
Abstract
Cumulative evidence shows a protective role for adenosine A1 receptors (A1R) in hypoxia/ischemia; A1R stimulation reduces neuronal damage, whereas blockade exacerbates damage. The signal transduction pathways may involve the mitogen-activated protein kinase (MAPK) pathways and serine/threonine kinase (AKT), with cell survival depending on the timing and degree of upregulation of these cascades as well as the balance between pro-survival and pro-death pathways. Here, we show
Introduction
Owing to their high basal oxygen requirements, neurons are extremely sensitive to ischemic insult. Any interruption in blood flow to the brain results in a rapid depletion of oxygen, which severely compromises ATP production and leads to failure of ionic conductance and excitotoxicity (Lee et al, 2000). Neurons respond to compromised oxygen availability by triggering a dynamic network of signaling pathways including the mitogen-activated protein kinases (MAPKs) and phosphatidylinositol 3-kinase (PI3-K/AKT). The MAPKs constitute a superfamily of proline-directed serine/threonine kinases that includes the extracellular signal-regulated kinases (ERK) and the stress-activated protein kinases (such as JNK/SAPK1 and p38/SAPK2). The MAPKs control numerous intracellular events in response to external stimuli, ultimately changing cell function through altered gene expression or the posttranslational modification of proteins (Martindale and Holbrook, 2002). Activation of the MAPKs and PI3-K/AKT involves phosphorylation and takes place in response to a variety of stressors including oxidative stress and anoxia (Clerk et al, 1998; Kamada et al, 2007).
The ERK signaling cascade has been extensively studied in several cell types; activation of ERK has been found to be protective in PC12 cell and rat ischemic models (Xia et al, 1995; Irving et al, 2000), though others have found ERK activation promotes cell death after ischemia (Wang et al, 2003; Zhuang and Schnellmann, 2006; Sawe et al, 2008). Activation of the ERK pathway is thus implicated in both neuronal survival and death and differences may be linked to the trigger, dynamics, and duration of its activation (Martindale and Holbrook, 2002; Ho et al, 2007). Rapid and transient activation of ERK seems to be pro-survival (Li et al, 2002), whereas long-term activation apparently promotes cell death (Wang et al, 2003). The role of other MAPK pathways is also under debate, with investigations reporting JNK and p38 as apoptosis inducers (Guan et al, 2006), whereas others showed protective effects of JNK and p38 in different mammalian ischemic models (Dougherty et al, 2002; Beguin et al, 2007).
The PI3-K/AKT is generally reported to be antiapoptotic (Zhao et al, 2006); in part through its interactions with the Bcl-2 family of proteins. Activation of the AKT pathway phosphorylates Bad and suppresses its proapoptotic activity in both
Determining whether a particular pathway is pro-survival or proapoptotic in mammalian models, however, is difficult, in part because they simultaneously exhibit both physiological and pathological responses to such stressors as hypoxia, ischemia, or oxidative stress. But such events can be dissected and analyzed in the anoxia tolerant freshwater turtle, which survives extended anoxia and postanoxic reoxygenation without brain damage (Lutz and Milton, 2004; Kesaraju et al, 2009). It is thus likely in such a model that modulation of a particular molecular signaling cascade is adaptive rather than pathological. Determining survival mechanisms in such an alternative animal might help to disentangle the complexities of vulnerable mammalian neurons and provide insights into the mechanisms of mammalian anoxic brain damage and survival, including critical upstream signals that regulate intracellular events.
One signaling molecule thought to affect AKT and the MAPK pathways is adenosine (AD), a neuroprotectant rapidly formed during ischemia as a result of the intracellular breakdown of ATP (Dunwiddie and Masino, 2001). AD has a role in preconditioning in the brain (Ciccarelli et al, 2007) and heart (Downey et al, 2007). Recent studies have shown that activation of AKT confers protection during adenosine mediated and myocardial ischemic preconditioning (Ban et al, 2008); however, the intracellular signaling pathways mediating the protective effects of adenosine are still under investigation (Brust et al, 2006). In turtles, it has been shown that AD is critical at the physiological level to anoxic neuronal survival, acting as a ‘retaliatory metabolite’ to balance energy supply and demand (Nilsson and Lutz, 1991). The AD levels increase as much as 12-fold over the initial 1 to 2 hours anoxia (Nilsson and Lutz, 1991) accompanied by an increase in A1 receptor (A1R) sensitivity (Lutz and Manuel, 1999). The ADR activation modulates extracellular levels of excitotoxins (Milton et al, 2002; Milton and Lutz, 2005) and is a key element of channel arrest (Pek and Lutz, 1997). We have also recently reported that AD affects MAPK and AKT levels in the turtle brain
Materials and methods
All experiments were approved by the Florida Atlantic University Institutional Animal Care and Use Committee. Juvenile freshwater turtles (4 to 6 inch carapace) were obtained from a commercial supplier (Clive Longdon, Tallahassee, FL, USA). Turtles were maintained in freshwater aquaria on a 12-hour light–dark cycle and were fed three times weekly. Animals are killed for cell culture harvest by decapitation in accordance with the animal care standards established by the American Veterinary Medical Association for reptiles.
Cell Culture Preparation
We have successfully isolated and maintained turtle primary neuronally enriched cell cultures using a density centrifugation method adopted from Brewer (1997) (Milton et al, 2007). Briefly, brain tissue was minced aseptically and digested in a cocktail of enzymes (collagenase (25 U/mL), dispase (0.32 U/mL), hyaluronidase (1300 U/mL) in 4 mL of minimal essential media containing 10% fetal bovine serum (FBS), 56 units each of penicillin and streptomycin with gentle rocking for 4 hours before centrifugation at 750
Immunostaining
To confirm the neuronal phenotype, cultured neurons were double immunostained with primary antimouse NeuN antibody, antirabbit neurofilament and antirabbit cysteine string protein neuronal markers. Alexa Fluor 568 goat antimouse or antirabbit antibodies (Invitrogen, Molecular probes, Eugene, OR, USA) were used as secondary antibodies. Briefly, cells were fixed with 1 to 2 mL of freshly prepared 4% paraformaldehyde for 10 minutes. Cells were washed three times with phosphate-buffered saline and further permeabilized with 0.1% Triton X-100 in phosphate-buffered saline at room temperature. Cells were then washed with 0.1% Triton X-100 and nonspecific binding was blocked with blocking solution (0.5% bovine serum albumin in phosphate-buffered saline for 15 minutes. Cells were incubated overnight at 4°C with primary antimouse NeuN and antirabbit neurofilament/cysteine string protein antibodies (Chemicon, Billerica, MA, USA). After washings, this was followed by Alexa Fluor 568 goat antimouse/rabbit-secondary antibodies used for 1 hour in a 1:1000 dilution. After further washings, cells were prepared with Vectashield mounting medium (Vector laboratories, Inc., Burlingame, CA, USA) and coverslipped. Cells were observed using a confocal microscope equipped for epifluorescence.
Experimental Treatments
Cell cultures were used at ~14 DIV and existing media was replaced with minimal essential media containing the specific A1R blocker 8-cyclopentyl-1,3-dihydropylxanthine (DPCPX, 1 nmol/L) or the A1R agonist 2-chloro-
Protein Extractions
For protein extraction, neurons were washed with ice-cold phosphate-buffered saline. Cells were further treated with 50 μL of RIPA lysis buffer (5 mmol/L EDTA, pH 8.0, 0.15 mol/L NaCl, 1% Triton X-100, 10 mmol/L Tris-Cl, 2 mmol/L protease inhibitor pH 7.4) and scraped into individual tubes on ice for 10 minutes. The samples were triturated and centrifuged for 10 minutes at 18,500
Immunodetection
Proteins were detected by standard Western blot as described earlier, using horseradish peroxidase (HRP)-conjugated secondary antibodies (Milton et al, 2008). The rabbit polyclonal antibodies for the nonphosphorylated MAPKs and rabbit Bax polyclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). These included ERK1/2 (SC-93), JNK1 (SC-571), p38MAPK (SC-535), and Bax (SC-526). Rabbit polyclonal antibody to AKT (#9272) and phosphorylated antibodies phospho-AKT (#9271), phospho-p38MAPK (#4631), phospho-JNK (#4671), phospho-ERK1/2 (#4376), all were from Cell Signaling Technology (Danvers, MA, USA). Antirabbit Bcl-2 antibody was from Chemicon (Temecula, CA, USA).
The protein-secondary antibody complexes were identified using a chemiluminescent system (GE Healthcare, Piscataway, NJ, USA). Densitometric analysis was conducted using National Institute of Health Image J 1.60 software and band densities expressed as a percentage of the β-actin signal. Actin was used as a loading control, and its expression remained unchanged in all experimental conditions. Earlier studies in our laboratory have also indicated that β-actin levels do not change with anoxia or reoxygenation in this model (Milton et al, 2008). Data are expressed as percent change of the control group.
Statistically significant differences between treatments were determined by analysis of variance with
Results
We performed double-labeled immunostaining using NeuN, neurofilament, and cysteine string protein neuronal markers. Figure 1A clearly indicates the neuronal nuclei (green) and localization of the neuronal filaments in cytoplasm (red), and Figure 1B shows neuronal nuclei (green) and cysteine string protein (red) in the neuronal cytoplasm thus confirming the neuronal identity of our cultures.
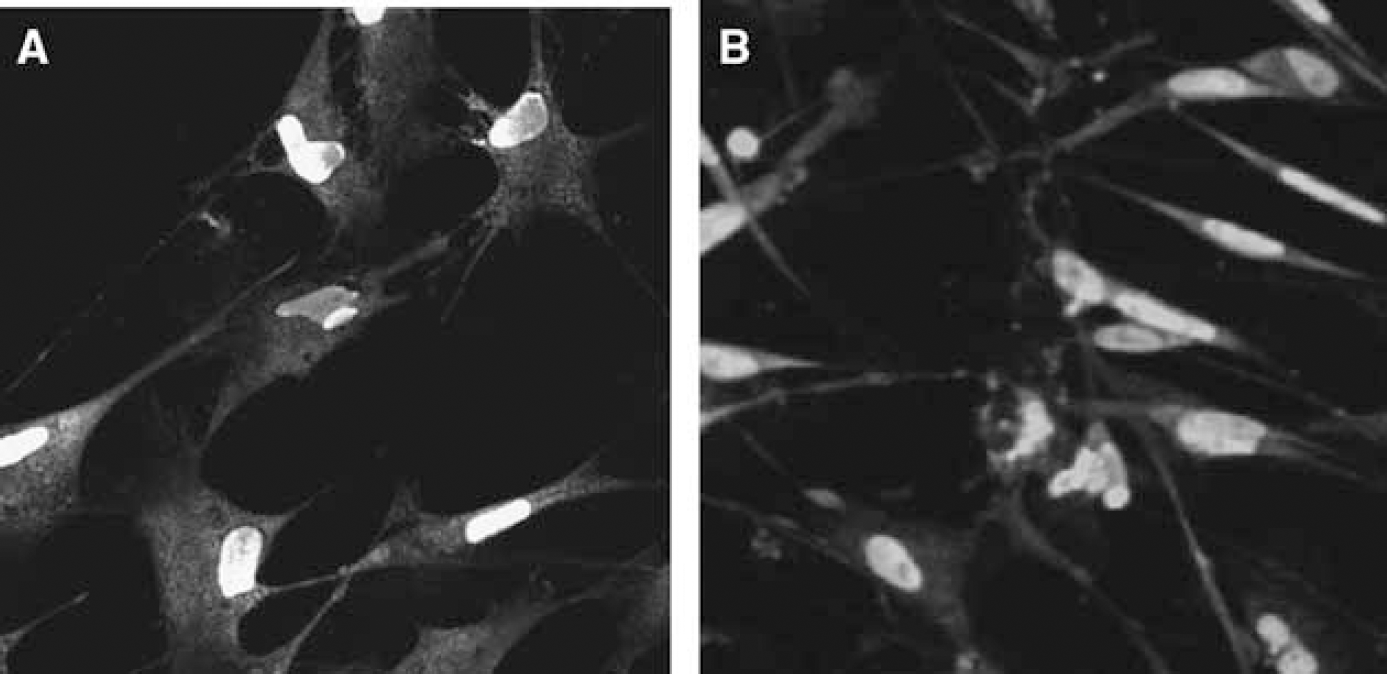
(
Alterations in Mitogen-Activated Protein Kinase/AKT Protein Levels in Normoxia and Anoxia Without Adenosine Receptor Agonists and Antagonists
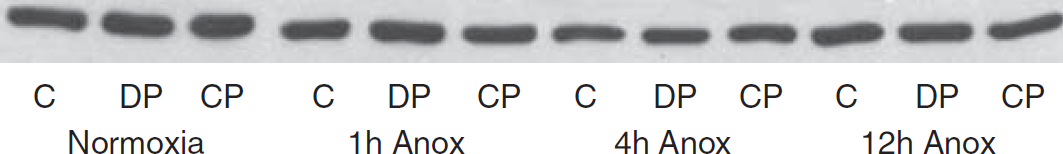
Representative Western blot showing expression of β-actin in turtle neuronally enriched cell cultures. Actin expression is unchanged in all experimental conditions. C, controls; CCPA, 2-chloro-
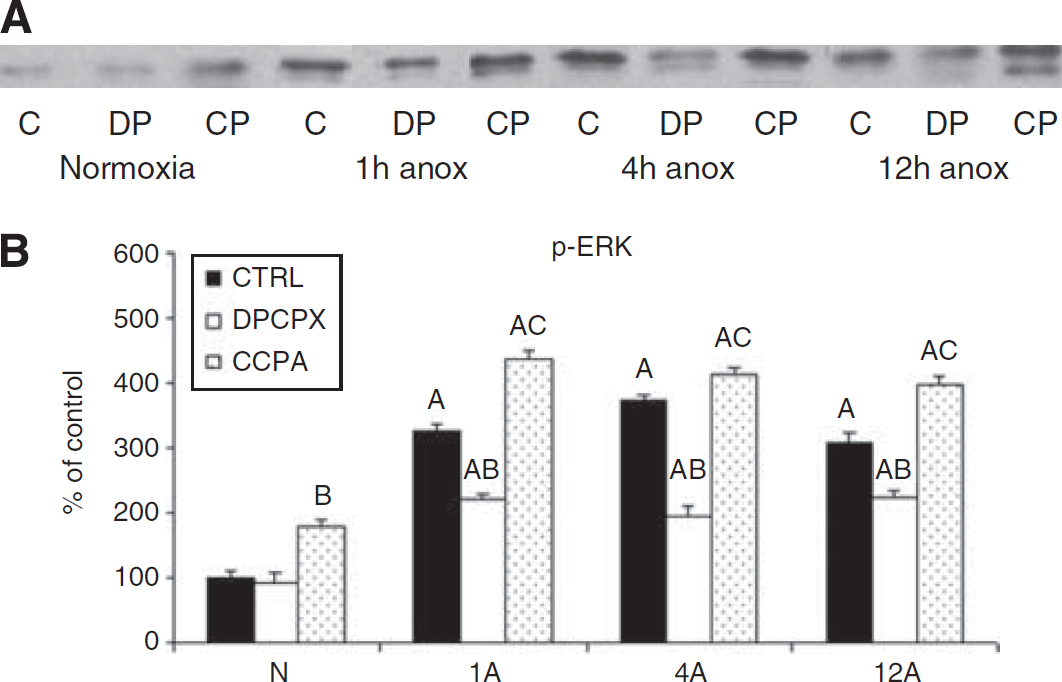
Representative Western blot of phosphorylated extracellular signal-regulated kinase (p-ERK) (
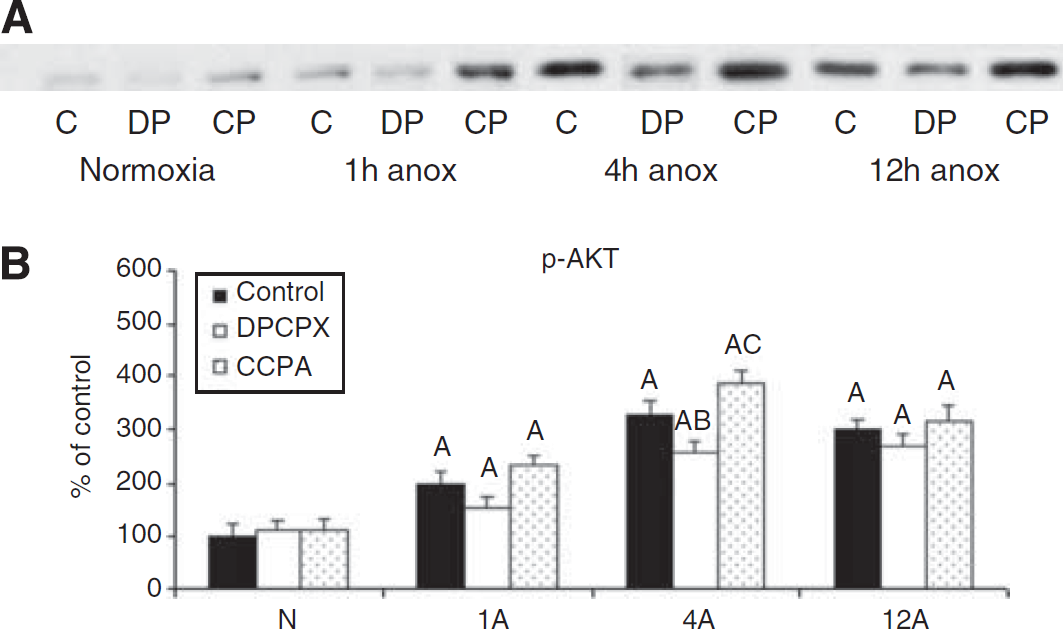
Representative Western blot of p-AKT (
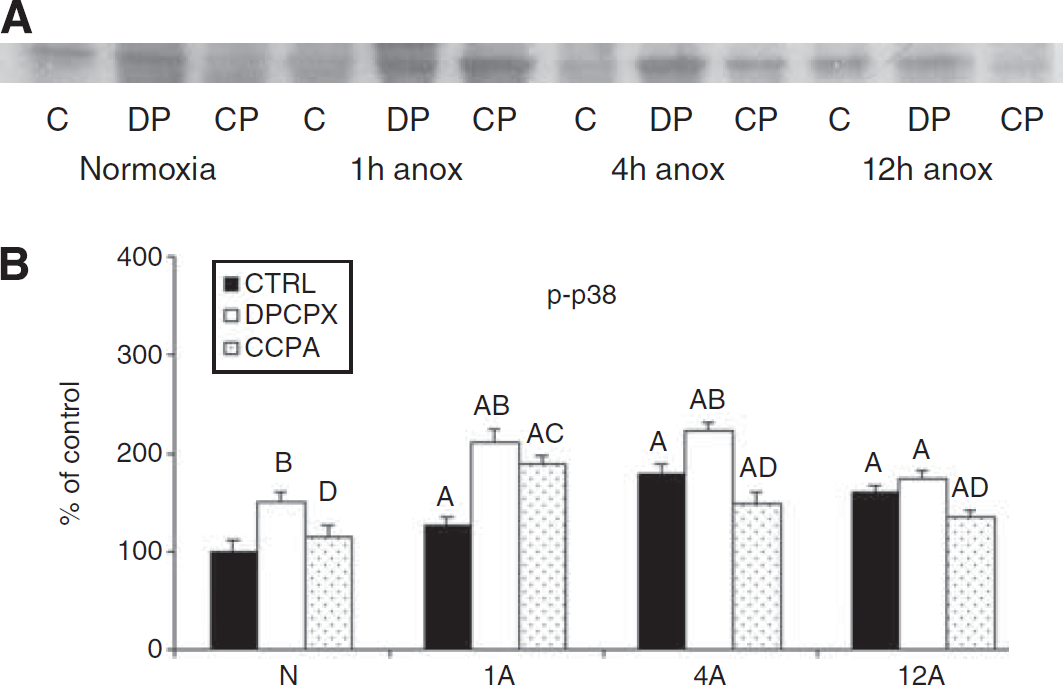
Representative Western blot of pp38 (
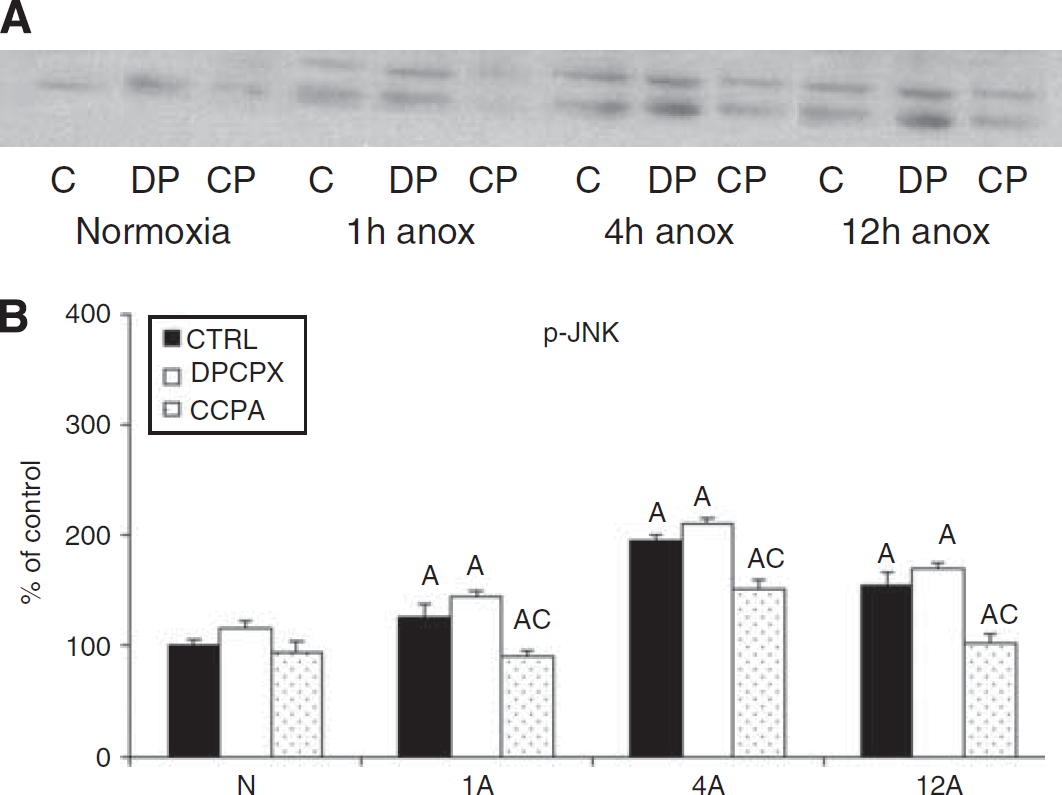
Representative Western blot of p-JNK (
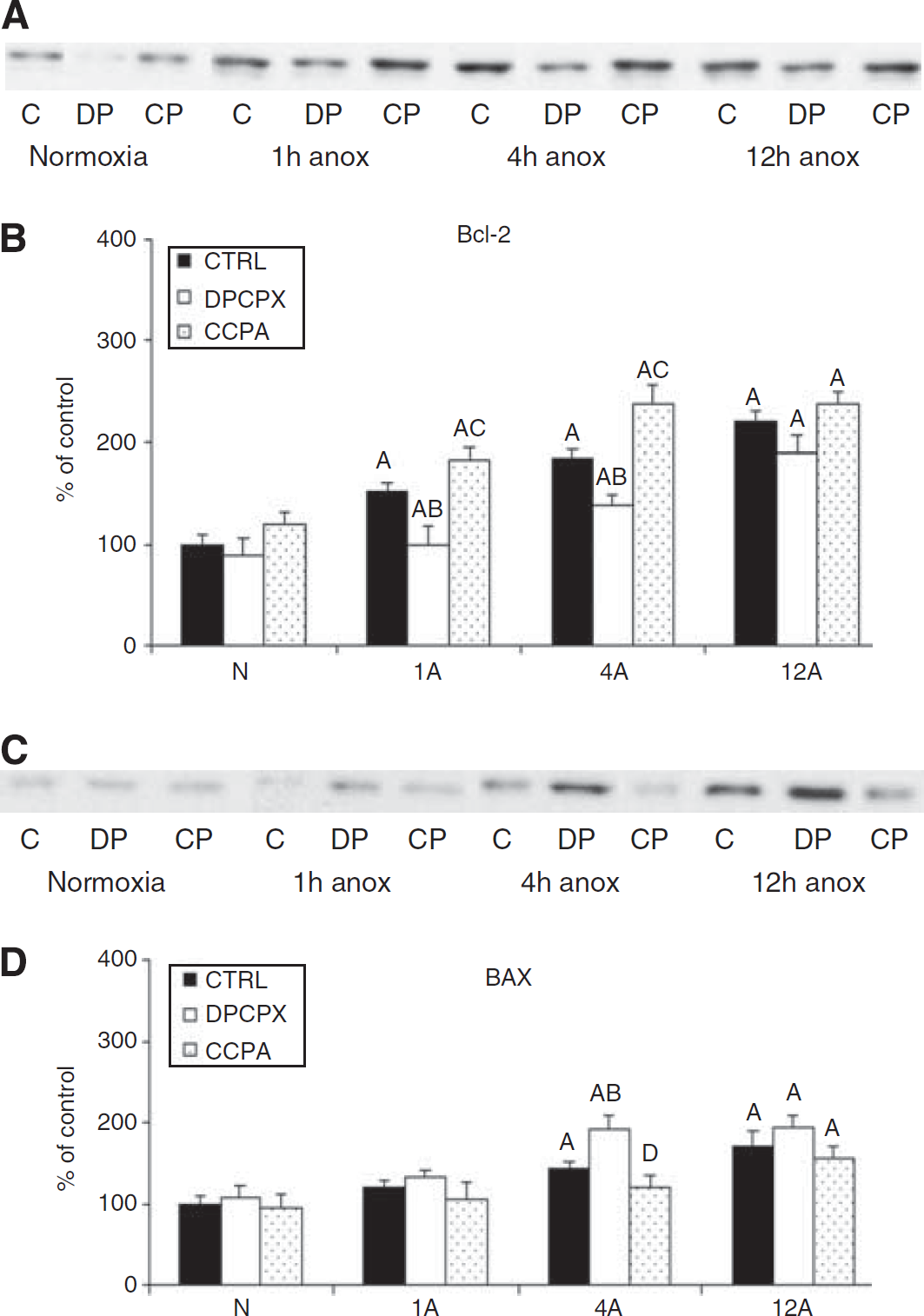
Representative Western blots of Bcl-2 and Bax (
Alterations in Mitogen-Activated Protein Kinase/AKT Pathway Components After Exposure to Adenosine Receptor Agonists and Antagonists
Alterations in Pro and Antiapoptotic Pathway Components After Exposure to Adenosine Receptor Agonists and Antagonists
Discussion
Although the physiology of anoxic survival in freshwater turtles such as
In contrast to the whole brain, we also observed a moderate increase in p38 and JNK activation in the cell cultures, though the increases were less than for the presumed pro-survival kinases. These results are similar to those observed in mammalian models where hypoxia/ischemia leads to the activation of all the MAPKs (Irving et al, 2000; Li et al, 2002; Wang et al, 2003), and together with the sustained MAPK activation suggests that the cells in culture are under more physiological stress during anoxia than is the intact brain, most likely due to the lack of glial support or such whole body adaptations as buffering capacity.
Mammalian hypoxia/ischemia experimental models to date have examined the role of MAPK and AKT signaling cascades in determining the outcome of cerebral ischemia. However, the triggers to the activation of these complex cascades in the brain have not been widely investigated. Fewer studies have focused on the immediate effects of adenosine on preventing neuronal cell death after ischemia in mammals, though these studies support our results showing A1R protection occurring through the upregulation of ERK and AKT and the suppression of p38MAPK (Gervitz et al, 2002; Ciccarelli et al, 2007; Ban et al, 2008). A reduction in blood flow to the mammalian brain during hypoxia/ischemia causes a steep elevation in the extracellular levels of adenosine as ATP breaks down; adenosine in turn acts as a neuroprotectant (Canals et al, 2005; Dunwiddie and Masino, 2001). Unlike mammals, however, the adenosine increase in the turtle brain is temporary (Nilsson and Lutz, 1991). By 4 hours anoxia, the turtle has entered a fully hypometabolic state, in which energy consumption is suppressed to match anaerobic energy production, allowing ATP levels to return to normal (see reviews Lutz and Milton, 2004; Milton and Prentice, 2007). Thus, in the turtle, the initial 1 to 2 hours anoxia is a crucial period of physiological stress, which it survives by applying a suite of adaptations, and it is during this transition phase that we see significant molecular changes, including increases in a variety of heat shock proteins (Kesaraju et al, 2009) and the MAPK reported here. The initial
As previously reported
In mammals, several studies have shown that neuroprotective AD effects even in acute ischemia result from downstream molecular events triggered by ADR activation, including activation of AKT (Gervitz et al, 2002) ERK1/2, and other MAP kinases (Brust et al, 2006). Activation of the ERK pathway leads to the phosphorylation of a wide range of cellular substrates and activates transcription of cAMP response element binding protein, which activates the pro-survival Bcl-2 and suppresses apoptotic proteins such as BAD, Bim, and the caspases (Chan, 2004; Sawe et al, 2008) and prevent apoptosis. Similarly, the protection offered by AKT is thought to be in part through its interactions with the Bcl-2 family of proteins. The AKT targets cAMP response element binding protein to activate pro-survival mechanisms (Shibata et al, 2002) and also phosphorylates BAD (Datta et al, 1997; Chan, 2004; Kamada et al, 2007), thus preventing its translocation to the mitochondria, inhibiting the release of apoptogenic proteins (Wang et al, 2007), and decreasing other stress pathways (Kamada et al, 2007; Wang et al, 2007). Ciccarelli et al (2007) have shown that AD1R reduces apoptosis by suppressing JNK and p38MAPK, simultaneously activating ERK and AKT, and decreasing the levels of pro-apoptotic proteins like Bad while elevating levels of antiapoptotic Bcl-X(L). The results of this study suggest that the links between AD and cell survival in anoxia/ischemia in turtles also occur through the MAPKs and their downstream effects. In the turtle cultures, both Bcl-2 and Bax increased along with ERK and AKT, but with Bcl-2 more strongly upregulated than Bax, suggesting that the molecular pathways are tilted towards cell survival even with accompanying (but smaller) increases in p38MAPK and JNK.
In addition, Bcl-2 has been shown to have a major role in suppressing cell death during apoptotic and oxidative stress cell injury by enhancing levels of antioxidants and suppressing the generation of free radicals (Lee et al, 2001). In this study, the anoxic increases in Bcl-2 expression above Bax were reversed by ADR blockade; ADR blockade is accompanied by an increase in ROS production and doubling of cell death (Milton et al, 2007), implicating that Bcl-2 protein levels can be altered by adenosine receptor activation, which might be directly linked to ROS suppression in species. These studies suggest that the pathway of ROS suppression in turtles is similar to that in mammals, but is expressed more robustly and hence is more successful in protecting turtle neurons against anoxia and reoxygenation damage.
Thus, in addition to its other myriad neuroprotective effects in the anoxic turtle brain; we have shown that activation of ADR receptor also increases cell survival by modulating molecular pathways in favor of cell survival. Pretreatment with CCPA increased activation of ERK and AKT, which further lead to elevation of Bcl-2, inhibition of Bax, and reduced activation of p-p38; these changes in turn are thought to stabilize the mitochondria and reduce ROS release (Gervitz et al, 2002; Brust et al, 2006). The overall observations from earlier investigations and this current study clearly indicate a link between the physiological mechanisms of survival (elevated AD, low ROS) and molecular adaptations in this anoxia tolerant model. Through utilization of an animal model of anoxia tolerance, honed for survival by millions of years of adaptation, and investigating the differences between