
Abstract
Ischemic tolerance is a phenomenon in which brief episodes of ischemia protect against the lethal effects of subsequent periods of prolonged ischemia. The authors investigated the activation of p38 mitogen-activated protein kinase (p38) in the gerbil hippocampus by Western blotting and immunohistochemistry to clarify the role of p38 kinase in ischemic tolerance. After the 2-minute global ischemia, immunoreactivity indicating active p38 was enhanced at 6 hours of reperfusion and continuously demonstrated 72 hours after ischemia in CA1 and CA3 neurons. Pretreatment with SB203580, an inhibitor of active p38 (0–30 μmol/L), 30 minutes before the 2-minute ischemia reduced the ischemic tolerance effect in a dose-dependent manner. Immunoblot analysis indicated that alteration of the phosphorylation pattern of p38 kinase in the hippocampus after subsequent lethal ischemia was induced by the preconditioning. These findings suggest that lasting activation of p38 may contribute to ischemic tolerance in CA1 neurons of the hippocampus and that components of the p38 cascade can be target molecules to modify neuronal survival after ischemia.
Keywords
The mitogen-activated protein kinases (MAPKs), which are characterized as proline-directed serinethreonine-protein kinases, in particular c-Jun amino-terminal kinases (JNK), p38 mitogen-activated protein kinases (p38), and extracellular signal-regulated kinases (ERK), act as key molecules in transducing stress-related signals in eukaryotic cells by phosphorylating intracellular enzymes and transcription factors (Kyriakis and Avruch, 1996). Recent studies demonstrate that the members of the MAPK family, in particular JNK and p38, are activated in heart, kidney, and brain after ischemia and ischemia/reperfusion (Herdegen et al., 1998; Mizukami et al., 1997; Sugino et al., 2000b; Walton et al., 1998; Yin et al., 1997), and suggest that this kinasesignaling pathway may be important and responsible for tissue injury after ischemia/reperfusion and in ischemic tolerance.
It has been reported that prior sublethal ischemia in brain tissue induces the phenomenon of ischemic tolerance to subsequent lethal ischemic stress in the hippocampal CA1 region (Kato et al., 1991; Kirino et al., 1991; Kitagawa et al., 1990). The neuronal tolerance to lethal ischemia also can be induced by a variety of sublethal stresses other than ischemia, such as the synthesis of 72-kd heat shock protein (Kirino et al., 1991), hyperthermia (Kitagawa et al., 1991), oxidative stress (Ohtsuki et al., 1992), spreading depression (Kobayashi et al., 1995), opening of sulfonylurea-sensitive ATP-sensitive K+ channels (Heurteaux et al., 1995), and intraperitoneal administration of 3-nitropropionic acid (Sugino et al., 1999). Recent studies have suggested that phosphorylation of tyrosine 182 of p38 correlates with the protective action of preconditioning in the heart (Nagarkatti et al., 1998; Weinbrenner et al., 1997). We recently reported the activation of MAPK cascades after the administration of 3-nitropropionic acid, which is a mitochondrial succinate dehydrogenase inhibitor, at a dose that induced ischemic tolerance in the gerbil hippocampus (Sugino et al., 2000a), but it remains to be shown whether the activations of MAPK cascades may affect the ischemic tolerance phenomenon induced by sublethal ischemia. In previous studies, we obtained evidence suggesting that p38 was activated in the gerbil hippocampus after 5-minute transient forebrain ischemia in vivo and that the inhibition of the activity of p38 protected against delayed neuronal death in CA1 pyramidal cells (Sugino et al., 2000b). In the current study, we investigated the phosphorylation of p38 kinase during transient forebrain ischemia in gerbil to clarify the role of p38 kinase in ischemic tolerance.
MATERIALS AND METHODS
Induction of global ischemia
Adult male Mongolian gerbils (Shimizu Laboratory Supplies, Kyoto, Japan) weighing 60 to 80g were used for all experiments. They were housed in a temperature-controlled (23° ± 1°C) and light/dark cycle–controlled animal room (lights on at 8:00 am, off at 8:00 pm). The animals were deprived of food overnight before the induction of ischemia to exclude the influence of hyperglycemia on ischemic brain damage (Ginsberg et al., 1987; Pulsinelli et al., 1982). All experiments were performed under spontaneous respiration. Anesthesia was induced with 4% halothane in a gas mixture of 30% oxygen and 70% nitrous oxide administered via a facemask and maintained at 2% halothane in the same gas mixture. Bilateral common carotid arteries were dissected and occluded with microaneurysmal clips and then the halothane concentration was reduced to 1%. During the operation, rectal temperature was monitored and kept at 37° to 38°C with a thermo regulator (Animal Blanket Controller ATB-1100, Nihon Kohden, Tokyo, Japan) and a heating pad and a lamp, and did not change significantly during the operative procedure and postoperative period (about 2 hours). In the sham-operated group, the same procedure was performed without carotid occlusion. For a combined ischemia model (tolerance model), the gerbils preconditioned with 2 minutes of global ischemia were subjected to 5 minutes of global ischemia after 48 hours of reperfusion. The gerbils used for the 5-minute ischemia model (lethal model) underwent sham operation and were subjected to 5 minutes of global ischemia after 48 hours.
Pretreatment with SB203580
To examine the effect of SB203580 (Calbiochem, San Diego, CA, U.S.A.), an inhibitor of active p38 (Cuenda et al., 1995; Lee et al., 1994), on ischemic tolerance, SB203580 (0, 1, 10, 30 μmol/L) was administered into the right lateral ventricle stereotaxically 30 minutes before the 2-minute ischemic insult under general anesthesia. The heads of the gerbils were secured in a stereotaxic frame, and a 27-gauge needle was inserted into the right lateral ventricle. Coordinates were 2 mm lateral and 2 mm ventral from the dural surface, with bregma used as a landmark (Thiessen et al., 1970). Twenty-five microliters of each drug solution was injected over a 10-minute period. SB203580 was dissolved in 1% dimethyl sulfoxide (DMSO; Nacalai tesque, Kyoto, Japan). After 48 hours, under general anesthesia, the gerbils preconditioned with 2 minutes of global ischemia were subsequently subjected to 5 minutes of global ischemia. Physiologic parameters (rectal temperature, mean arterial blood pressure, Pco2, Po2, pH, and glucose) were not significantly changed after injection of SB203580 (data not shown; Sugino et al., 2000b).
Tissue and sample preparation
For the immunohistochemical study, at 0, 6, 18, or 72 hours (each n = 3) after 2 minutes of ischemia, the animals (n = 12) were killed and perfused transcardially with a buffer (0.1-mol/L phosphate-buffered saline [PBS], pH7.4, containing 0.2-mmol/L sodium orthovanadate) followed by 4% buffered paraformaldehyde. To examine the effect of SB203580 (0, 1, 10, 30 μmol/L) on p38 cascade after 2 minutes of ischemia, we perfused gerbils (n = 3 for each dose) at 0 minutes and 18 hours after 2-minute ischemia. The brains were rapidly removed and cryoprotected in 25% sucrose in 0.1-mol/L PBS overnight at 4°C. For Western blot analysis of the total amounts and active form of p38 in the hippocampus, the gerbils were killed at 0 or 30 minutes or at 2 or 12 hours after 5 minutes of ischemia (each n = 3). The whole hippocampus was rapidly excised and frozen in liquid nitrogen.
Antibodies
Anti -JNK1 (FL) antibody, anti-p38 (c-20) antibody, anti-ERK2 (c-14) antibody, anti–p-ATF-2 (F-1) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), and antibodies for the active forms of (Anti-ACTIVE) JNK, p38 and MAPK (ERK) (Promega, Madison, WI, U.S.A.) were used in this study.
Western immunoblot analysis for p38 and activated p38
Whole hippocampal homogenates were collected and stored at −80°C. Proteins were isolated according to standard techniques, separated on a 12% sodium dodecyl sulfate/polyacrylamide gel electrophoresis, and transferred onto polyvinylidene diflouride membranes. Subsequently, the blots were incubated with the rabbit polyclonal antibodies described in Antibodies section. A donkey peroxidase-conjugated antirabbit antibody was used as the second antibody, and the binding was revealed by chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.). For quantitative analysis of the densities of the immunoblot bands, the densities were measured by use of NIH image analyzer 1.61 (Dr. Wayne Rasband, National Institutes of Health, U.S.A.).
Immunohistochemistry
Frozen coronal sections (40-μm thick) of the brains were prepared with a microtome and processed by the free-floating method. The sections were washed three times in 0.1-mol/L PBS, immersed in 2% H2O2 in 60% methanol for 60 minutes to quench endogenous peroxidase activity, blocked with 5% goat serum, and incubated overnight with the primary antibodies. Next, the sections were washed with 0.1-mol/L PBS and incubated with biotinylated anti–rabbit immunoglobulin G antibody for 2 hours and thereafter with an avidin-biotin complex for 60 minutes. Peroxidase was demonstrated with a diaminobenzidine substrate kit. For evaluation of morphologic change, adjacent sections were stained with cresyl violet.
Measurement of surviving CA1 neurons
To examine the effect of intraventricular administration of SB203580 30 minutes before the 2-minute ischemia on the development of delayed neuronal death in the CA1 region after 5-minute ischemia (5-minute ischemia preconditioned with 2-minute ischemia), the animals were anesthetized with diethyl ether and perfused transcardially with a buffer (0.1-mol/L PBS, pH 7.4, containing 0.2-mmol/L sodium orthovanadate) followed by 4% buffered paraformaldehyde 7 days after the operation. Thirty-four gerbils were used (n = 8, 7, 12, 7 for vehicle and doses of 1, 10, 30 μmol/L, respectively). Frozen coronal sections (20-μm thick) of the brains were prepared and stained with cresyl violet. The severity of neuronal damage in the right CA1 region was evaluated by the number of surviving neurons. Normal CA1 pyramidal cells showed round and pale stained nuclei in cresyl violet staining. The shrunken cells with pyknotic nuclei after ischemia were counted as dead cells. The mean number of surviving neurons of the pyramidal cell layer per 450-μm length was calculated in right CA1 region for each group. Independent observers performed cell counting in a blind fashion using a light microscope equipped with a × 10 objective (Tokime et al., 1996). The data were statistically analyzed by analysis of variance (ANOVA) followed by post hoc Bonferroni test between groups using Stat View II (Abacus Concepta, Berkeley, CA, U.S.A.). Data are presented as mean ± SD; differences were considered significant at the P < 0.05 level.
TUNEL staining
Terminal deoxynucleotidyl transferase (TdT)-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining was performed using a kit from Boehringer Mannheim (Indianapolis, IN, U.S.A.), according to the manufacturer's instructions. Briefly, sections were incubated for 1 hour at 37°C with TdT buffer (30-mmol/L Tris, pH 7.2; 140-mmol/L sodium cacodylate; and 1-mmol/L cobalt chloride) containing TdT enzyme (0.5 U/mL) and biotin-16-dUTP (0.04 mmol/L). Incubating the sections in 300-mmol/L NaCl and 30-mmol/L sodium citrate for 15 minutes terminated the reaction. The sections were washed with 50-mmol/L Tris-HCl (pH 7.7) and incubated with an avidin-biotin complex for 60 minutes. Peroxidase was demonstrated with a diamino benzidine substrate kit. The total number of TUNEL-positive cells was counted for each brain. The data were statistically analyzed as described above. Data are presented as mean ± SD; differences were considered significant at the P < 0.05 level.
RESULTS
Two-minute global ischemia does not cause neuronal death in the hippocampus
Cresyl violet staining showed no obvious morphologic changes in the hippocampus 72 hours after 2 minutes of global ischemia (data not shown).
Immunolocalization for active p38 after sublethal ischemia in the hippocampus
Few neurons in the hippocampus showed immunoreactivity indicating phosphorylated p38 in the sham-operated animals (data not shown) or immediately after 2-minute ischemia (Figs. 1A and 1B). By 6 hours of reperfusion after the 2-minute global ischemia, however, the level of phosphorylated p38 was enhanced in neurons in CA1 (Fig. 1C) and in CA3 (Fig. 1D), and the immunoreaction in both CA1 and CA3 was even stronger at 18 (Figs. 1E and 1F) and 72 (Figs. 1G and 1H) hours of reperfusion after the ischemia.
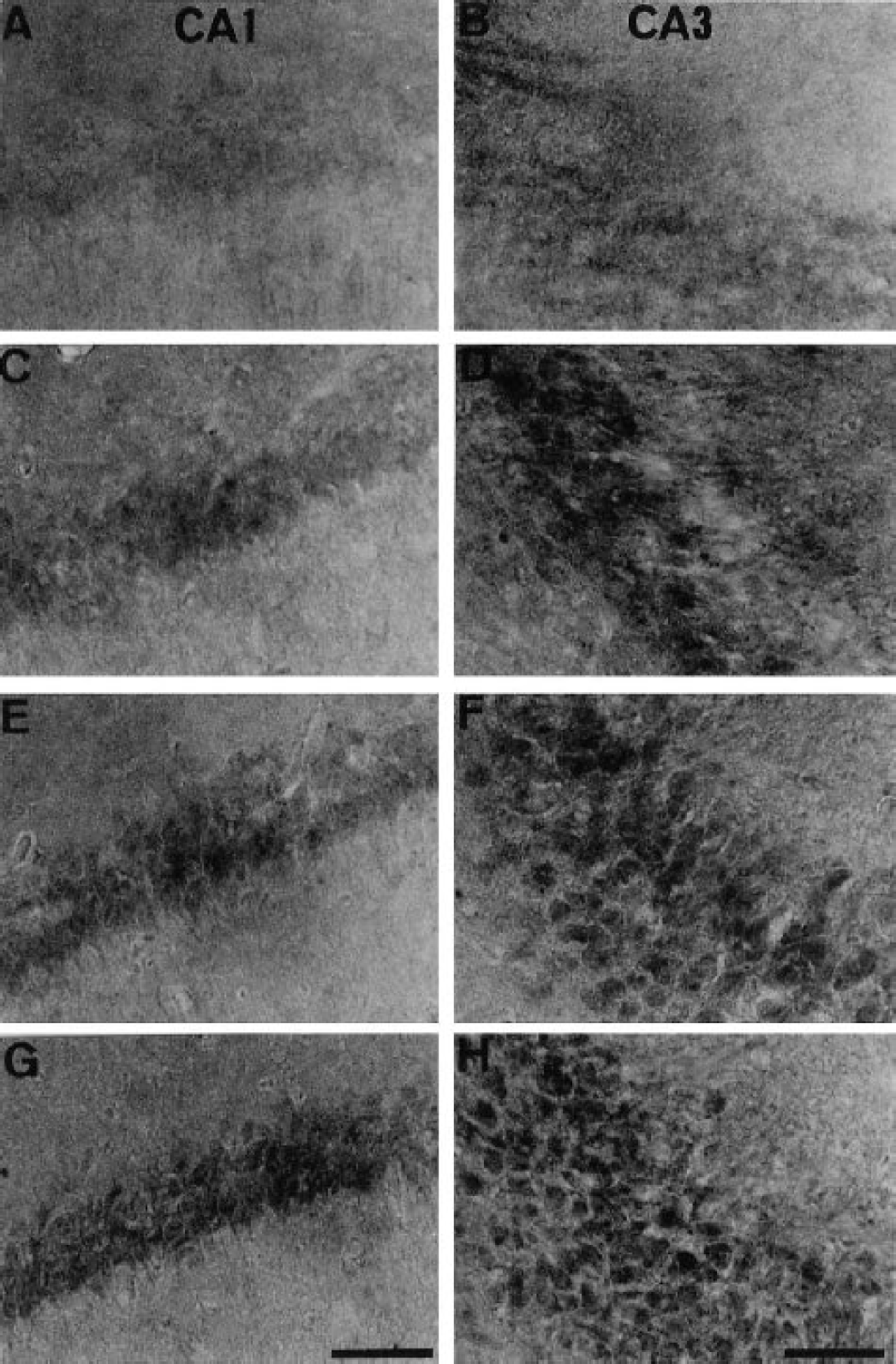
Immunohistochemical staining for active form of p38 in CA1
Effect of SB203580, an inhibitor of active p38, on MAPK cascades
SB203580 is not able to inhibit the phosphorylation of p38 (Larsen et al., 1997), but does inhibit the activity of the phosphorylated form of p38. Therefore, we evaluated the effect of SB203580 by investigating the immunoreactivity for phosphorylated ATF-2 (p-ATF-2). ATF-2 is a substrate of phosphorylated p38. Few neurons showed immunoreactivity for active p-ATF-2 in sham-operated animals (Fig. 2A). Consistent with the phosphorylation of p38, immunoreactivity for p-ATF-2 was observed 18 hours after 2-minute ischemia in the CA1 region (Fig. 2B). Intraventricular administration of SB203580 (0, 1, 10, 30 μmol/L) 30 minutes before the 2-minute ischemic insult could not reduce the immunoreactivity for active p38 at 18 hours after the ischemia (data not shown), but inhibited p-ATF-2 immunoreactivity (Fig. 2C). SB203580 did not inhibit JNK immunoreactivity (Figs. 2D and 2E), nor did it reduce the activity of active ERK immediately or 18 hours after the 2-minute ischemia (data not shown).
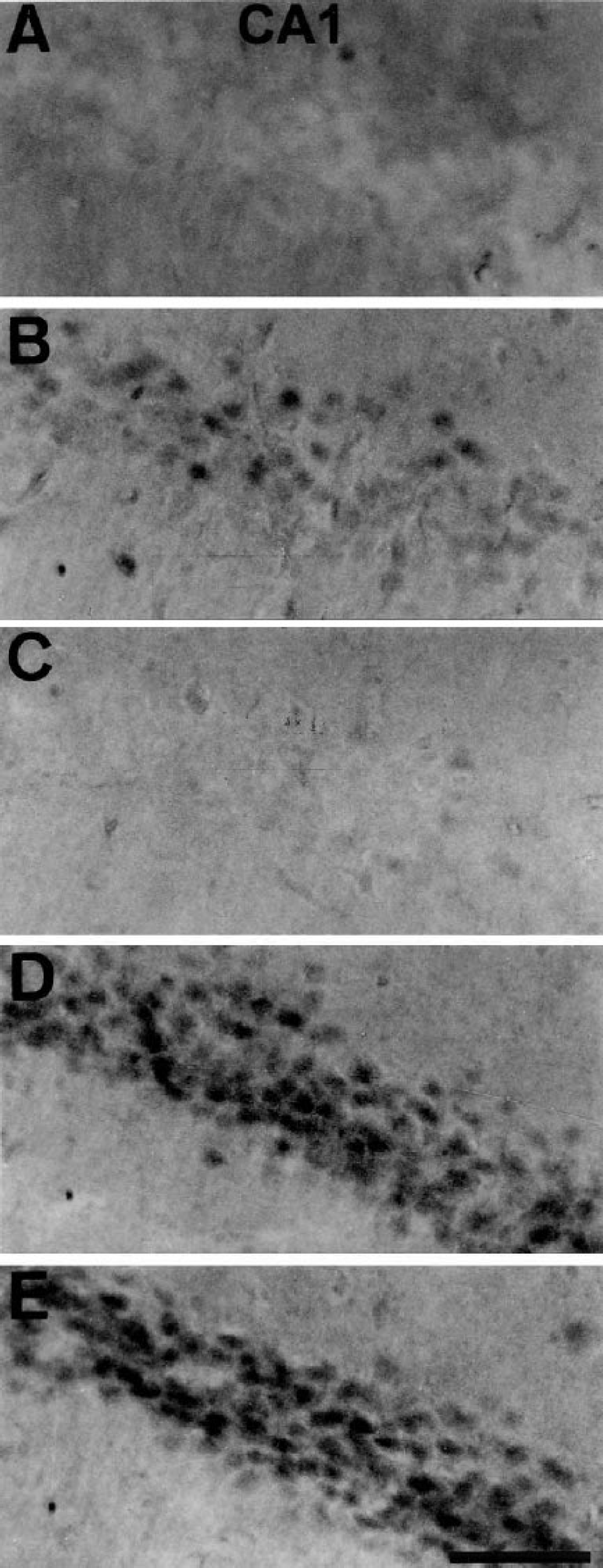
Representative photomicrographs of immunohistochemical study on the active forms of ATF-2
SB203580 reduces ischemic tolerance effect in CA1
In the group that underwent sham operation without carotid occlusion, no significant neuronal damage was detected. The mean number of surviving neurons in the CA1 region was 121.8 ± 9.7 (n = 8). In the lethalischemia group, the mean number of surviving neurons in this region was 3.1 ± 1.8 (n = 8) 7 days after 5 minutes of global ischemia. By preconditioning the animals with 2-minute ischemia, the mean number of surviving neurons in the CA1 subfield was 89.0 ± 11.6 (n = 7) 7 days after the subsequent 5 minutes of global ischemia. Intraventricular administration of SB203580 (0, 1, 10, 30 μmol/L) 30 minutes before the 2-minute ischemic insult significantly reduced the ischemic tolerance effect in the hippocampal CA1 region 7 days after the subsequent 5 minutes of ischemia in a dose-dependent manner, and the maximum effect was obtained at a dose of 30 μmol/L (Figs. 3G and 3I). In our preliminary study, we tested a 100 μmol/L or greater concentration of SB203580, but the effect of the inhibitor was not increased significantly. The rectal temperature was not significantly different between groups during the operative procedure or postoperative period (data not shown).
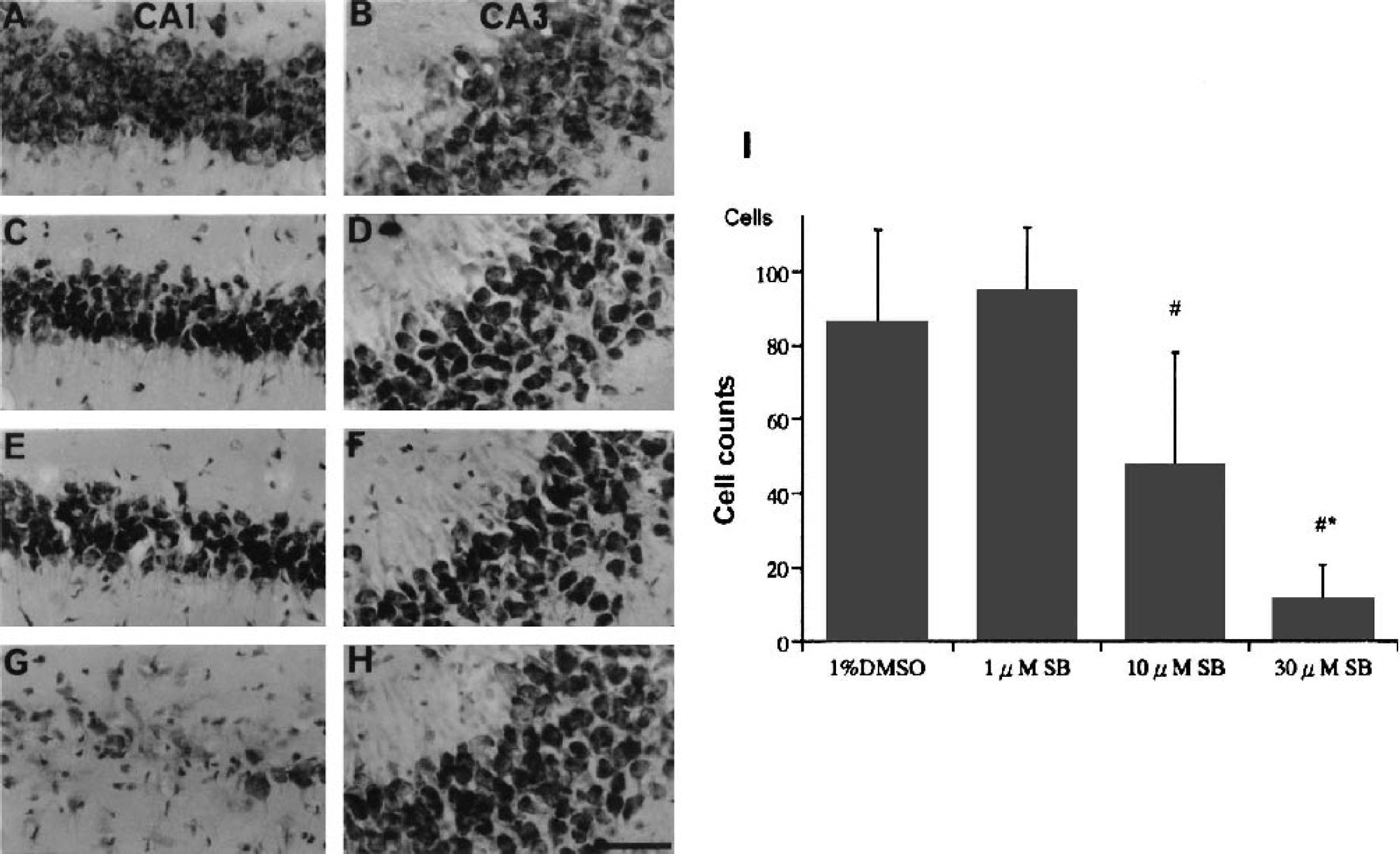
Cresyl violet staining in CA1
SB203580 increased the number of TUNEL-positive cells after lethal ischemia in the CA1 region
Lethal ischemia induced TUNEL-positive cells in the CA1 region 72 hours after 5-minute ischemia (Fig. 4A), but the combined ischemia (tolerance model) obviously decreased the number of cells 72 hours after the second (5-minute) global ischemia (Fig. 4B). The total number of TUNEL-positive cells was counted for each brain. Pretreatment with SB203580 30 minutes before the 2-minute ischemia increased the number of TUNEL-positive cells 72 hours after the subsequent 5-minute ischemia (Figs. 4C and 4D).
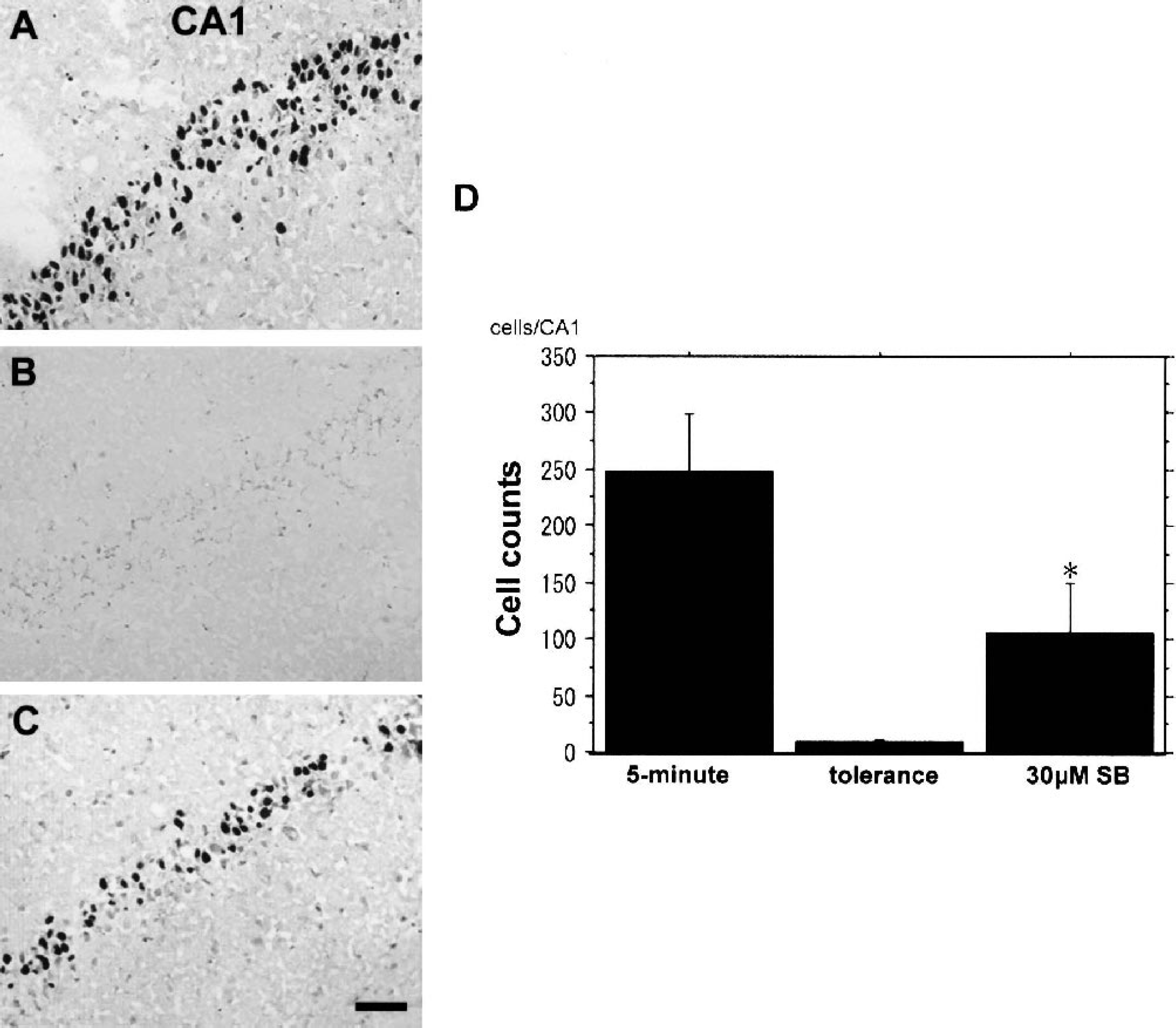
TUNEL staining in the CA1 region 72 hours after 5-minute ischemia. Without the induction of ischemic tolerance, 5-minute ischemia induces TUNEL-positive cells in CA1
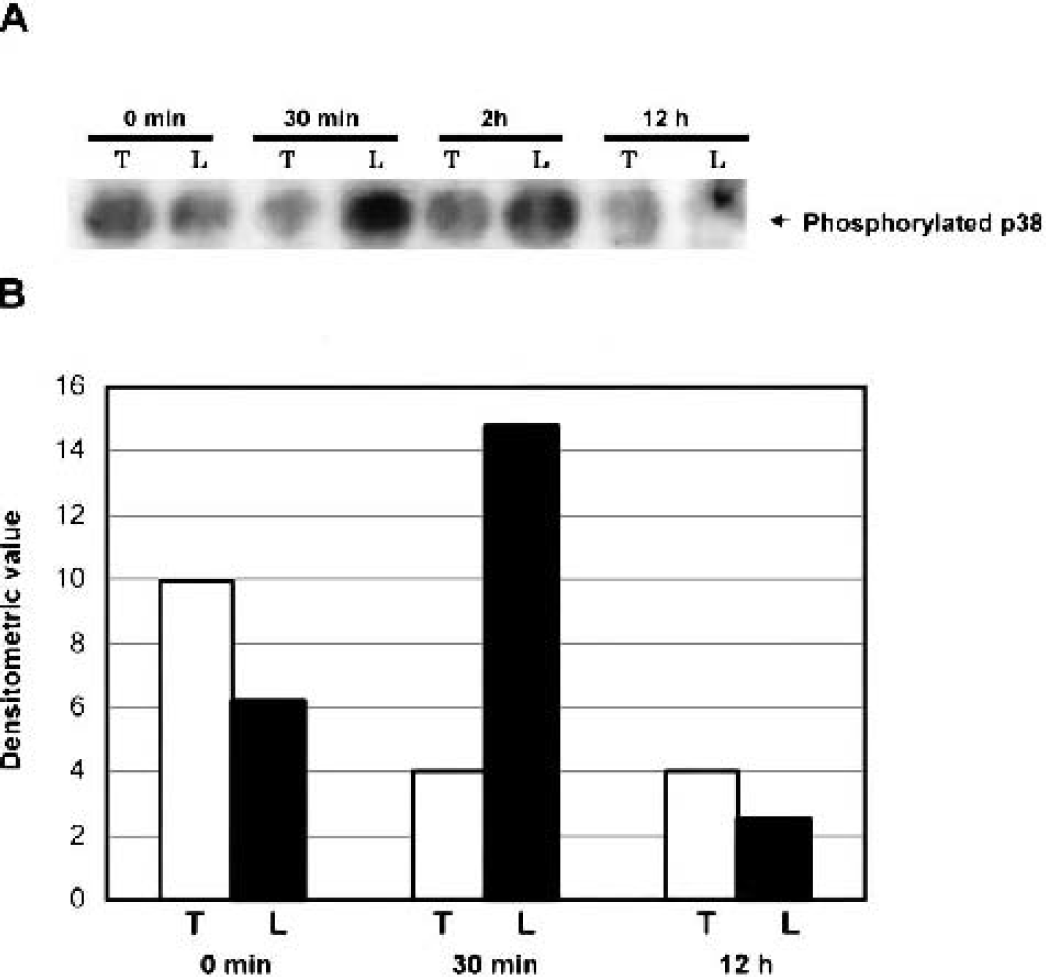
Effect of preconditioning on phosphorylation of p38 after lethal ischemia in the hippocampus
To determine whether preconditioning altered the levels of the phosphorylated forms of p38 after 5 minutes of ischemia, we subjected hippocampal extracts to Western blot analysis using antibodies against the active forms of p38. When the ischemic preconditioning was not induced, p38 was strongly phosphorylated 30 minutes after the 5-minute ischemia (Fig. 5). But in the tolerance model, p38 forms were already phosphorylated immediately after the subsequent 5-minute ischemia and decreases in the levels of phosphorylated p38 were observed 30 minutes after the subsequent 5-minute ischemia compared with their levels measured at the same time in the lethal ischemia model (Fig. 5). No significant alteration in the total protein level of p38 after 5-minute ischemia was found (data not shown). These results indicate that p38 kinase was phosphorylated by 48 hours after 2-minute ischemia and that alteration of the phosphorylation pattern of p38 kinase in the hippocampus after subsequent lethal ischemia was induced by the preconditioning.
DISCUSSION
A brief period of cerebral ischemia destroys neurons in specific neuronal populations, such as those in the CA1 subfield of the hippocampus (Araki et al., 1989; Kato et al., 1991). Preconditioning of the gerbil brain with 2-minute ischemia followed by 1 to 7 days of reperfusion protects against neuronal damage after a longer period of ischemia, which normally damages the CA1 neurons (Kato et al., 1991). This phenomenon has been termed ischemic tolerance and has received much attention because the clarification of its mechanism may afford a clue for protection against ischemic brain damage. However, the mechanism of ischemic tolerance still remains to be elucidated, although a role for stress proteins (Kirino et al., 1991; Nowak et al., 1993), bcl-2 oncoprotein (Shimazaki et al., 1994), Jun-related protein (Kato et al., 1995), opening of K+ channels (Heurteaux et al., 1995), and adenosine receptor activation (Kawahara et al., 1998) has been suggested.
Phosphorylation of tyrosine 182 of p38 in the heart is correlated with the protective action of preconditioning (Armstrong et al., 1998; Nagarkatti et al., 1998; Weinbrenner et al., 1997). Baines et al. (1999) suggested that p38 and/or JNK played important roles in ischemic preconditioning in the rabbit heart, based on the results of experiments using anisomycin, a p38/JNK activator. Haq et al. (1998) demonstrated that activation of p38 was involved in adenosine-mediated ischemic preconditioning in the perfused rat heart. These observations indicate that JNK and p38 play important roles in ischemic preconditioning in the heart.
Because the acquisition of tolerance occurs in a wide variety of cells, it is not surprising that p38 can be a component of ischemic tolerance in the brain. We previously demonstrated that the activation of JNK and p38 in CA1 was induced 15 minutes after 5 minutes of global ischemia and that pretreatment with SB203580, a p38 inhibitor, reduced ischemic cell death in this region after 5 minutes of global ischemia by inhibiting the activity of p38 (Sugino et al., 2000b). We hypothesized that p38 activation plays also an important role in ischemic tolerance, and investigated the activations of p38 kinase during transient forebrain ischemia in the gerbil to clarify the roles of this kinase in ischemic tolerance.
The current study demonstrated that the activation of p38 mainly occurred in CA1 and CA3 regions up to 72 hours after 2 minutes of global ischemia. Because no histologic change was observed in the hippocampus after 2 minutes of global ischemia, we considered this ischemic insult to be sublethal. In the brain, the time course of ischemic tolerance apparently follows the delayed pattern, and neuronal protection by ischemic tolerance is only a transient phenomenon, which leads to an assumption that activation of p38 may be necessary for the development of ischemic tolerance. To evaluate the significance of the activated p38 in ischemic tolerance, we tested the effect of SB203580, an inhibitor of active p38, on delayed neuronal death in the CA1 region of the hippocampus. Intraventricular administration of SB203580 30 minutes before 2-minute ischemic insult significantly reduced neuronal survival and increased the number of TUNEL-positive cells in the CA1 region after the subsequent 5-minute ischemia. In previous studies, we reported that p38 was activated in the gerbil hippocampus after 5-minute ischemia and that the inhibition of the activity of p38 protected against delayed neuronal death in CA1 pyramidal cells (Sugino et al., 2000b). These data imply that p38 kinase may be involved in both neuronal survival and death.
There are two possible explanations for the two opposite roles of p38: One is the timing of p38 expression and the other is the quantity of activated p38. Although there is a general consensus that p38 pathways induce neuronal death, some reports show that the activation of p38 pathways might be involved in neuronal cell survival. Roulston et al. (1998) demonstrated that TNF induced activation of p38 pathway and that the inhibition of early TNF-induced p38 kinase activation using the p38 inhibitor SB203580 increased TNF-induced apoptosis. They concluded that early activation of p38 kinase initiated by TNF might be necessary to protect cells from TNF-mediated cytotoxicity. The roles of p38 cascade might depend on the timing of expression in a given condition. However, we showed that after 2-minute global ischemia, the immunoreactivity indicating active p38 was enhanced at 6 hours of reperfusion and continuously demonstrated 72 hours after ischemia in CA1 and CA3 neurons, but could not detect phosphorylation of p38 after 2-minute ischemia by Western blot analysis. These results indicate that p38 was phosphorylated after 2-minute ischemia, but that the amount of phosphorylated p38 was smaller than that which was observed 30 minutes after 5-minute ischemia in the lethal ischemia model. This weak but lasting activation of p38 cascade that does not induce apoptotic signal seems to be important for acquisition of ischemic tolerance.
Cellular destiny is affected by both strength and duration of p38 activation, which must therefore be strictly controlled (Marshall, 1995). Recently, this control has been reported to be mediated in part by MAPK phosphatases (MKPs), which inactivate MAPKs by dephosphorylation of both threonine and tyrosine residues within the activation motif. MKP-1 may be catalytically activated by p38, and MKP-1 efficiently inactivates p38 (Hutter et al., 2000). These observations suggest the participation of MKP-1 in a complex feedback loop that modulates p38 function. If it is assumed that the first sublethal ischemia induced MKP-1 expression, the second peak of p38 activity may be controlled. We demonstrated that the activation of p38 was followed by the activation of ATF-2, a substrate of p38, but did not examine MKPs expression in this study. Concerning the upstream and downstream cascades of p38, Maulik et al. (1998) demonstrated that MAPK-activated protein kinase 2, the downstream enzyme of p38, was activated after sublethal ischemic insult in isolated perfused rat hearts. The upstream and downstream molecules of p38 cascades responsible for ischemic tolerance in the brain still remain to be elucidated.
In conclusion, p38 was activated after a sublethal 2-minute transient forebrain ischemia in the gerbil hippocampus, and an inhibitor of active p38 reduced ischemic tolerance in the CA1 hippocampal region. In this study, we demonstrated for the first time that lasting activation of p38 after sublethal 2-minute transient forebrain ischemia is one of the important mechanisms for acquisition of ischemic tolerance. Therefore, the p38 kinase cascade might play an important role in neuronal survival and death during brain ischemia, and its components can be considered possible target molecules for affording neuronal protection in ischemic brain damage.