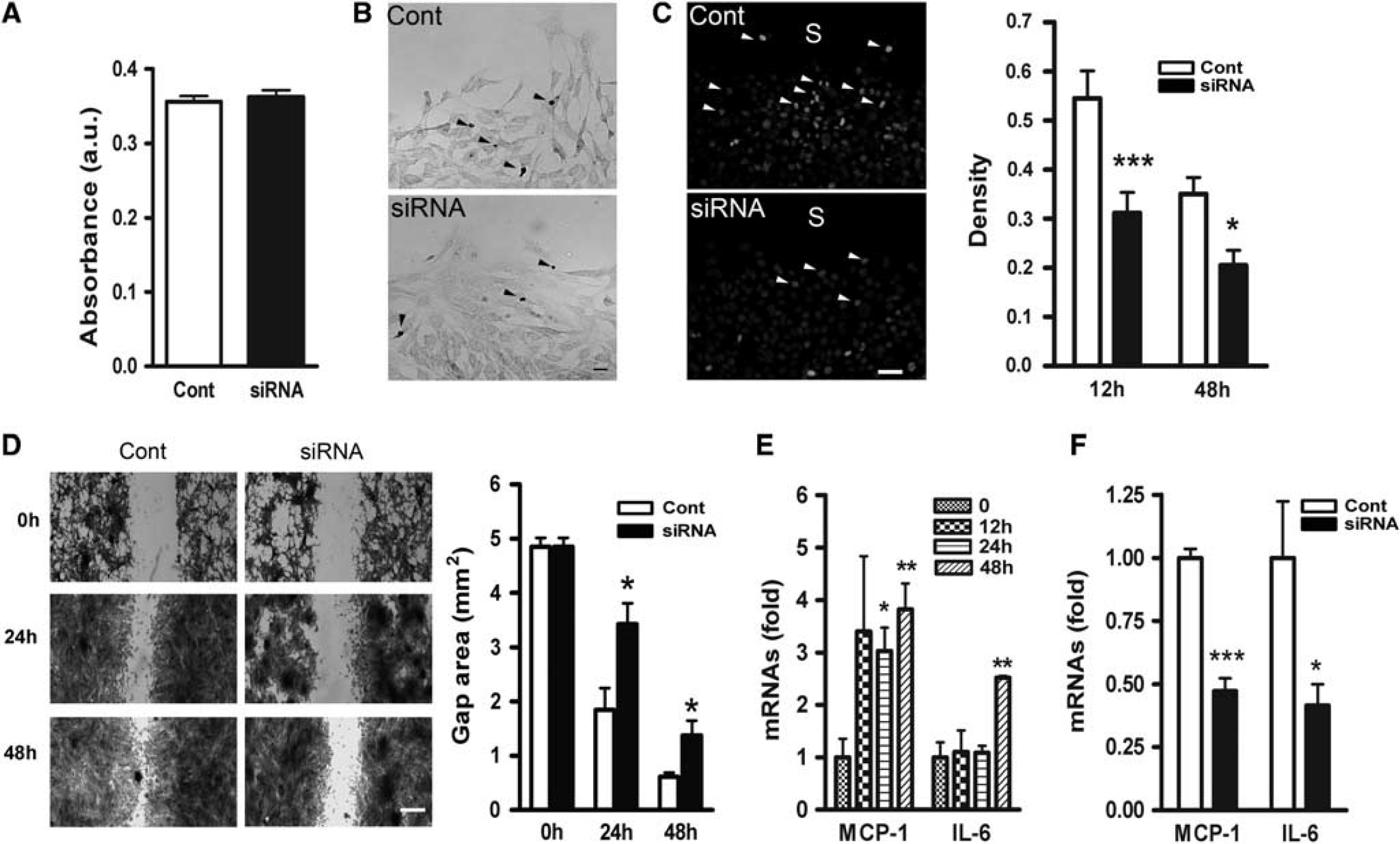
Delayed wound healing and inflammation in the absence of CD36. (
Inflammation is tightly associated with glial scar formation in multiple brain injury models, and CD36 deficiency significantly reduced the levels of CD36-associated pro-inflammatory molecules such as MCP-1 and IL-6 (Glabinski et al, 1996; Hughes et al, 2002; Kim et al, 2008; Okada et al, 2004; Penkowa et al, 2000; Sawada et al, 1992). Following the mechanical injury, MCP-1 mRNA levels in astrocyte cultures were rapidly increased and sustained until 48 hours postinjury, whereas IL-6 expression increased at 48 hours postinjury (Figure 3E). Silencing CD36 significantly reduced both MCP-1 and IL-6 mRNA levels at 48 hours (Figure 3F). Reduced expression of inflammatory factors in the absence of CD36 suggests a mechanism by which CD36 influences glial scar formation through the expression of the inflammatory factors.
CD36 and Glial Fibrillary Acidic Protein Expression in the Brain Occur in a Coordinated Manner During Development
Glial fibrillary acidic protein expression changes during brain development and peaks around 1 week after birth in mice (Kim et al, 2011). To confirm the coordinated expression of CD36 and GFAP
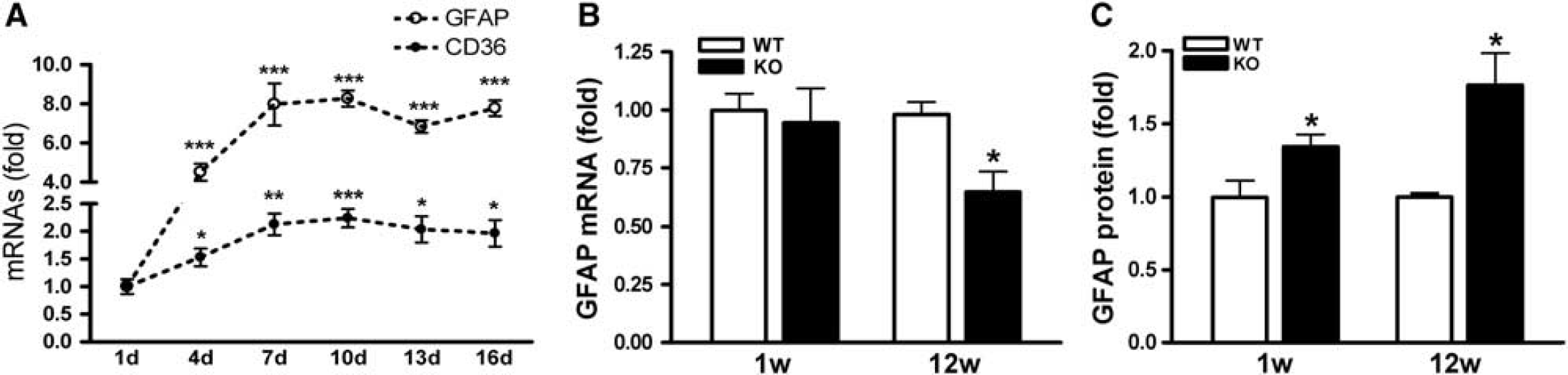
Coordinated expression of CD36 and glial fibrillary acidic protein (GFAP) expression in normal brain. (
Coordinated Expression of CD36 and Glial Fibrillary Acidic Protein Expression in Postischemic Brain
Previously, we reported that CD36 proteins were elevated in the ipsilateral side of the brain and the expression occurred in GFAP+ astrocytes in the peri-infarct area (Cho et al, 2005). Temporal changes of CD36 mRNA and GFAP mRNA expression showed significant increases in the ipsilateral hemisphere at 3, 7 days and 2 weeks after ischemia (Figures 5A and 5B). The expression was relatively unchanged in the contralateral side. The similar gene expression profiles of CD36 and GFAP following stroke suggest their coordinated expression in response to injury. Stroke-induced GFAP protein expression was significantly higher at 3, 7, days and 2 weeks after ischemia in the WT mice (Figure 5C). CD36 KO mice displayed consistently less stroke-induced GFAP expression at 3, 7, days and 2 weeks, suggesting that the absence of CD36 resulted in sustained GFAP reduction in postischemic brain (Figure 5D).
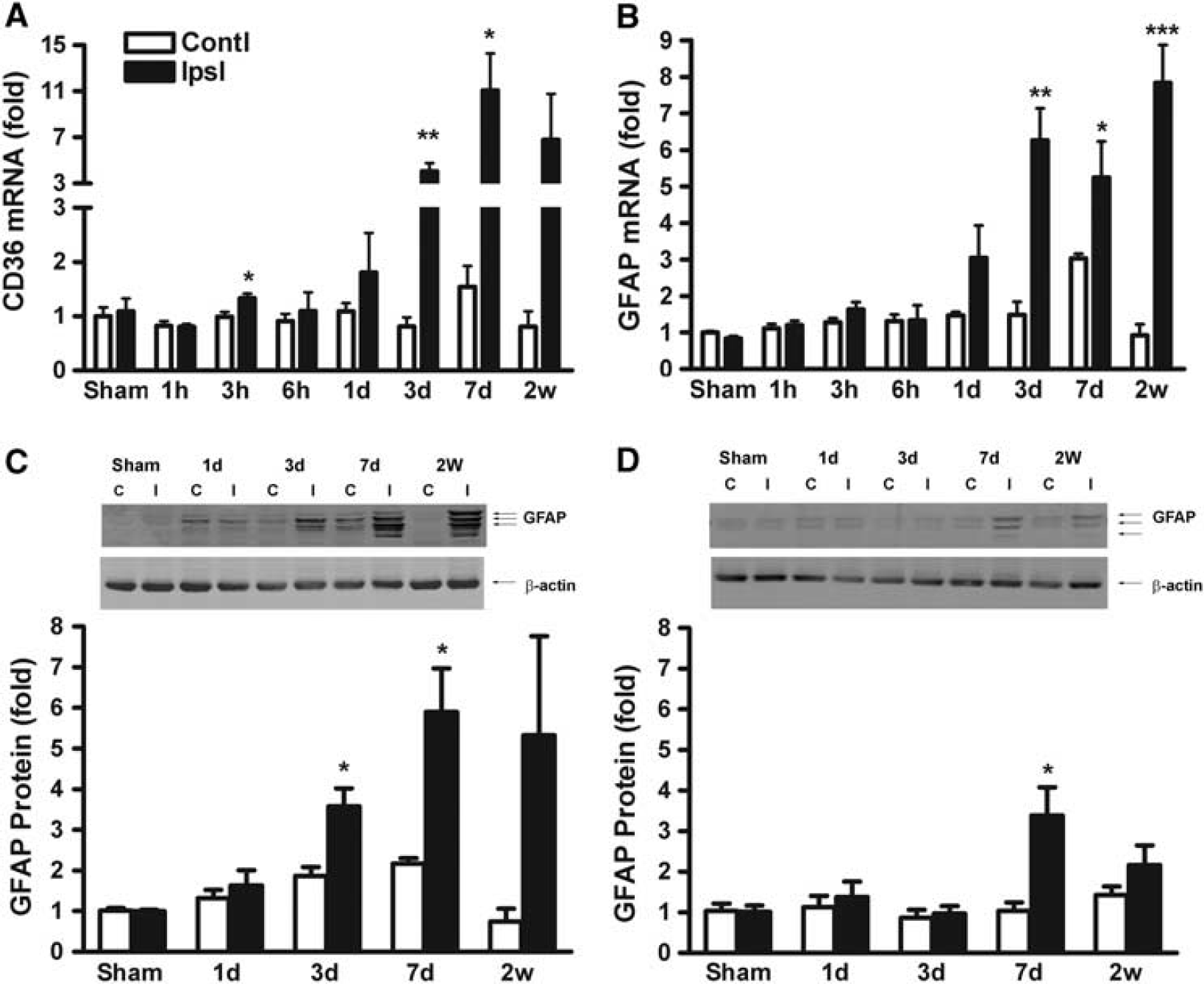
Coordinated expression of stroke-induced CD36 and glial fibrillary acidic protein (GFAP) expression in the postischemic brain. Fold changes of CD36 mRNA (
CD36 Is Required for Injury-Induced Glial Fibrillary Acidic Protein Expression and Glial Scar Formation
To further define the role of CD36 in scar formation
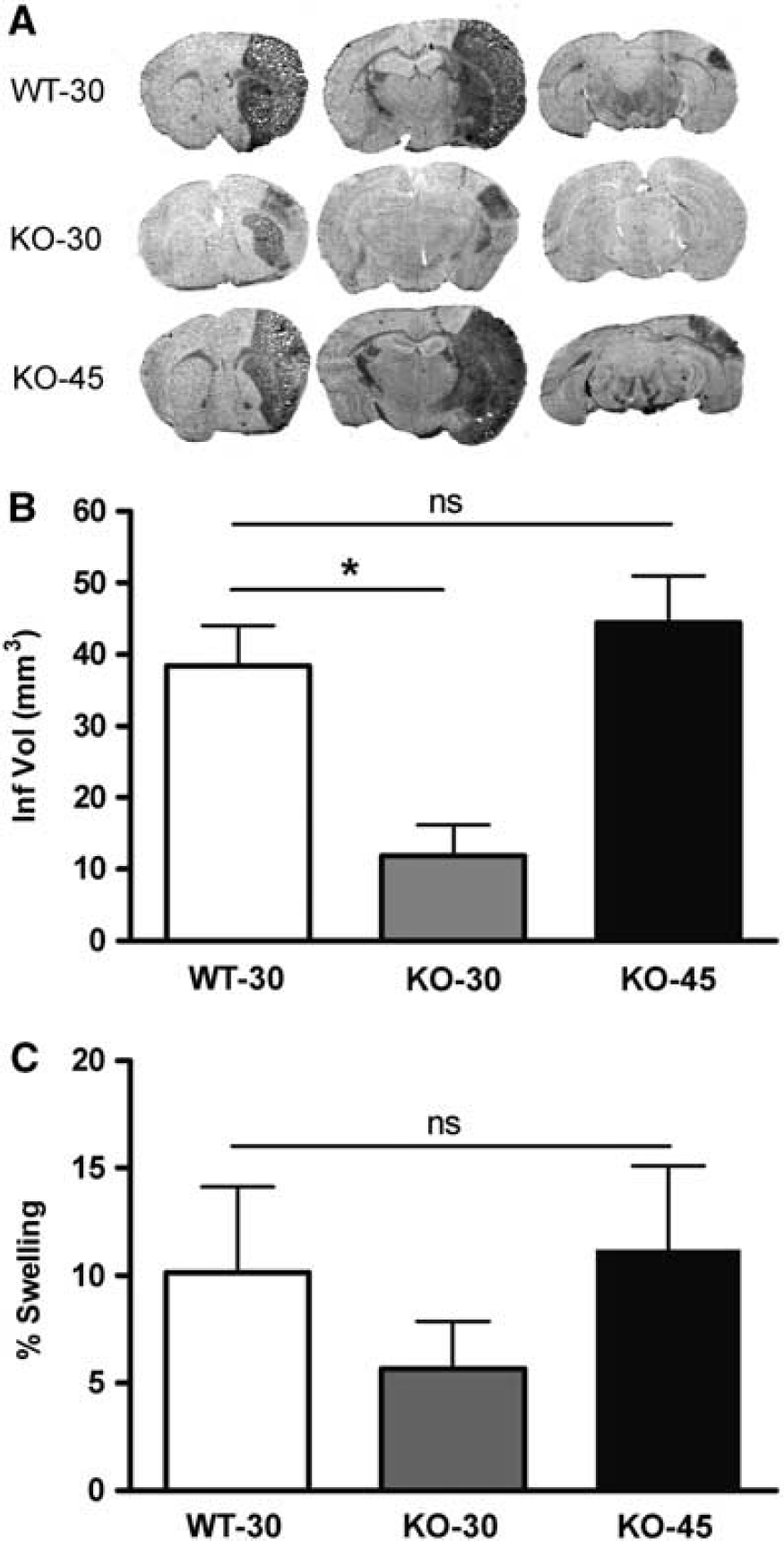
Effect of CD36 on stroke outcome. (
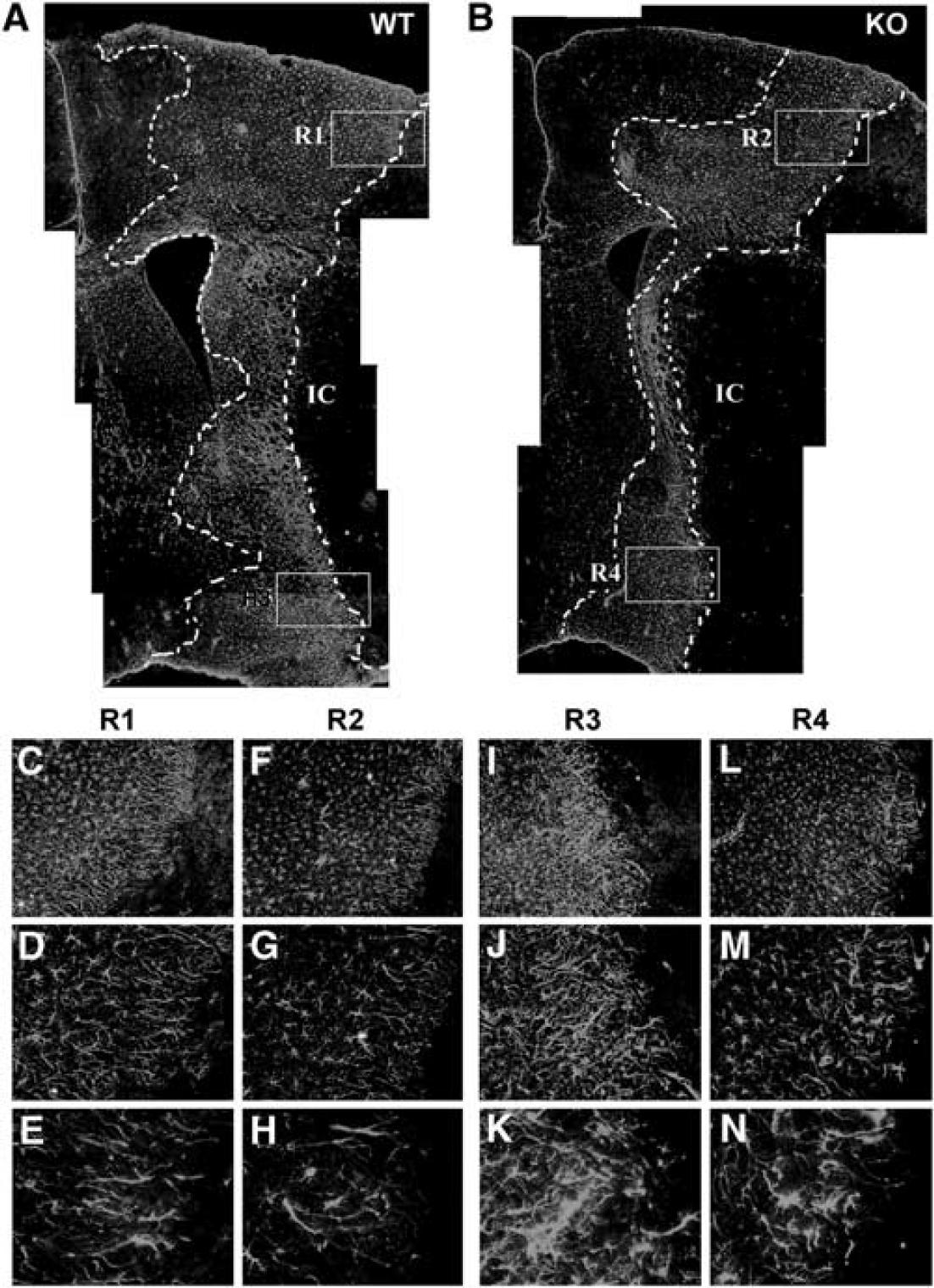
Stroke-induced glial scar formation is attenuated in CD36 knockout (KO) mice. Composite of low magnification of glial fibrillary acidic protein (GFAP) immunohistochemistry micrographs (× 4) from wild-type (WT) (
Glial fibrillary acidic protein mRNA levels in CD36 KO mice revealed a moderate but significant attenuation at day 3, but not day 7, postischemia (Figures 8A and 8B). Glial fibrillary acidic protein mRNA levels in the contralateral side were not different between genotypes, suggesting that ischemia–reperfusion and longer MCA occlusion in CD36 KO mice did not have an impact on GFAP gene expression in the contralateral hemisphere. We also found that GFAP protein levels in the stroked side (expressed as ratio of ipsilateral to contralateral) were significantly attenuated in CD36 KO mice at both 3 and 7 days postischemia (Figures 8C and 8D). Further correlation analyses between GFAP mRNA and protein levels showed a significant correlation in the ipsilateral side of the brain (
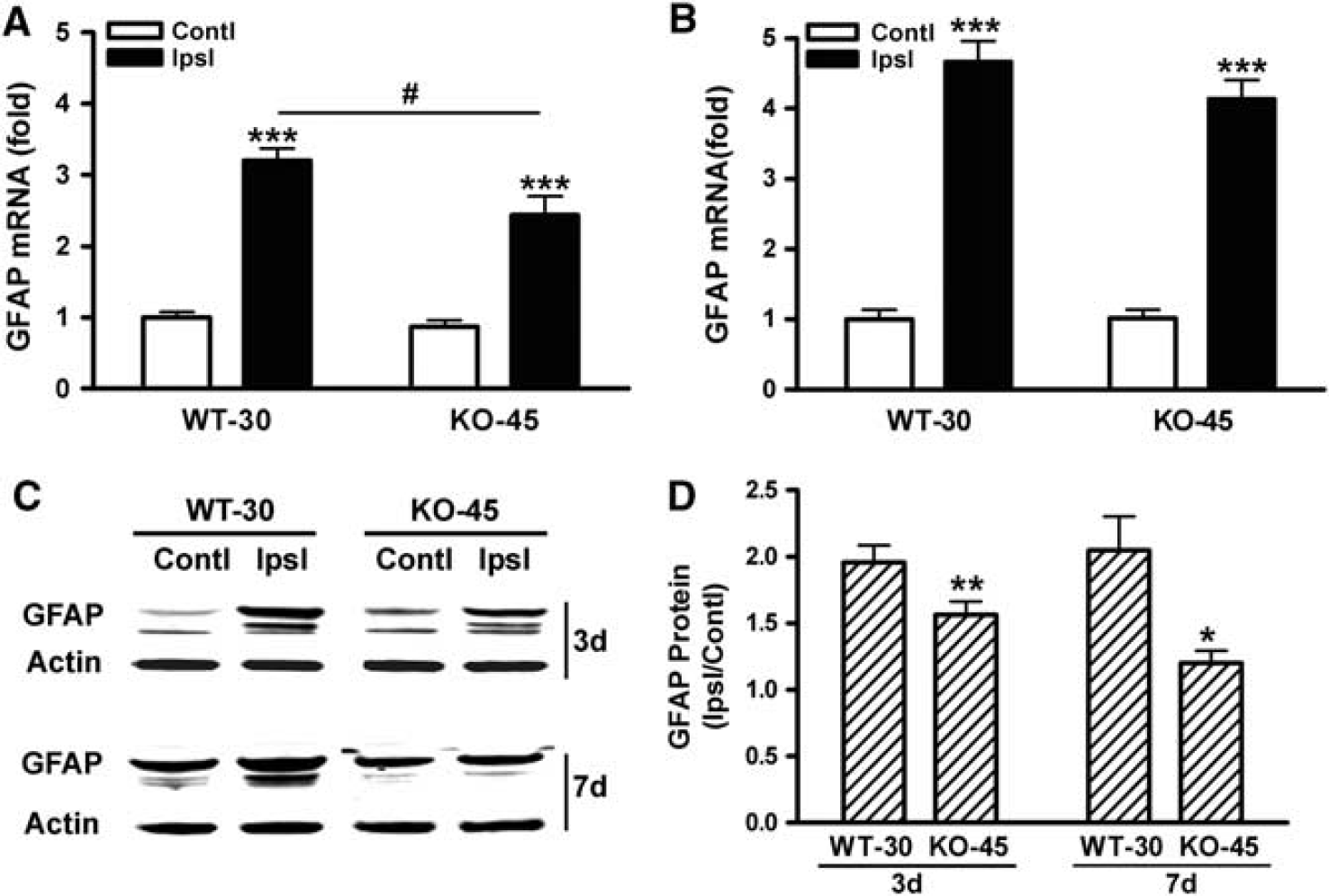
CD36 deficiency reduces glial fibrillary acidic protein (GFAP) expression in the postischemic brain. Fold changes in GFAP mRNA levels in the brains of wild-type (WT) and CD36 knockout (KO) mice at 3 (
Discussion
Despite an initial protective role, glial scar formed in the CNS hinders outgrowth of regenerating axons. Several studies have aimed at degrading or suppressing the production of inhibitory components from the scar tissue to overcome this nonpermissive milieu (Desclaux et al, 2009; Snow et al, 1990). Our study identifies CD36 as a novel mediator of glial scar formation in stroke as CD36 is coordinately expressed with GFAP, and genetic or molecular suppression of CD36 reduces the expression of the intermediate filaments after injury.
Functionally, the absence of CD36 reduces cellular proliferation, delays the wound closure in an
Multiple signaling pathways are involved in scar formation. The literature suggests several links by which CD36 may regulate these pathways. Pro-inflammatory factors, such as IL-1, IL-6, MCP-1, and IFN-
Ischemic injury size depends in part on the severity of blood flow reduction and the duration of occlusion (Pedrono et al, 2010; Zhu and Auer, 1995). Compared with WT mice, CD36 KO mice subjected to 30 minutes MCAO had a significantly smaller injury in this study, consistent with a previous report (Cho et al, 2005). Since a larger infarct results in a greater glial response, the current study uniquely addressed the specific contribution of CD36 on the degree of astrogliosis and glial scar formation by normalizing infarct size between the genotypes. Reduced GFAP expression and less glial scar formation in CD36 KO mice with similar infarct size confirmed the necessary and specific role of CD36 in astrocyte activation and glial scar formation.
The formation of glial scar following injury can be damaging as well as beneficial. In the absence of CD36, we observed reduction in the injury-induced GFAP expression, cellular proliferation, and wound healing rate. The astrocytes also exhibited less intricate and sparse processes in the CD36 KO postischemic brain. Because physical hindrance and a nonpermissive milieu are associated with scar tissue, these morphological changes in the absence of CD36 may be viewed as beneficial. Caveats to this view are reports showing increased CNS inflammation and injury when scar formation is inhibited. In STAT3 KO mice whose scar formation was impaired, inflammation and injury following spinal cord injury were increased (Herrmann et al, 2008). Similarly, the ablation of intermediate filaments in GFAP and vimentin double KO mice increased stroke- or stab wound-induced injury size (Li et al, 2008). Unlike these mice, CD36 KO mice, however, display reduced infarct size with better neurological outcome (Cho et al, 2005) while the glial scar formation was attenuated even infarct size was normalized. Since CD36 KO mice exhibited a shift to a less inflammatory state in the postischemic brain (Kim et al, 2008), reduced inflammation at the level of the receptor may dampen intracellular signalings that lead glial responses. Although the mechanistic distinction to reduce scar formation between CD36 KO versus other means such as STAT3 KO or GFAP/vimentin double KO mice warrants further investigations, the inflammatory property of CD36 and its involvement in GFAP expression suggest that CD36 may serve as an ideal target to suppress intrinsic inflammation and subsequently reduce astrogliosis/scar formation following stroke.
Footnotes
Acknowledgements
The authors thank Jimmy Payappilly for technical assistance on astrocyte cultures and Nancy Geibel and Eric Shipp for editing the manuscript.
Disclosure/conflict of interest
The authors declare no conflict of interest.