
Abstract
Bcl-2 plays a pivotal role in the control of cell death and is upregulated by ischemic tolerance. Because Bcl-2 expression is regulated by the transcription factor cyclic AMP response element-binding protein (CREB), we investigated the role of CREB activation in two models of ischemic preconditioning: focal ischemic tolerance after middle cerebral artery occlusion (MCAO) and in vitro ischemic tolerance modeled by oxygen–glucose deprivation (OGD). After preconditioning ischemia (30 minutes MCAO or 30 minutes OGD), phosphorylation of CREB was increased, and there was an increased interaction between the bcl-2 cyclic AMP-responsive element (CRE) promoter and nuclear proteins after preconditioning ischemia in vivo and in vitro. Chromatin immunoprecipitation revealed an increased interaction between CREB-binding protein and the bcl-2 CRE rather than CREB, after preconditioning ischemia. Ischemic tolerance was blocked by a CRE decoy oligonucleotide, which also blocked Bcl-2 expression. The protein kinase A inhibitor H89, the calcium/calmodulin kinase inhibitor KN62, and the MEK inhibitor U0126 blocked ischemic tolerance, but not the phosphatidylinositol 3-kinase inhibitor LY294002. H89, KN62, and U0126 reduced CREB activation and Bcl-2 expression. Taken together, these data suggest that after ischemic preconditioning CREB activation regulates the expression of the prosurvival protein Bcl-2.
Keywords
Introduction
Ischemic tolerance is the phenomenon whereby sublethal ischemia or other stressors protect the brain from subsequent severe ischemia (Dirnagl et al, 2003; Kirino, 2002; Schaller and Graf, 2002). The mechanisms responsible for tolerance are still unclear, although in the brain evidence favors a gene-based mechanism for tolerance, given that there is a delay before tolerant effects are observed: 1 to 3 days in vivo (Chen et al, 1996; Shimizu et al, 2001) or 24 hours in vitro (Bossenmeyer-Pourie and Daval, 1998; Gonzalez-Zulueta et al, 2000; Tauskela et al, 1999). Further, ischemic tolerance is blocked by inhibitors of protein synthesis (Barone et al, 1998; Gonzalez-Zulueta et al, 2000). While many genes are upregulated by ischemia, not all are relevant to the induction of tolerance. For example, although expression of heat shock proteins (HSP) increases with a temporal profile comparable to tolerance (Chen et al, 1996; Yagita et al, 2001), ischemic tolerance may occur in the absence of HSP expression (Abe and Nowak, 1996). Perhaps a more convincing gene candidate for mediating tolerance is the cell death repressor protein, Bcl-2.
The Bcl-2 family of genes are involved in the regulation of cell death processes (Adams and Cory, 1998, 2001). Bcl-2 plays a pivotal role in the control of cell death via the stabilization of the mitochondria membrane potential, thereby preventing cytochrome c and APAF-1 release into the cytosol, which activates proapoptotic pathways (Shimizu et al, 1999; Yang et al, 1997). Bcl-2 expression increases in neurons that survive ischemic infarcts (Chen et al, 1995), whereas reductions in Bcl-2 by antisense oligonucleotides exacerbate neuronal loss (Chen et al, 2000). Overexpression of Bcl-2 in transgenic mice, or by viral vectors, reduces neuronal cell death during development or after neuronal injury, such as ischemia and facial nerve axotomy (Alberi et al, 1996; Linnik et al, 1995; Martinou et al, 1994; Sagot et al, 1995). Recently, it has been shown that regulation of Bcl-2 expression may play a critical role in ischemic preconditioning (Bossenmeyer-Pourie and Daval, 1998; Shimizu et al, 2001).
Bcl-2 gene expression is controlled by multiple promoters in the 5′-regulatory region. Of particular interest, bcl-2 contains a cyclic AMP-responsive element (CRE) located between −1640 and −1529, which controls bcl-2 levels via the activation of a transcription factor, CRE-binding protein (CREB) (Freeland et al, 2001; Ji et al, 1996; Pugazhenthi et al, 2000; Riccio et al, 1999; Wilson et al, 1996).
Cyclic AMP-responsive element-binding protein mediates cyclic AMP-induced gene expression via its binding to a CRE in the gene promoter region (Montminy and Bilezikjian, 1987). The transcriptional activation of CREB involves several key steps, but is crucially dependent on phosphorylation of Ser 133 (Shaywitz and Greenberg, 1999; Walton and Dragunow, 2000) and the recruitment of CREB-binding protein (CBP) (Cardinaux et al, 2000; Chrivia et al, 1993). Cyclic AMP-responsive element-binding protein is a substrate for phosphorylation by multiple protein kinases, including protein kinase A (PKA), calcium/calmodulin dependent kinase II and IV (CAMK), p42/p44 mitogen-activated protein kinase (p42/p44 MAPK: otherwise known as ERK1/2), and Akt (also known as protein kinase B) (Beitner-Johnson and Millhorn, 1998; Bonni et al, 1999; Pugazhenthi et al, 2000; Sun et al, 1994; Walton and Dragunow, 2000; Zanassi et al, 2001). As such, CREB is a convergence point for many intracellular signaling pathways, some of which are activated after preconditioning ischemia. Indeed, hypoxia and ischemia increase CREB phosphorylation (Jin et al, 2001; Tanaka, 2001; Tanaka et al, 1999; Walton et al, 1999; Walton and Dragunow, 2000), although the protein kinase responsible is not yet clear (Beitner-Johnson and Millhorn, 1998). Furthermore, CREB activation has been shown to be required for the acquisition of ischemic tolerance in gerbil CA1 neurons after global ischemia (Hara et al, 2003).
Upregulation of gene expression is an important mechanism for cell survival in tolerance; however, the relationship between CREB transcription activity and tolerance is not yet clear. Furthermore, it is unclear via which intracellular signaling pathway CREB activation is regulated after ischemic preconditioning. Here, we use a combination of in vitro and in vivo studies to suggest that the neuroprotective effect of preconditioning is regulated by an increase in CREB-mediated Bcl-2 expression and involves multiple intracellular protein kinase pathways.
Materials and methods
Ischemic Tolerance Model In Vivo
All procedures were performed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), in accordance with protocols approved by the Legacy Institutional Animal Care and Use Committee and the principles outlined in the National Institute of Health Guide for the Care and Use of Laboratory animals. Middle cerebral artery occlusion (MCAO) was induced in adult male Sprague–Dawley rats (280–350 g) using the intraluminal filament technique, as described previously (Chen et al, 1996). Rats were anesthetized with 4% isoflurane and ventilated with 1.5% isoflurane (70% N2O, 30% O2) to maintain blood gas variables within physiological range. Rectal and temporalis muscle temperature were kept at 37.0°C to 37.5°C. A 3-0 surgical monofilament nylon suture with rounded tip was introduced into the left internal carotid artery and advanced 20 to 21 mm past the carotid artery bifurcation to occlude the middle cerebral artery (MCA). After completion of the MCAO (30 or 100 mins), the suture was withdrawn to allow reperfusion. Second MCA occlusions were performed 72 hours after preconditioning with 30-min MCAO. As a control, sham-operated rats underwent identical surgery but did not have the suture inserted. Animals were euthanized (phenobarbitone overdose) at various time points after MCAO or after 24 hours in nonischemic controls. For Western blotting and electrophoretic mobility shift assay, brains were rapidly removed, placed on ice and ipsilateral cortex was dissected, and stored at −80°C until assay. For immunohistochemistry studies, whole brains were removed and then frozen at −30°C in methylbutane.
Infarct Volume Measurement
Infarct volume was assessed using a triphenyltetrazolim hydrochloride (TTC) method, as described previously (Chen et al, 1996). Animals were subject to (1) 30-min MCAO, (2) 100-min MCAO, or (3) 30-min MCAO followed by 72 hours reperfusion and 100-min MCAO. Animals were killed 24 hours after the last period of MCAO by an overdose of phenobarbitone (i.p.); brains were rapidly removed and sectioned coronally at 2-mm intervals. Sections were immersed in TTC (2%) at 37°C for 20 mins, followed by formaldehyde (4%) for 15 mins. The hemispheric infarct area in each section was calculated by subtracting the area of normal TTC-stained brain in the ipsilateral cortex from the contralateral area, as described previously (Swanson et al, 1990).
Western Blotting
Western blotting was performed as described previously (Meller et al, 2003). Tissue samples were lysed in a nondenaturing buffer containing the protease inhibitors phenylmethylsulfonylfluoride (100 μg/mL), aprotinin (1 μg/mL), leupeptin (1 μg/mL), pepstatin (1 μg/mL), NaF (50 mmol/L), Na3VO4 (2 mmol/L) and phosphatase cocktail inhibitor (Sigma, St Louis, MO, USA). Protein concentration was determined by Bradford reagent spectrophotometrically at A595. Protein samples (50 μg) were denatured in a gel-loading buffer at 100°C for 5 mins and then loaded onto 12% SDS-polyacrylamide gels to detect Bcl-2, CREB, or phosphorylated CREB proteins or onto 7.5% SDS-polyacrylamide gels to detect CBP protein. Proteins were transferred to polyvinylodene difluoride membranes and incubated with primary antibodies at 4°C overnight: anti-CREB and phospho-CREB (Ser 133) (Cell Signaling Technology, Beverly, MA, USA), anti-Bcl-2 and CBP (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were incubated with anti-rabbit IgG conjugated to horseradish peroxidase (Cell Signaling Technology, Beverly, MA, USA) followed by chemiluminescense detection (NEN Life Science Products, Boston, MA, USA) and then exposed to film. Images were captured using a Dage 72 camera.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear protein for EMSA assay was prepared by lysing brain or cell samples in a high-salt buffer containing Hepes (20 mmol/L; pH 7.9), NaCl (420 mmol/L), NaF (20 mmol/L), Na3VO4 (1 mmol/L), Na4P2O7 (1 mmol/L), EDTA (1 mmol/L), EGTA (1 mmol/L), DTT (1 mmol/L), glycerol (20%), PMSF (0.5 mmol/L), leupeptin (1 μg/mL). The samples were freeze-thawed on dry ice and the cell lysate was centrifuged at 14,000g for 20 mins. The bcl-2 CRE probe sense (5′-GAACCGTGTGACGTTACGC-3′) and antisense (5′-TGCGTAACGTCACACGGTTC-3′) oligonucleotides (200 pmol) were annealed in 100 μL of an annealing buffer containing Tris (10 mmol/L; pH 8.0), NaCl (100 mmol/L) and EDTA (1 mmol/L) by heating at 90°C for 5 mins and then reducing the temperature to 25°C over 30 mins. The annealed oligonucleotides (30 pmol) were labeled by TdT kinase reaction and purified using a G-25 column.
Nucleic proteins (10 μg) were incubated with oligonucleotides and poly(dI-dC)-poly(dI-dC) in a buffer containing Hepes (12 mmol/L; pH 7.9), NaCl (60 mmol/L), DTT (1 mmol/L), EDTA (0.2 mmol/L), and glycerol (8%) at room temperature for 20 mins. The DNA–protein complexes were resolved by a 5% polyacrylamide gel containing 0.5 × TBE and 2.5% glycerol, at 200 V for 2 hours. The gel was dried and exposed to an X-ray film at −80°C with an intensifying screen. In some experiments, an excess of cold (unlabeled) oligonucleotide or CREB antibody was added to the reaction mixture.
Neuronal Cell Culture
Cortical neuronal cultures were prepared from 1- to 3-day-old Sprague–Dawley rat pups using the method of Goslin (1998) as described previously (Stenzel-Poore et al, 2003). Briefly, cortices from 10 to 12 rat pups were dissected, dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA) and plated out at a density of 400,000 cells per coverslip in Neurobasal-A/B27 media (Invitrogen, Carlsbad, CA, USA). The cells were used after 7 to 10 days in culture, when cultures consist of approx. 80% to 90% neurons. For Western blotting, EMSA and chromatin immunoprecipitation assays, cells were plated out onto 10-cm polylysine-coated culture dishes (Primara; Becton Dickinson, San Jose, CA, USA) and samples were collected at various time points after oxygen–glucose deprivation (OGD).
In Vitro Ischemic Tolerance Model
Oxygen–glucose deprivation was performed by washing the cells with phosphate-buffered saline (PBS) (0.5 mmol/L CaCl2, 1 mmol/L MgCl2; pH 7.4) and placing culture dishes in an anaerobic chamber (Forma Scientific, Marjetta, OH, USA) (85% N2, 5% H2, 10% CO2; 35°C). Anaerobic conditions in the chamber were monitored using Gaspack anaerobic indicator strips (Becton Dickinson, San Jose, CA, USA). Oxygen–glucose deprivation was terminated by removing cells from the chamber, replenishing with Neurobasal A media and placing them back into the normoxic incubator. Cultured cells were subjected to the following treatments: (1) control cells were washed with PBS and maintained in Neurobasal A media, (2) 30-min OGD (preconditioning) and 48-hour recovery in Neurobasal A media, (3) 120-min OGD (toxic ischemia) followed by 24-hour recovery in Neurobasal A media, (4) 30-min OGD followed by 24-hour recovery, then 120-min OGD followed by 24-hour recovery in Neurobasal A media.
Some cells were incubated with a CRE decoy oligonucleotide based on the bcl-2 CRE sequence or control oligonucleotides, similar to those previously described by Park et al (1999) (all 0.5 μmol/L) (CRE decoy oligonucleotide 5′-TGACGTCAGAGAGCGCTCTCTGACGTCA-3′; Control CRE mis-match sequence 5′-TAGCTGCAGAGAGCGCTCTCTGCAGCTA-3′; (all linkages phosphorothioate protected) (Invitrogen Carlsbad, CA, USA or Sigma-Genosys, The Woodlands, TX, USA). Oligonucleotides were heated to 95°C and then slowly cooled to room temperature before incubation with the cells. Some cells were incubated with the following protein kinase inhibitors for 24 hours after 30-min OGD preconditioning and before the 120-min OGD challenge; PKA inhibitor H89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride) 0.5 μmol/L, CAMK inhibitor KN 62 (1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-
Cell Viability Assay
For cell viability assays, coverslips containing cortical cells were incubated with propidium iodide (1.5 μg/mL) for 2 mins, washed with PBS and fixed with 4% formaldehyde. Cells were permeabilized with 0.1% Triton X-100 and then mounted onto glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA, USA). Cell viability was determined as the ratio of propidium iodide-stained cells, to the total number of DAPI-stained cells visualized using a fluorescence microscope.
Immunohistochemistry and Immunocytochemistry
Fresh frozen brain sections (12 μm) or cultured cells on coverslips were permeabilized with 3% Triton X-100. The samples were preblocked with 2% goat serum and 1% BSA, followed by incubation with primary antibody at 4°C overnight. The samples were washed three times with PBS and incubated with secondary antibody for 1 hour at 37°C (Jackson Labs). Slides were washed and mounted in a medium containing DAPI (Vector Laboratories, Burlington, CA, USA). Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling was performed according to manufacturer's instructions (Roche, Indianapolis, IN, USA). Immunolabeling was studied using a Leica microscope under Ex/Em wavelengths of 340/425 nm (blue), 500/550 nm (green) and 580/630 nm (red), respectively. Images were collected using an Optronics DEI-750 3-chip camera equipped with a BQ 8000 sVGA frame grabber and analyzed using Bioquant (Nashville, TN, USA).
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed according to the method described by Impey et al (2002). After preconditioning OGD, cells were fixed with paraformaldehyde (2.0%, 2 mins) and lysed using the following buffer; Tris (10 mmol/L, pH 8.0), NaCl (140 mmol/L), glycerol (5%), sodium deoxycholate (0.1%), SDS (0.1%), Triton X-100 (1%), and Protease inhibitors with EDTA (Roche Complete™, Roche, Indianapolis, IN, USA). Samples were cleared (12,000g; 15 minutes) and 800 μg of tissue was subjected to immunoprecipitation with anti-CREB (Abcam, Cambridge, MA, USA) or anti-CBP (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in a total volume of 1.0 ml. After precipitation with protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA), the DNA crosslinked to the CREB was released by heating in 0.1 mol/L NaHCO3 for 16 hours at 65°C, and purified using a PCR clean-up kit (Qiagen, Valencia, CA, USA). The DNA was subject to PCR with primers specific to the bcl-2 CRE promoter region (antisense 5′-TCGCACACTCACTGGGTTACG-3′ and sense 5′-GCGCCTGATTTCCTGTACG-3′) for 38 cycles using Fast start Taq (Roche). The bcl-2 promoter region was identified on the rat genbank (http://www.hgsc.bcm.tmc.edu) as a part of a BAC Sequence (Rat CH236 194D24 contig 15-18). The start site and promoter region were predicted using the Mat-Inspector software (Genomatix, Germany). BlastN searches show low homology with rat DNA sequences, except for the rattus norvegicus bcl-2 gene and promoter (accession number AF531426). (Note there is a single base mismatch between the rat GenBank sequence and AF531426 for the sense oligonucleotide.) Amplicons were run on 2% agarose or 10% acrylamide gels and stained with ethidium bromide. Amplicons for bcl-2 CRE ran as a single band with a weight of 250 bp. The coding bcl-2 region (accession number NM_016993) was amplified with the following primers (sense 5′-TTCCAGCCTGAGAGCAACC-3′; antisense 5′-CCACAGAGCGATGTTGTCC-3′); the amplicon ran as a single band at a weight of approximately 370 bp.
Data Analysis
Data are presented as mean±standard deviation of n determinations. Data were analyzed using repeated-measures one-way analysis of variance (ANOVA) with Bonfferoni's post hoc test (Graphpad Prism, Graphpad Software, San Diego, CA, USA). Significance was accepted at P<0.05.
Results
Ischemic Preconditioning Reduces Neuronal Damage after Harmful Ischemia and Increases Bcl-2 Expression In Vivo
We investigated the role of CREB in mediating the increase of Bcl-2 expression after preconditioning in a focal model of ischemic tolerance (Figure 1A). Ischemic infarct size in rats after MCAO was determined using the vital dye TTC. Cortical infarction after 100 mins MCAO was reduced by preconditioning the animals with 30-min MCAO, 72 hours before the injurious period of ischemia (Figure 1B). In contrast, 30-min MCAO did not cause infarction. We also assessed DNA damage using TUNEL. There was no TUNEL labeling in sham control or animals subject to 30-min MCAO. In contrast, there was a robust increase in TUNEL-positive cells in the brains of animals subjected to 100-min MCAO. The increase in TUNEL-positive cells was reduced in animals subjected to preconditioning 30-min MCAO, 72 hours before 100-min MCAO (Figure 1C).
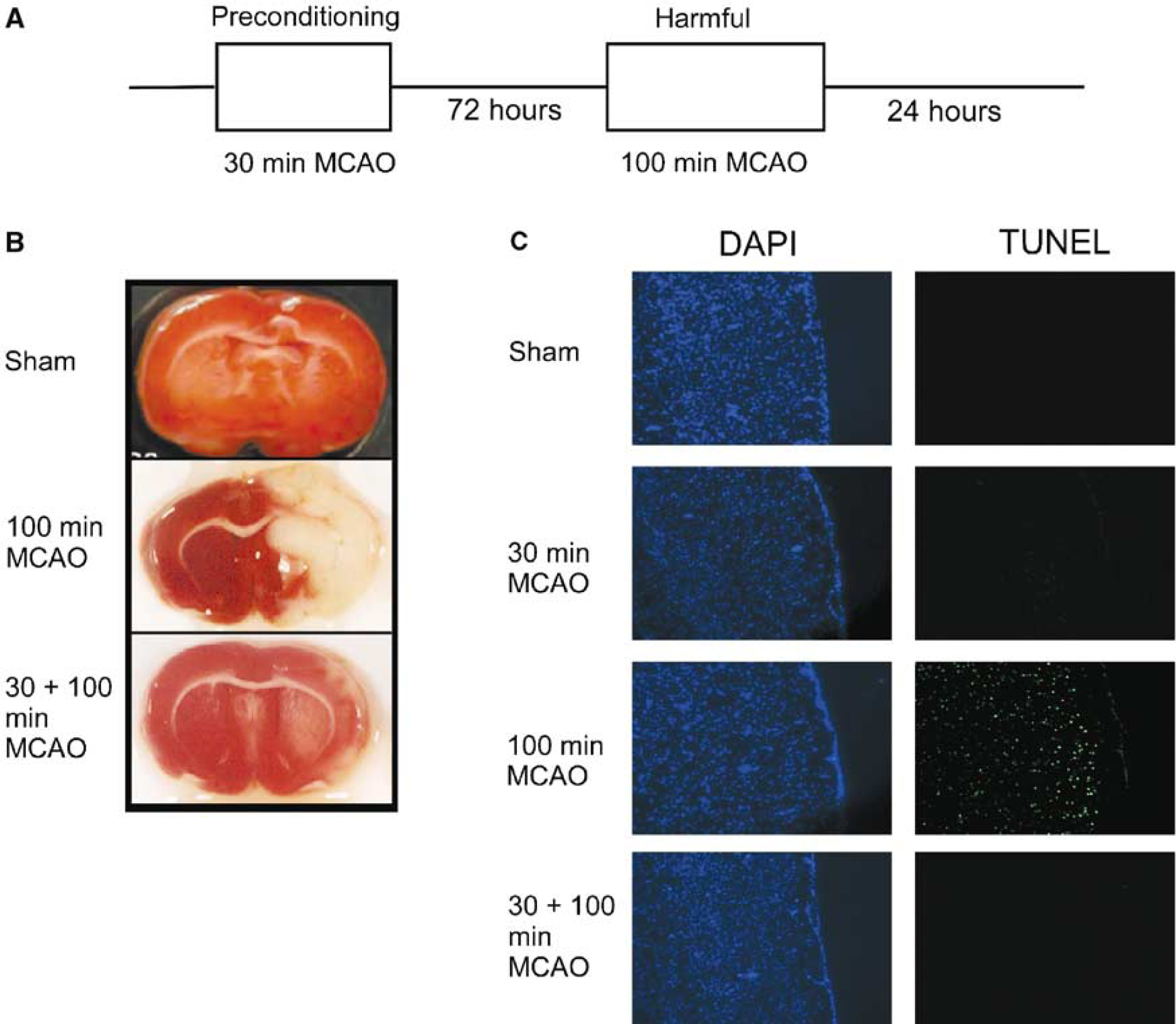
Preconditioning the brain with mild ischemia results in the brain being tolerant to harmful ischemia. (
Protein expression in the protected region of the cortex was measured using Western blotting. Consistent with previous reports (Bossenmeyer-Pourie and Daval, 1998; Shimizu et al, 2001), we detected an increase in Bcl-2 expression after 30-min MCAO and levels remained elevated for up to 72 hours (Figure 2A). In comparison, after harmful ischemia (100-min, MCAO), there was no increase in Bcl-2 expression (not shown).
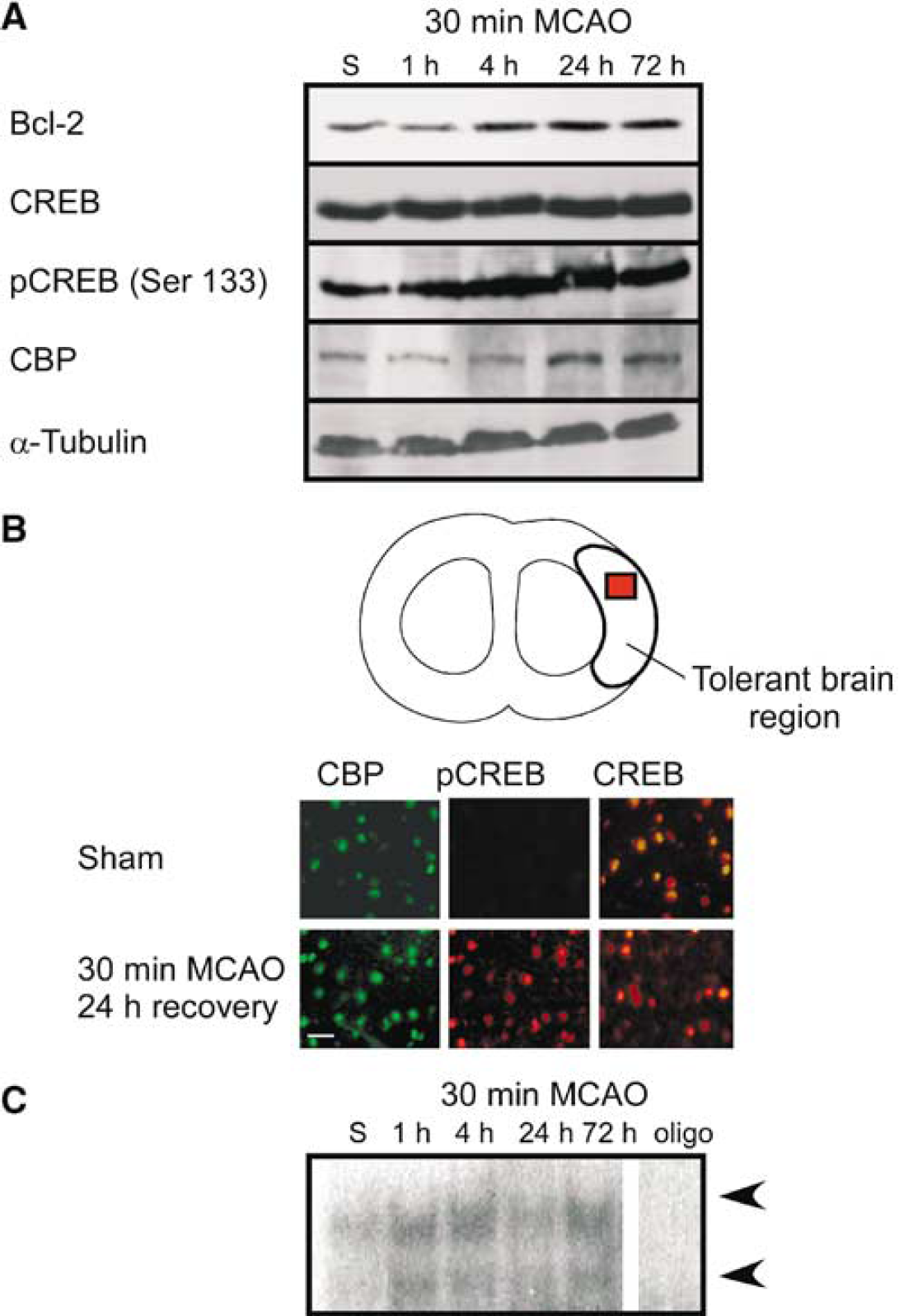
Preconditioning ischemia activates CREB in the cortex. (
Preconditioning Ischemia Increases CREB Activation In Vivo
It has been shown that Bcl-2 expression might be regulated by the transcription factor CREB (Wilson et al, 1996); hence, we investigated the effect of preconditioning ischemia on CREB expression and phosphorylation, using Western blotting. CREB was expressed as a 43-kDa protein in control (sham) animals. CREB expression did not change significantly after either 30-min MCAO (Figure 2A) either. CREB phosphorylated on Ser 133 has been shown to increase its activity (Walton and Dragunow, 2000), and phosphorylation of CREB (Ser 133) levels increased 1 hour after 30-min MCAO and remained elevated for up to 72 hours (Figure 2A). CREB-binding protein (CBP) is expressed at low levels as a 300-kDa protein in control cortex. CREB-binding protein expression was slightly increased at 24 and 72 hours after 30-min MCAO (Figure 2A). These results suggest that, after preconditioning ischemia, CREB is phosphorylated at Ser 133, which may increase its activity.
We used immunohistochemistry to investigate the expression of CBP, CREB, and phosphorylation of CREB after preconditioning. Sections taken from the cortical area protected by 30-min MCAO (as shown in Figures 1B and 2B) show an increase in phospho-CREB (Ser 133) immunoreactivity after 30-min MCAO and 24 hours reperfusion (Figure 2B). CREB-binding protein and CREB immunoreactivity show no difference between sham control and animals treated with 30-min MCAO with reperfusion for 24 hours (Figure 2B).
To investigate the regulation of bcl-2 by CREB, we used an EMSA to assess the binding of transcription factors to the bcl-2 CRE site after preconditioning. Nuclear extracts were prepared from brain samples of rats subjected to 30 minute MCAO and various reperfusion times (1 to 72 hours). We observed low basal interaction of nuclear proteins with the bcl-2 CRE oligonucleotide (Figure 2C). The interaction was increased after 30-min preconditioning ischemia for up to 72 hours (Figure 2C). Incubating the extracts with excess cold oligonucleotide (Figure 2C) abolished the interaction of the nuclear extracts with the bcl-2 CRE. These results suggest that increased binding of transcription factors to the bcl-2 CRE region may regulate the increase in Bcl-2 expression after preconditioning.
Preconditioning Ischemia Increases CREB Activation In Vitro
The role of CREB in regulating ischemic tolerance was further investigated in an in vitro model of ischemia (Figure 3A). Preconditioning cells with 30-min OGD reduces 120-min OGD-induced cell death when the interval between preconditioning and harmful OGD is 24 hours (Figure 3B). The total number of cells in the cultures did not vary after either 30- or 120-min OGD (Figure 3C). The protective effect of the preconditioning was eliminated in the presence of the protein synthesis inhibitor cycloheximide (0.5 μmol/L) but not vehicle control (0.1% DMSO) (not shown).
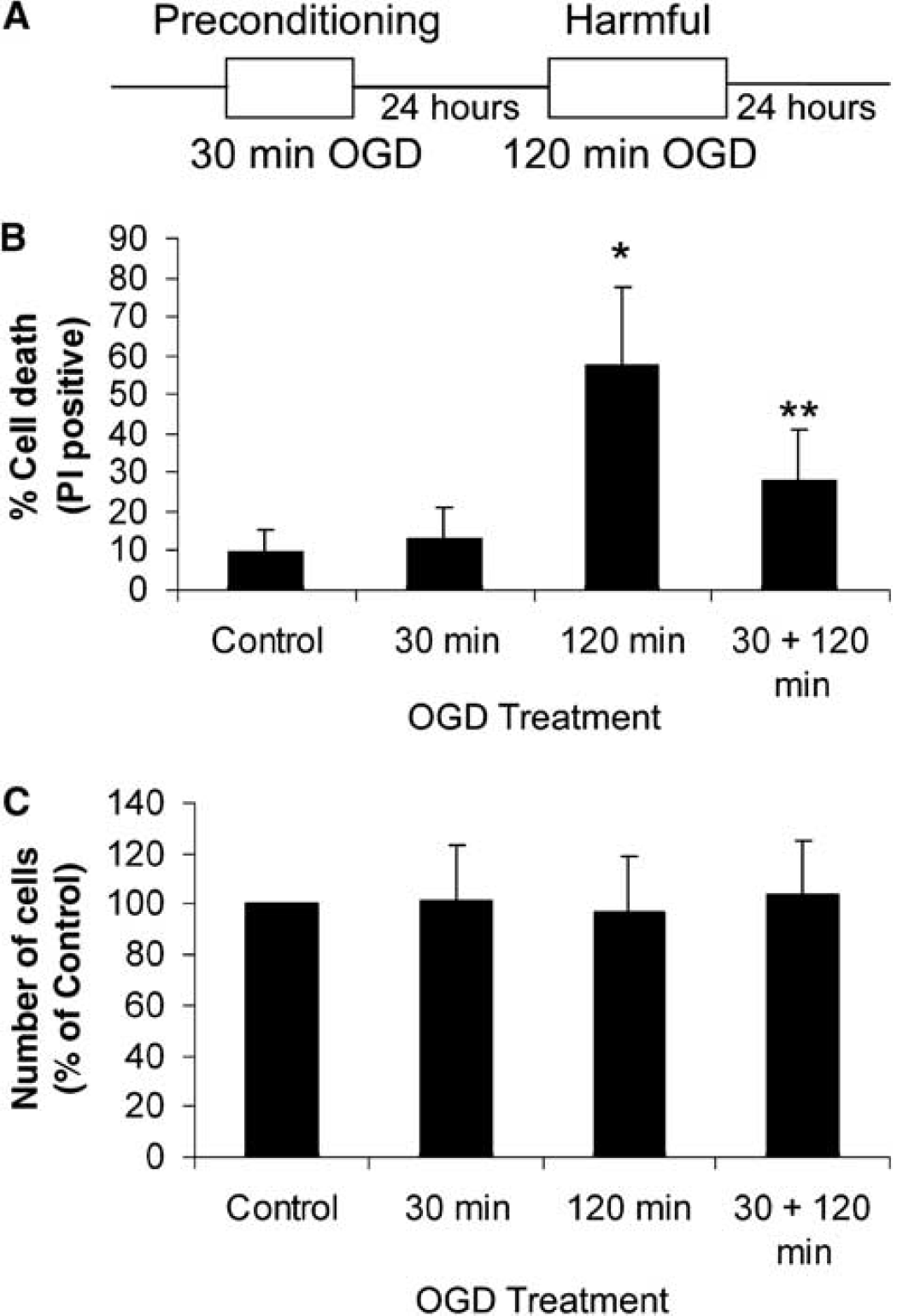
Model of ischemic tolerance in cultured cortical cells. (
Consistent with our in vivo results, we saw an increase in Bcl-2 expression and CREB phosphorylation, after 30-min OGD (preconditioning) (Figure 4A), but not after 120-min OGD (injurious) (not shown). Levels of CBP and CREB were not affected by 30 mins (Figure 4A) either. These data support the in vivo data suggesting that there is a concurrent increase in Bcl-2 expression and CREB phosphorylation after ischemic preconditioning.
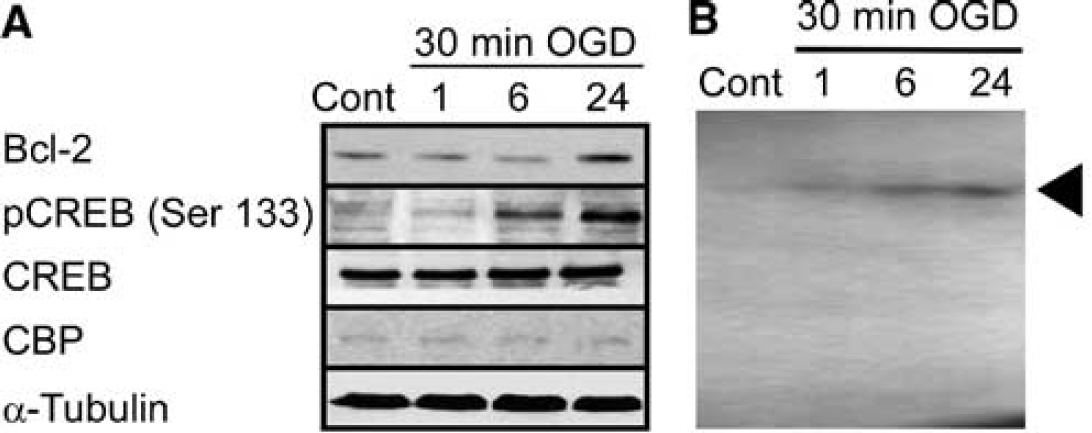
Increased expression of Bcl-2 and activation of CREB after preconditioning ischemia in vitro. (
Interaction of CREB and CREB-Binding Protein with the bcl-2 Cyclic AMP-Responsive Element after Ischemia In Vitro
To show an increased interaction of CREB with the bcl-2 CRE, nuclear extracts were prepared from cultured cells after 30- and 120-min OGD and subjected to EMSA with a bcl-2 CRE oligonucleotide, as used in in vivo experiments. A very faint interaction was detected in control cell extracts (Figure 4B). After preconditioning 30-min OGD, the interaction was increased at 6 and 24 hours. Interestingly, we only observed a single band in these EMSA experiments compared with gels from in vivo tissue (Figure 2C). The significance of this is unclear.
We further investigated the interaction of CREB with the bcl-2 CRE site by ChIP. After 30-min preconditioning, cells were subjected to ChIP with anti-CREB (Abcam) antibodies and subjected to PCR using primers specific for the rat bcl-2 CRE or the coding region of the bcl-2 gene (Figure 5A). Initial experiments showed that the bcl-2 CRE was immunoprecipitated when anti-CREB antibody was added (Figure 5B). In contrast, the coding region of the bcl-2 gene was not amplified by coding region-specific primers (Figure 5B). Genomic DNA was used as a positive control for all experiments (Figure 5B). These data suggest that the ChIP conditions used allow the precipitation of specific promoter regions of the bcl-2 gene and not total genomic DNA.
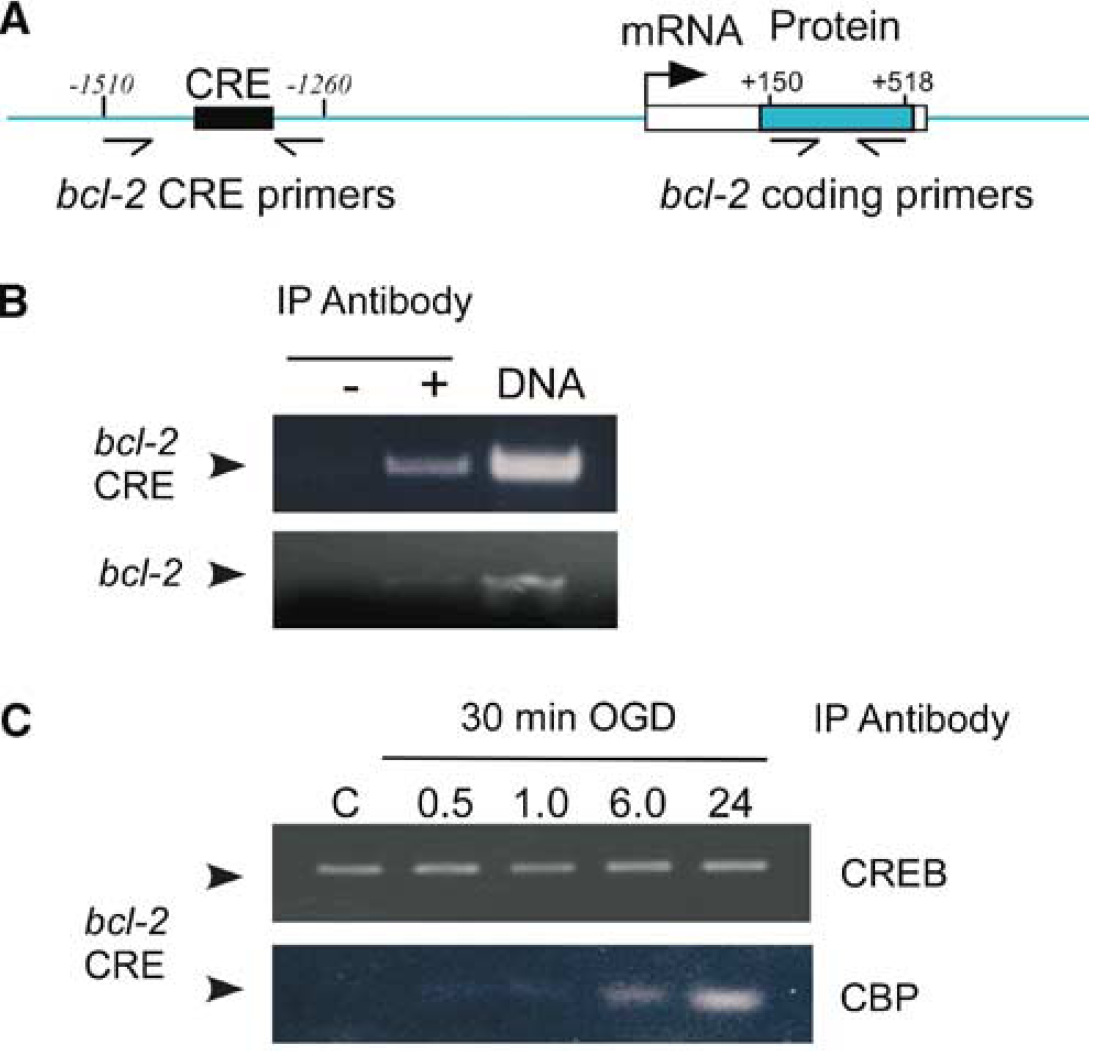
Chromatin immunoprecipitation reveals recruitment of CBP but not CREB to bcl-2 CRE after preconditioning. Cells were subjected to 30-minute OGD, followed by various recovery times. Cells were fixed with 2% formaldehyde, and DNA precipitated by either anti-CREB or anti-CBP antibodies. After clean-up, DNA was subjected to PCR with primers specific for the rat bcl-2 CRE. (
Under basal conditions, the CREB antibody precipitated the bcl-2 CRE region (Figure 5C). However, after 30-min OGD, there was no change in CREB immunoprecipitation of the bcl-2 CRE (Figure 5C). Because CREB requires the recruitment of CBP to activate transcription machinery, we investigated the ability of anti-CBP antibodies to immunoprecipitate the bcl-2 CRE. In comparison with the CREB antibody, the levels of DNA immunoprecipitated with the CBP antibody were very low in control samples (Figure 5C). After 30-min OGD, bcl-2 CRE immunoprecipitated by CBP was increased at 6 and 24 hours (Figure 5c). These data suggest that after 30-min OGD, the binding of CREB to the CRE site does not change, but rather the recruitment of CBP to CREB already bound to the bcl-2 CRE site increases after preconditioning.
Blocking CREB Binding to the bcl-2 Cyclic AMP-Responsive Element Site Inhibits Ischemic Tolerance
To investigate the role of CREB in ischemic preconditioning, we used a CRE decoy oligonucleotide to block the binding of CREB to the bcl-2 CRE, as described by Park et al (1999). Cells were incubated with a hairpin loop of DNA containing the bcl-2 CRE, which differs from the consensus CRE site by one base (TGACGT
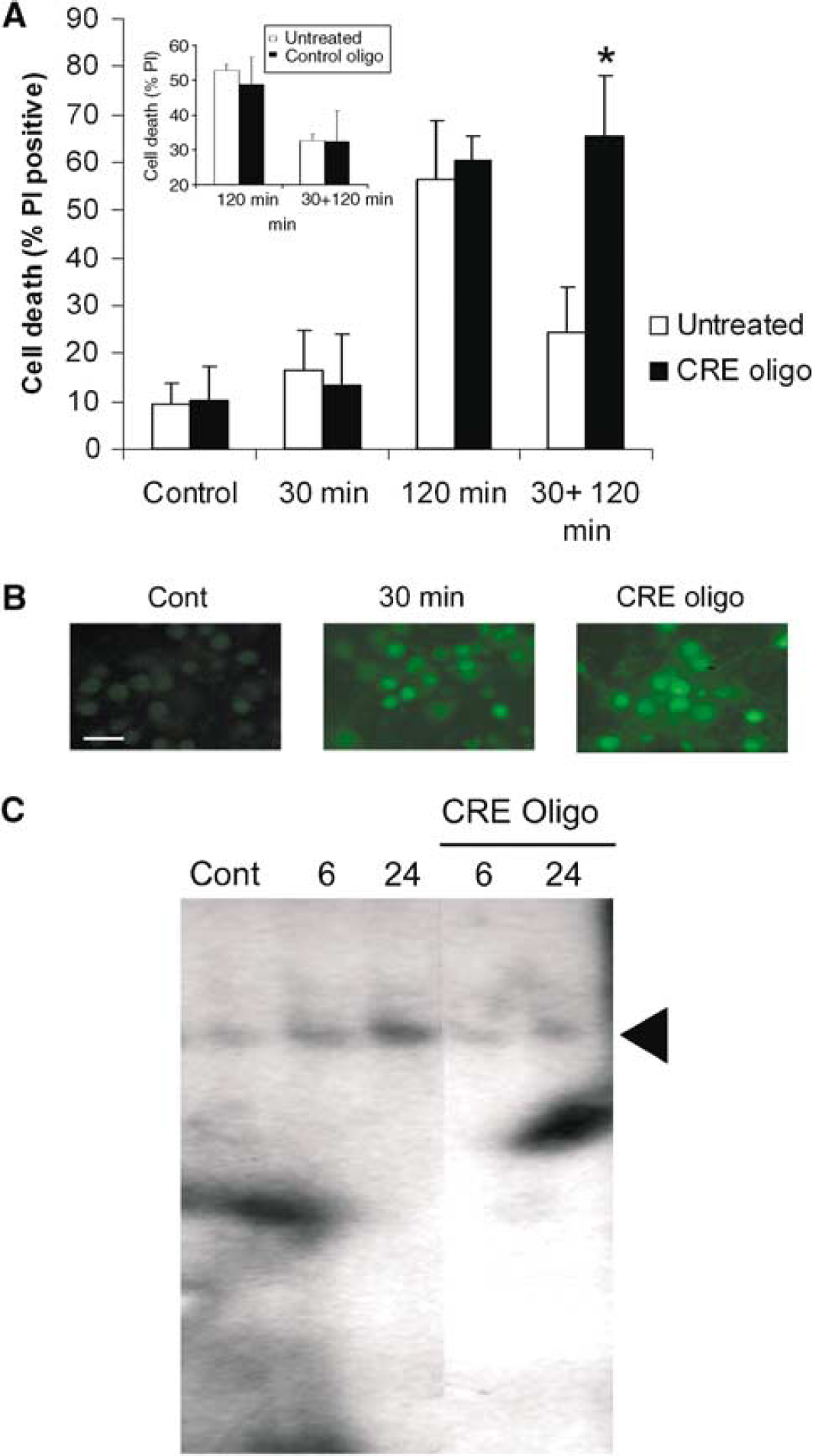
Blocking CREB function inhibits ischemic tolerance. Cells were preconditioned with 30-minute OGD and then incubated with a CRE decoy oligonucleotide (0.5 μmol/L) for 24 hours before 120-minute OGD. The neuroprotective effect of preconditioning was blocked by the CRE decoy oligonucleotide, but not the control oligonucleotide (inset). Data shown are mean±s.d. (n=4). ∗P<0.01 versus untreated cells. (
To elucidate the mechanism of action of the CRE decoy oligonucleotide, we investigated the activation of CREB. The CRE decoy oligonucleotide did not inhibit phosphorylation of CREB (Ser 133) after 30-min OGD preconditioning (Figure 6B). The CRE oligonucleotide blocked the CREB bcl-2 CRE interaction after 30-min OGD, as demonstrated by EMSA (Figure 6C). Hence, these data confirm previous reports that blocking CREB function with a CRE decoy oligonucleotide blocks ischemic tolerance (Hara et al, 2003).
Multiple Protein Kinases Regulate Ischemic Tolerance and CREB Phosphorylation
CREB is a substrate for many protein kinases (Beitner-Johnson and Millhorn, 1998; Bonni et al, 1999; Pugazhenthi et al, 2000; Sun et al, 1994; Walton and Dragunow, 2000; Zanassi et al, 2001); hence, we investigated the role of multiple protein kinases in the regulation of ischemic tolerance. Specifically, we examined the effect of the PKA inhibitor H89 (0.5 μmol/L), the calcium/calmodulin kinase inhibitor KN62 (1.0 μmol/L), blocking p42/p44 MAP kinase activation with the Mek inhibitor U0126 (10 μmol/L) and blocking the activation of Akt with the PI3K inhibitor LY294002 (10 μmol/L). After preconditioning with 30-min OGD, cells were incubated with H89, KN62, U0126, or LY294002 for 24 hours and then subjected to a further 120-min OGD. Compared with control untreated cells, H89, KN62, U0126, or LY294002 had no effect on basal cell viability (9.3±4%, 17.5±9%, 11.7±8%, 9.3±6% and 7.7±7% cell death, respectively, all P>0.05) or cell death after 120-min OGD (Figure 7A). Cells treated with H89, KN62 or U0126 showed a significant inhibition of the neuroprotective effect of 30-min preconditioning (Figure 7A). LY294002 did not block ischemic tolerance, consistent with previous reports (Namura et al, 2000). LY294002 had no effect on the induction of tolerance, hence we did not study this compound further (Figure 7A).
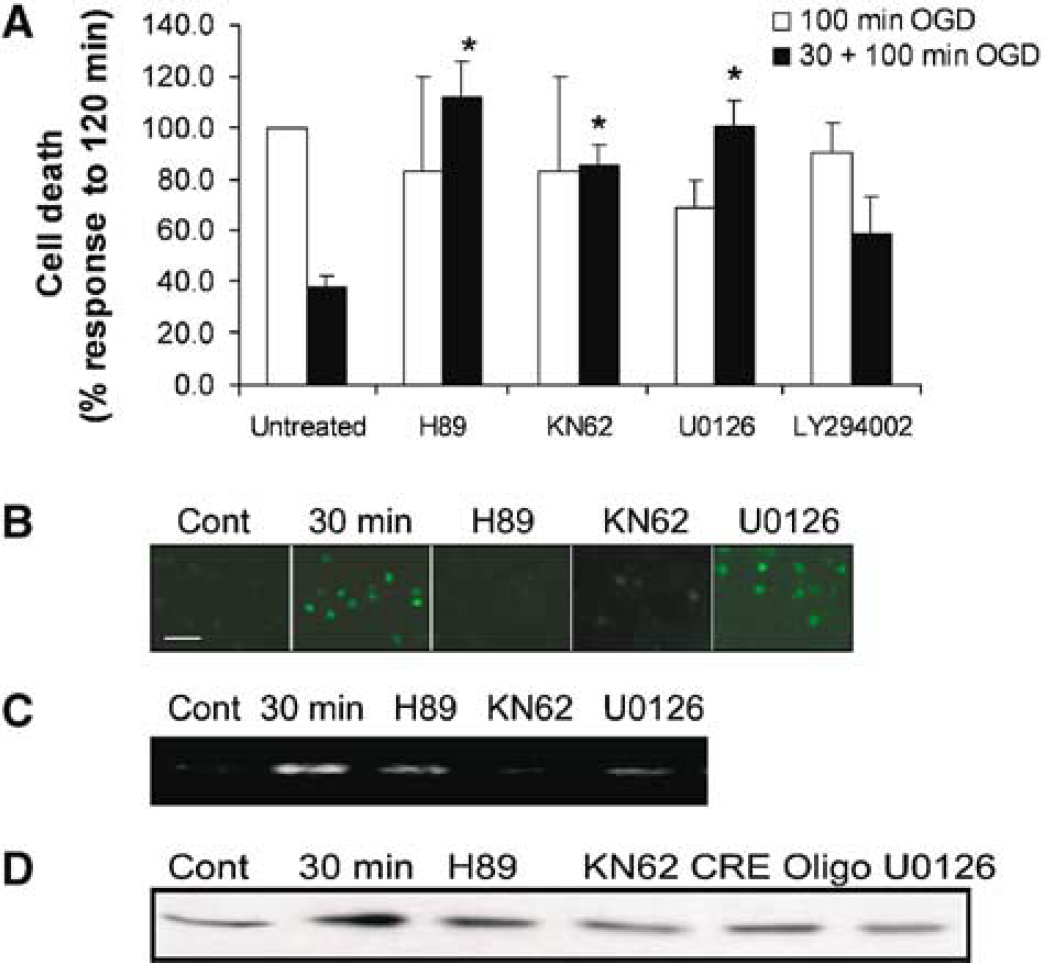
Blocking protein kinases inhibits ischemic tolerance, CREB function and Bcl-2 expression. (
Since H89, KN62, and U0126 blocked ischemic tolerance, we investigated the effects of these compounds on CREB phosphorylation. After 30-min OGD, there was an increase in the nuclear expression of phosphorylated CREB (Figure 7B). In cells treated with KN62 and H89, the increase in CREB phosphorylation (Ser 133) after 30-min OGD was reduced (Figure 7B). In contrast, in cells subjected to 30-min OGD and then recovered in the presence of U0126, CREB phosphorylation was still observed.
We decided to further investigate the effect of the protein kinase inhibitors using a ChIP assay. The immunoprecipitation of the bcl-2 CRE with anti-CBP antibody was increased after 30-min OGD preconditioning (Figure 7C). The increase in bcl-2 CRE immunoprecipitation after preconditioning was reduced by H89, KN62, and U0126 (Figure 7C). These data suggest that protein kinase inhibitors that block either the phosphorylation of CREB or the recruitment of CBP to the bcl-2 CRE block ischemic tolerance.
Inhibition of Bcl-2 Protein Expression after Preconditioning Reduces Ischemic Tolerance
Finally, we examined the effect of H89, KN62, and U0126 on Bcl-2 protein expression after preconditioning. Bcl-2 protein expression was increased 24 hours after 30-min preconditioning (Figure 7D). The increase in Bcl-2 expression after preconditioning was reduced by H89, KN62, and U0126. Furthermore, the CRE decoy oligonucleotide, which blocks CREB activation, also reduced Bcl-2 expression (Figure 7D). Taken together, these results suggest that blockade of CREB function prevents the increase in Bcl-2 expression after preconditioning, inhibiting ischemic tolerance.
Discussion
Here we present evidence to suggest that the activation of the transcription factor CREB leads to increased Bcl-2 expression as an essential step of ischemic preconditioning. A tolerance-inducing ischemic challenge in vivo and in vitro increased CREB phosphorylation and Bcl-2 protein levels. Furthermore, preconditioning ischemia increased the interaction between the bcl-2 CRE region and CBP, a bridging factor between CREB and other transcription proteins. The protective effect of preconditioning was reduced by blocking the interaction of CREB with the bcl-2 CRE using a CRE decoy oligonucleotide. Blocking CREB phosphorylation on Ser 133 with H89 and KN62, or CBP binding to the bcl-2 CRE with U0126 reduced Bcl-2 expression and ischemic tolerance (see Figure 8). These data suggest that after preconditioning, activation of the transcription factor CREB increases expression of the prosurvival protein Bcl-2 leading to ischemic tolerance.
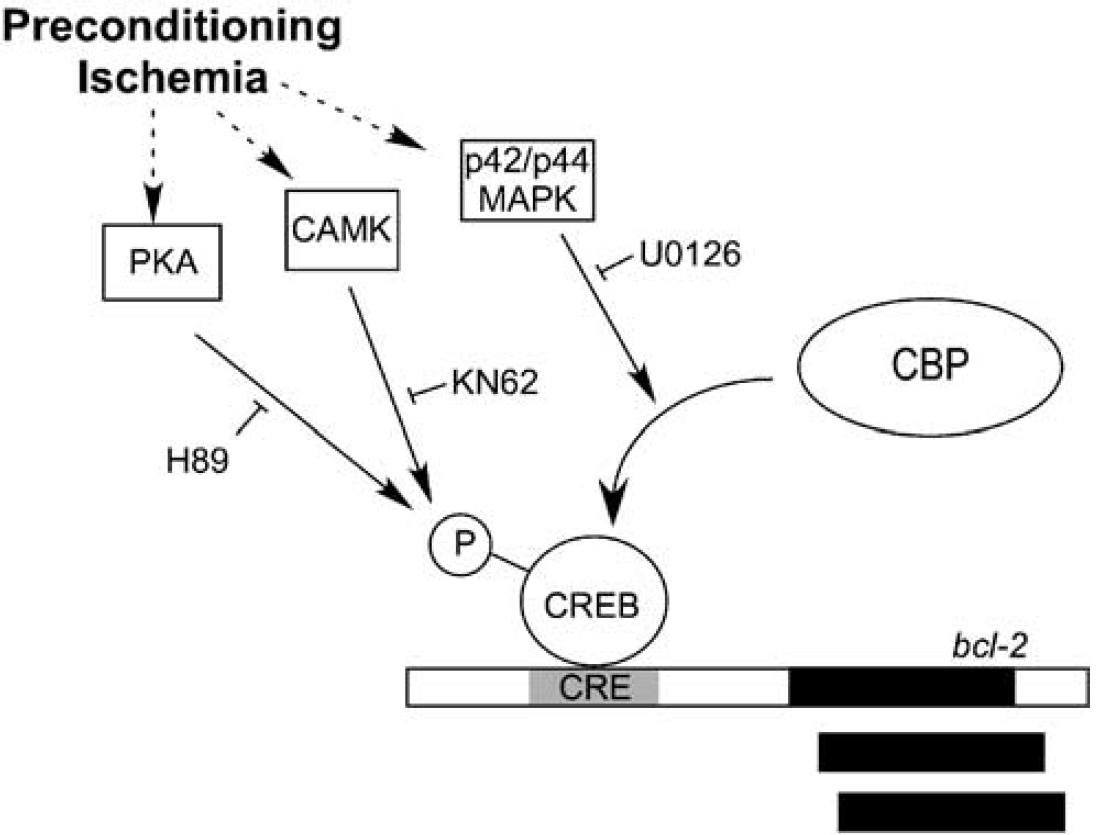
Activation of CREB-mediated Bcl-2 expression after preconditioning ischemia. Preconditioning ischemia activates PKA, Ca2+/calmodulin-regulated protein kinase (CAMK) and p42/p44 MAPK. Protein kinase A and CAMK both regulate the phosphorylation of CREB on the Ser 133 residue. This leads to the recruitment of CBP to the bcl-2-CRE. The recruitment of CBP is also mediated by p42/44 MAPK. The effects of PKA, CAMK, and p42/p44 MAPK are inhibited by H89, KN62, and U0126, respectively. Inhibition of these protein kinases prevents ischemic tolerance.
Our models of ischemic tolerance show that brief noninjurious ischemic challenges (preconditioning) induce the activation of neuroprotective processes, which render the brain tolerant against a more harmful ischemic insult. After preconditioning, we observed an increase in Bcl-2 protein expression, consistent with other reports (Bossenmeyer-Pourie and Daval, 1998; Shimizu et al, 2001). It is of note that the time course for ischemic tolerance was shorter in the in vitro model (24 hours) compared with the in vivo model of ischemic tolerance (3 days). We focused on the activation of CREB as a regulatory step for the induction of tolerance, due to the previous reports of Bcl-2 regulation by CREB and reports showing an increase in CREB phosphorylation after ischemia (Hara et al, 2003; Jin et al, 2001). An accepted model of CREB activation is that kinases phosphorylate CREB on Ser 133, which recruits the coactivators CBP and P300, leading to the recruitment of transcription machinery. Using a phospho-specific antibody to CREB Ser 133, we show that CREB phosphorylation increased after a 30-min MCAO or 30-min OGD, which suggests that, after preconditioning ischemia, increased phosphorylation of Ser 133 may regulate CREB activity.
The results obtained using the ChIP assay suggest that the rate of CREB binding to the bcl-2 CRE site may not change after preconditioning. Indeed, it was the binding of CBP to the bcl-2 CRE site that increased after preconditioning ischemia (Figure 5). This would suggest that preconditioning leads to conditions that favor CBP binding to the bcl-2 CRE, presumably due to the recruitment of CBP to phosphorylated CREB, and thus increases Bcl-2 expression. Indeed, it has recently been shown that binding of CBP to the fos-CRE promoter increases after glutamate stimulation of cells (Impey et al, 2002).
The importance of activating the CREB pathway after preconditioning was demonstrated by the blockade of ischemic tolerance by a CRE decoy oligonucleotide. This approach has also been used to show that CREB activation is required in a global ischemia model of tolerance in CA1 neurons in gerbils (Hara et al, 2003). To control against nonspecific effects, a control oligonucleotide was used, containing a mismatched CRE sequence. The CRE decoy oligonucleotide, but not the control sequences, blocked ischemic tolerance, suggesting that inhibition of CREB function blocks ischemic tolerance (Figure 5A and inset). As an important control, we show that the CRE decoy oligonucleotide did not effect CREB phosphorylation after preconditioning ischemia. The CRE decoy oligonucleotide blocked CREB binding to the bcl-2 CRE and the expression of the Bcl-2 protein after preconditioning, consistent with its hypothesized biological effect (Hara et al, 2003; Park et al, 1999).
CREB is the substrate for phosphorylation by many protein kinases. Brief periods of ischemia have been shown to increase intracellular cyclic AMP levels and activate PKA (Tanaka, 2001), which can phosphorylate CREB. A rise in calcium can activate calcium calmodulin kinases that phosphorylate CREB (Sun et al, 1994; Zanassi et al, 2001). An important role of PKA and CAMK in mediating the effects of preconditioning is suggested, given that the PKA inhibitor H89 and the CAMK inhibitor KN62 inhibited CREB phosphorylation, Bcl-2 expression and ischemic tolerance (Figure 8). Indeed, it has previously been shown that KN62 blocks glutamate-mediated CREB phosphorylation (Mabuchi et al, 2001), and ischemic tolerance in vitro (Tauskela et al, 2003).
Brief ischemia activates p42/p44 MAP kinase and blocking p42/p44 MAP kinase activation blocks ischemic tolerance (Figure 7). In our studies, U0126 did not block CREB phosphorylation, but inhibited the binding of CBP to the bcl-2 CRE. This suggests that p42/p44 MAP kinase may regulate CREB activation, downstream of CREB phosphorylation on Ser 133. Indeed, phosphorylation of CREB on Ser 133 alone is not synonymous with CREB-mediated transcription. Mayr et al (2001) showed that both PKA and protein kinase C phosphorylate CREB on Ser 133; however, only PKA activates CREB-mediated expression of inducible cAMP early repressor gene (ICER). Furthermore, CBP-dependent transcription is also regulated by phosphorylation (Impey et al, 2002) and CBP activity is enhanced by p42/p44 MAPK (Gusterson et al, 2002; Liu et al, 1998). Hence, U0126 may inhibit p42/p44 MAP kinase and block the recruitment of CBP to the bcl-2 CRE, thereby inhibiting ischemic tolerance (Figure 8). Clearly, the role of CBP in ischemic tolerance requires further investigation.
These initial data hint at the complexity of ischemic tolerance, whereby multiple protein kinases are activated after preconditioning; yet, inhibition of just one protein kinase is sufficient to block ischemic tolerance. One potential point of convergence of these protein kinases is the transcription factor CREB (see Figure 8). As such, there are many genes with prosurvival effects that are regulated by CREB, for example, brain-derived neurotrophic factor (Shieh and Ghosh, 1999), as well as the prosurvival protein Bcl-2. Increased Bcl-2 expression has profound neuroprotective effects (Alberi et al, 1996; Linnik et al, 1995; Martinou et al, 1994; Sagot et al, 1995) and blocking the increase in Bcl-2 expression after preconditioning blocks ischemic tolerance (Shimizu et al, 2001). Our study supports the importance of Bcl-2 expression in ischemic tolerance, by showing that pharmacological and molecular manipulations that reduce Bcl-2 expression after preconditioning block ischemic tolerance. Specifically inhibition of PKA, CAMK, p42/p44 MAPK, and CREB activation all reduced Bcl-2 expression and ischemic tolerance.
Our data support the therapeutic approaches investigated by other groups, which suggest that increased levels of prosurvival proteins may be an effective therapeutic strategy to reduce cell damage after ischemia (Cao et al, 2002). Further, the role of post-translational modification of Bcl-2 after brief ischemia may also contribute to its prosurvival effects; for example, phosphorylation of Bcl-2 effects its stability (Dimmeler et al, 1999) or antiapoptotic abilities (Tamura et al, 2004). Clearly, the elucidation of the biochemical pathways involved in regulating Bcl-2 expression and function may help in the application of therapeutic approaches to protect against cell death after ischemia.
Footnotes
Acknowledgements
This work was supported by NIH grants NS24728 and NS35965 (RPS).