
Abstract
Mild or moderate hypothermia is generally thought to block all changes in signaling events that are detrimental to ischemic brain, including ATP depletion, glutamate release, Ca2+ mobilization, anoxic depolarization, free radical generation, inflammation, blood—brain barrier permeability, necrotic, and apoptotic pathways. However, the effects and mechanisms of hypothermia are, in fact, variable. We emphasize that, even in the laboratory, hypothermic protection is limited. In certain models of permanent focal ischemia, hypothermia may not protect at all. In cases where hypothermia reduces infarct, some studies have overemphasized its ability to maintain cerebral blood flow and ATP levels, and to prevent anoxic depolarization, glutamate release during ischemia. Instead, hypothermia may protect against ischemia by regulating cascades that occur after reperfusion, including blood—brain barrier permeability and the changes in gene and protein expressions associated with necrotic and apoptotic pathways. Hypothermia not only blocks multiple damaging cascades after stroke, but also selectively upregulates some protective genes. However, most of these mechanisms are addressed in models with intraischemic hypothermia; much less information is available in models with postischemic hypothermia. Moreover, although it has been confirmed that mild hypothermia is clinically feasible for acute focal stroke treatment, no definite beneficial effect has been reported yet. This lack of clinical protection may result from suboptimal criteria for patient entrance into clinical trials. To facilitate clinical translation, future efforts in the laboratory should focus more on the protective mechanisms of postischemic hypothermia, as well as on the effects of sex, age and rewarming during reperfusion on hypothermic protection.
Introduction
Neuroprotection provided by induced hypothermia has been documented for several decades (Colbourne et al, 1997; Maher and Hachinski, 1993). Deep hypothermia (less than 20°C) is effective in reducing cerebral ischemic damage in patients after cardiac arrest; however, it can produce deleterious complications (Krieger et al, 2005). Since Busto et al, (1987) reported in 1987 that even a 1 to 2°C temperature reduction profoundly protects against experimental stroke in rats, the protection of mild (33 to 36°C) to moderate (28 to 32°C) hypothermia against central nervous system injury and its potential mechanisms have received extensive study in the past 20 years (Corbett and Thornhill, 2000; Krieger and Yenari, 2004).
Cerebral ischemia results from a reduction or complete loss of cerebral blood flow (CBF), followed by depletion of ATP and occurrence of anoxic depolarization (AD) and/or spreading depression (SD)-like depolarization (Colbourne et al, 1997; Nedergaard and Hansen, 1993; Obrenovitch and Urenjak, 1997; Ohta et al, 1997; Figure 1). A large amount of glutamate is released from the intracellular space into the extracellular space (Colbourne et al, 1997; Obrenovitch and Urenjak, 1997; Zhao et al, 1997), which stimulates N-methyl-
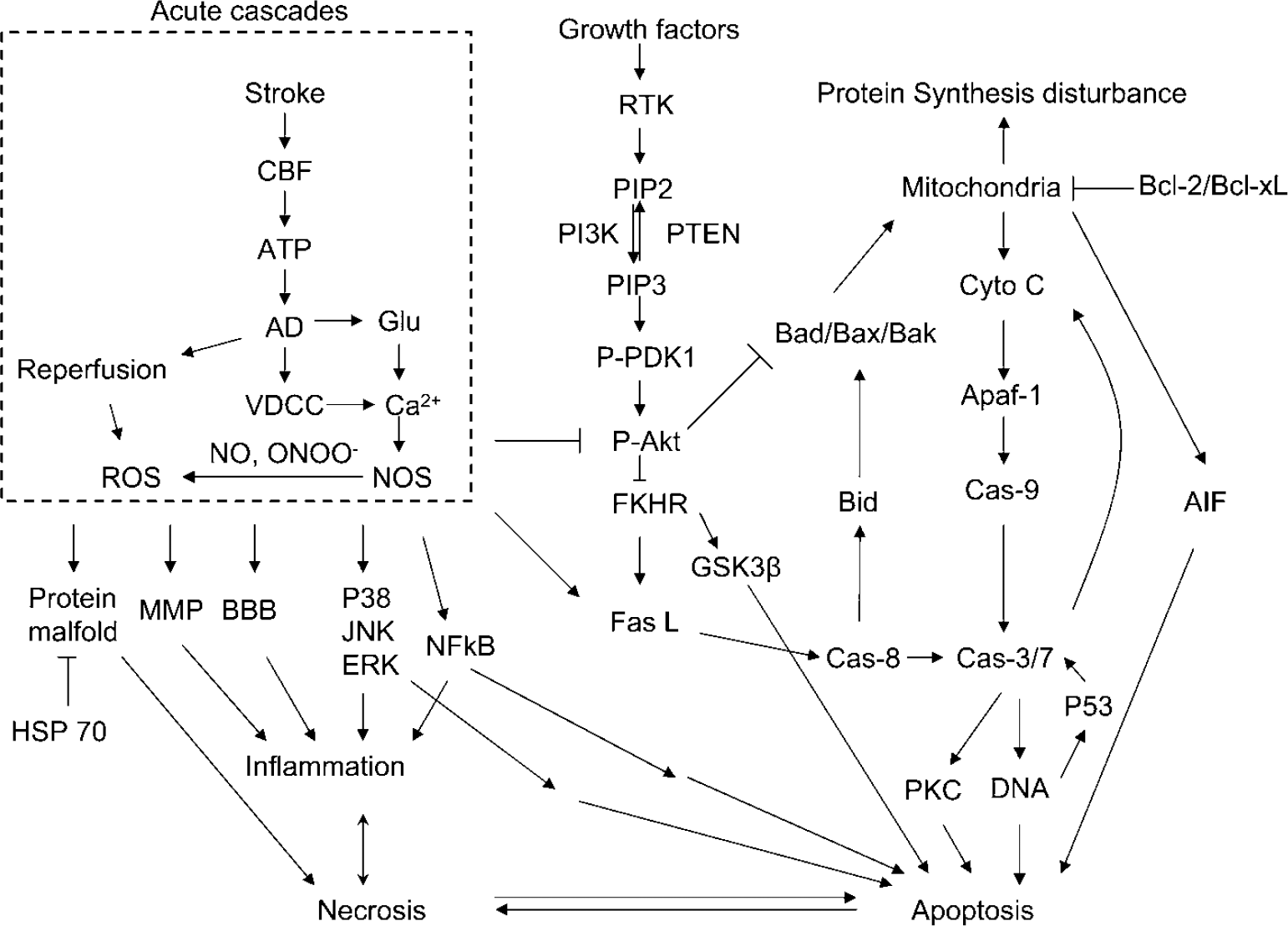
A diagram showing the major cascades that occur after stroke reviewed in this article. AD, anoxic depolarization; AIF, apoptosis-inducing factor; BBB, blood—brain barrier; Cas, caspase; CBF, cerebral blood flow; cyto c, cytochrome c; Fas L, Fas ligand; FKHR, forkhead homologue in rhabdomyosarcoma; Glu, glutamate; GSK3β, glycogen synthase kinase 3β; MMP, matrix metalloprotease; NOS, nitric oxide synthesis; NO, nitric oxide; ONOO−, peroxynitrite; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidyliositol-4,5-bisphosphate; PIP3, phosphatidyliositol-3,4,5-bisphosphate;PKC, protein kinase C; P-Akt, phosphorylated Akt; PTEN, phosphatase and tensin homologue deleted on chromosome 10; P-PDK1, phosphorylated phosphoinositide-dependent protein kinase-1; ROS, reactive oxygen species; RTK, receptor tyrosine kinase; VDCC, voltage-dependent calcium channel.
Understanding the mechanisms by which hypothermia protects brain tissue may lead to advances in stroke treatment. We review the effects of mild to moderate hypothermia on various events according to the ischemic cascade depicted in Figure 1. Unless otherwise specified, the term hypothermia in this review refers to mild to moderate hypothermia.
The Ostensible Pan-Inhibitive Effects of Hypothermia
We summarized some studies on the ‘pan-inhibiting’ effects of mild to moderate hypothermia on detrimental cascades caused by stroke in Table 1. Intraischemic hypothermia delays or attenuates both ATP depletion (Ibayashi et al, 2000; Sutton et al, 1991; Welsh et al, 1990) and anoxic depolarization (Bart et al, 1998; Nakashima and Todd, 1996; Takeda et al, 2003), it also blocks glutamate release (Busto et al, 1989b; Patel et al, 1994; Winfree et al, 1996), suppresses inflammation (Kawai et al, 2000; Wang et al, 2002), maintains the integrity of the BBB (Dietrich et al, 1990; Huang et al, 1999; Kawanishi, 2003), reduces free radical production (Maier et al, 2002), inhibits protein kinase C translocation (Cardell et al, 1991; Shimohata et al, 2007a, b ; Tohyama et al, 1998), inhibits matrix metalloproteinase expression (Hamann et al, 2004), and blocks both necrosis and apoptosis. Intraischemic hypothermia also preserves the base-excision repair pathway, which repairs oxidative damage (Luo et al, 2007). In addition to those cascades directly associated with neuronal injury, hypothermia further blocks astrocyte activity and inhibits white matter injury (Colbourne et al, 1997; Dempsey et al, 1987; Kimura et al, 2002). Similarly, postischemic hypothermia blocks free radical generation (Horiguchi et al, 2003), attenuates inflammation (Horstmann et al, 2003; Ohta et al, 2007), prevents BBB permeability (Preston and Webster, 2004), and suppresses caspase activities (Van Hemelrijck et al, 2005). Indeed, a browse through the literature gives an overwhelming impression that hypothermia seems to block every damaging event associated with necrosis or apoptosis. One reason for this impression of pan-inhibition may lie in the causality of ischemic damage. For example, is the inflammatory response the cause of tissue damage or is it induced by brain injury? If it is the latter, then since hypothermia prevents tissue damage, it certainly also prevents the inflammatory response.
Hypothermia blocks changes in many detrimental cascades occurring after stroke
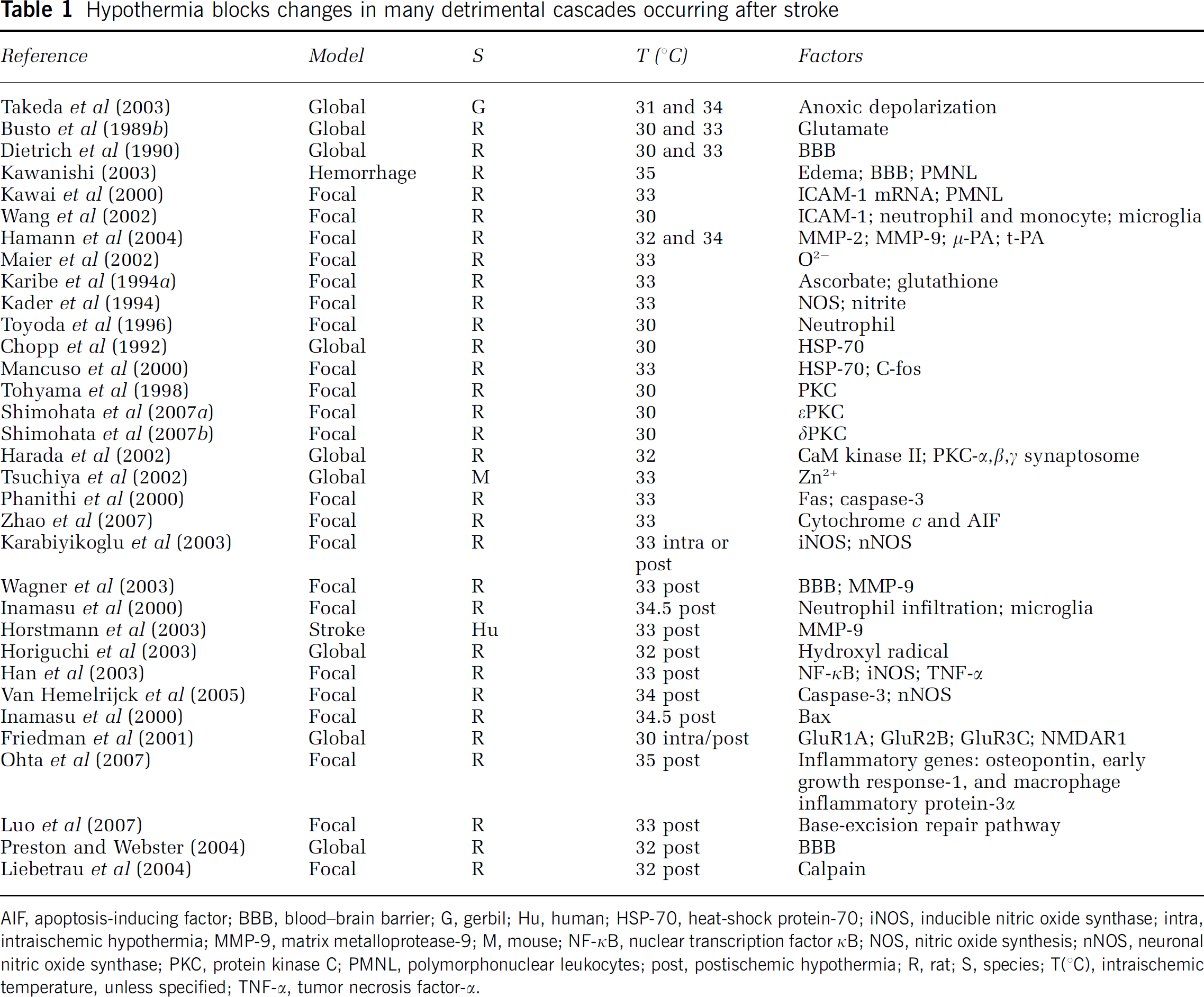
AIF, apoptosis-inducing factor; BBB, blood—brain barrier; G, gerbil; Hu, human; HSP-70, heat-shock protein-70; iNOS, inducible nitric oxide synthase; intra, intraischemic hypothermia; MMP-9, matrix metalloprotease-9; M, mouse; NF-κB, nuclear transcription factor κB; NOS, nitric oxide synthesis; nNOS, neuronal nitric oxide synthase; PKC, protein kinase C; PMNL, polymorphonuclear leukocytes; post, postischemic hypothermia; R, rat; S, species; T(°C), intraischemic temperature, unless specified; TNF-α, tumor necrosis factor-α.
The ‘pan-inhibiting’ effect of hypothermia seems to apply not only to detrimental cascades, but also to some protective genes as well. For instance, overexpression of heat-shock protein-70 (HSP-70) protects neurons (Kelly et al, 2002), but hypothermia blocks HSP-70 expression after stroke; therefore, it was believed that protection by hypothermia is independent of HSP-70 (Chopp et al, 1992; Mancuso et al, 2000). However, the truth may be more complex. It is possible that neurons saved by hypothermia do not require HSP-70 expression; HSP-70 may only be upregulated in neurons that are injured. Furthermore, neurons may not overexpress HSP-70 even if they need it, because hypothermia generally inhibits protein translation. Nerve growth factors also improve neuron survival after cerebral ischemia; however, hypothermia does not enhance expression of nerve growth factors, brain-derived neurotrophic factor, neurotrophin 3, and tyrosine kinase receptor B in global ischemia, suggesting that the protective effect of hypothermia is not achieved this way (Boris-Moller et al, 1998). The argument that applies to HSP-70 expression may also apply here.
Findings such as these ‘pan-inhibiting’ effects may lead one to assume that hypothermia simply blocks, inhibits, or reverses any ischemia-induced damaging cascade, even including some beneficial cascades that occur, thereby preventing cell damage. By extension, hypothermia's actions may be so broad that it may provide no real clues for understanding the mechanisms of cell death due to ischemia. Nevertheless, such an assumption is oversimplified and misleading. Although it is true that hypothermia has multiple effects on various cascades after ischemia, and this is almost certain why hypothermia has such profound protective effects, hypothermia does not simply block every detrimental downstream reaction after stroke.
In fact, the mechanisms underlying hypothermic protection are controversial, and a bias appears to favor results showing that hypothermia blocks all damaging cascades after ischemia, whereas results to the contrary have often been under appreciated. We focus on the more controversial issues surrounding hypothermia's effects on CBF, glutamate release, and survival signaling pathways, and do not dwell on hypothermia's effects on BBB permeability and inflammation, because there is somewhat more of a consensus in the literature on these issues. However, first, we summarize the protection of mild to moderate hypothermia in experimental stroke models.
Hypothermia Does Not Always Attenuate Stroke Even in the Laboratory
To translate hypothermia to the clinic, it is important to recognize that its neuroprotective effects are limited in the laboratory as well. Intraischemic hypothermia is most effective in transient ischemia or ‘reperfusion’ models (Busto et al, 1989a; Carroll and Beek, 1992; Colbourne and Corbett, 1995); the effects of intraischemic hypothermia in permanent ischemia models are far less uniform (Baker et al, 1991; Kader et al, 1992; Kozlowski et al, 1997; Lo et al, 1993; Morikawa et al, 1992; Moyer et al, 1992; Ridenour et al, 1992; Zhao et al, 2007). In addition, it will be most meaningful clinically if hypothermia protects when applied postischemically; however, therapeutic time windows for postischemic hypothermia are limited. For example, mild hypothermia applied 15 mins after reperfusion failed to improve neurologic function in global ischemia, whereas it protected when applied immediately after reperfusion (Kuboyama et al, 1993). This therapeutic time window might be similar to what occurs in the penumbra after focal ischemia. In addition to the onset time of hypothermia, duration and depth of hypothermia and animal species/strains also determine the protective effects of postischemic hypothermia (Busto et al, 1989a; Chopp et al, 1991; Coimbra and Wieloch, 1992, 1994; Colbourne and Corbett, 1994, 1995; Colbourne et al, 1997; Doerfler et al, 2001; Huh et al, 2000; Maier et al, 2001; Ren et al, 2004; Yanamoto et al, 1996). Such limitations suggest that hypothermia cannot alter some events that inevitably lead to ischemic damage.
The Effects of Hypothermia on Cerebral Blood Flow and Metabolism
Cerebral Blood Flow
Hypothermia reduces CBF in anesthetized animals and human patients in the absence of cerebral ischemia, as reviewed recently by Erecinska et al (2003). Hypothermia does not affect CBF during complete global ischemia (Busto et al, 1987; Jiang et al, 1994; Kil et al, 1996; Sonn et al, 2002; Sugimura et al, 1998); global ischemia reduces CBF to less than 5% of normal (Busto et al, 1987), making it unlikely that hypothermia would have a further significant effect on CBF. However, there are a wide range of findings on CBF after reperfusion in global ischemia: hypothermia increases (Jenkins et al, 2001; Jiang et al, 1994; Karibe et al, 2000), decreases (Huang et al, 1999; Mori et al, 1998), or has no effect on CBF (Baldwin et al, 1991; Hoffman and Thomas, 1996; Horiguchi et al, 2003; Kil et al, 1996; Sonn et al, 2002; Sugimura et al, 1998).
Focal ischemia reduces CBF in the penumbra to various levels (e.g., 50 to 60% of baseline), and one would expect that hypothermia might further reduce CBF in the penumbra. However, most reports suggest that mild to moderate hypothermia has no effect on the CBF level during ischemia in the penumbra (Kawai et al, 2000; Morikawa et al, 1992; Yanamoto et al, 2001). Nevertheless, one study reported that moderate hypothermia (30°C) significantly reduced CBF using a diffusion-weighted imaging technique (Jiang et al, 1994). Some studies suggest that hypothermia does not affect CBF after reperfusion in focal ischemia (Kawai et al, 2000; Morikawa et al, 1992), whereas others suggest that hypothermia blocks the postischemic hyperperfusion and the delayed, sustained hypoperfusion (Huang et al, 1998; Jiang et al, 1994; Karibe et al, 1994b).
Although the discrepancies may be due to animal species, anesthesia, temperature controls, or other variables, we speculate that the techniques used by most of these studies to measure CBF are the key source of variation. Laser Doppler flowmetry only reflects the relative changes in CBF levels, and it is unreliable to compare such levels across different temperature groups using this technique. Other potential factors affecting CBF results are differences in cooling techniques, the rewarming process, and shivering. Rapid rewarming not only exacerbates axonal injury (Suehiro and Povlishock, 2001), but it also causes dysfunction of blood vessels after brain trauma. Furthermore, shivering during rewarming, especially when anesthesia is withdrawn, is a common phenomenon that significantly affects CBF and ATP recovery (Ueda et al, 2004). However, previous studies usually ignored this issue and so the degree to which rewarming impacts CBF after ischemia/reperfusion is not known. More stringently designed experiments are needed to clarify the effect of hypothermia on CBF in cerebral ischemia.
Hypothermia Promotes Energy Recovery after Reperfusion
After stroke onset, ATP depletes immediately after the reduction of CBF, and the extent of ATP depletion should be proportional to the extent of CBF reduction at a fixed temperature. ATP production and cerebral metabolism closely associate with brain temperature. A 10° decrease in temperature reduces ATP consumption and the cerebral metabolic rate of glucose, oxygen, and lactate two- to four-fold (Erecinska et al, 2003). Hypothermia may not significantly improve energy levels during global ischemia, as the CBF level is already significantly reduced (Busto et al, 1987).
Whether mild hypothermia protects against ischemia in adult animal models by maintaining ATP levels is controversial. One report showed that mild hypothermia attenuated phosphate depletion and lactate accumulation during global ischemia, and attributed the neuroprotective effect of hypothermia to preserved ATP levels (Ibayashi et al, 2000). However, other studies disagree with this conclusion (Busto et al, 1987; Kimura et al, 2002; Kozlowski et al, 1997; Shimizu et al, 1997; Sutton et al, 1991). At the end of 20 mins of global ischemia, # Busto et al (1987) reported that phosphocreatine, glucose, and pyruvate all decreased to a similar degree among groups treated with different temperatures from 30 to 36°C. Similarly, during 15 mins of global ischemia, the hypothermic effect (34°C) did not differ from that of normothermia (37°C) on the amount of high-energy phosphate metabolites including ATP, phosphocreatine, and inorganic phosphate (Kimura et al, 2002). Taken together, it is reasonable to assume that under severe ischemia in which CBF is almost or completely cut off, energy would be depleted within several minutes even with hypothermia, but mild hypothermia would still significantly reduce ischemic damage. Thus, hypothermia's effect on energy depletion during ischemia is unlikely to play a significant role in its neuroprotective effects.
Almost all the studies mentioned above agree that after severe ischemia, hypothermia accelerates ATP recovery during reperfusion (Haraldseth et al, 1992; Kimura et al, 2002; Kozlowski et al, 1997; Shimizu et al, 1997; Sutton et al, 1991). For example, mild hypothermia (34°C) increased the rate of metabolic recovery relative to normothermia by 10 to 20% for the first 10 to 25 mins of reperfusion after 15 mins of transient global ischemia in gerbils (Kimura et al, 2002); metabolism recovered more at 20 to 60 mins after the onset of reperfusion in hypothermic rats subjected to 60 mins of global ischemia (Shimizu et al, 1997); in hippocampal slices of adult guinea pigs, mild hypothermia improved ATP recovery at 12 h after oxygen-glucose deprivation, although mild hypothermia had no evident effect at 2 h after oxygen-glucose deprivation (Berger et al, 1998).
In conclusion, mild to moderate hypothermia may not maintain ATP levels during ischemia. Rather, a more likely mechanism of neuroprotection by hypothermia in adult animal models is the acceleration of ATP recovery after reperfusion.
Hypothermia's Effect on Anoxic Depolarization and Glutamate Release
Anoxic Depolarization
Almost all studies agree that hypothermia merely delays the occurrence of AD in global ischema. These reports consistently show a brief delay in the onset of AD after ischemia in mild to moderate hypothermic animals (e.g., 1 to 2 mins at 33 to 34°C; Table 2). However, few have studied the effects of hypothermia on AD in focal ischemia. Unlike in global ischemia, both AD- and SD-like depolarization occur in the ischemic core and penumbra, respectively, in focal ischemia. In addition, AD- or SD-like depolarization occurs along with redistribution of ions such as Ca2+, K+, and Na+. Changes in these ions' extracellular or intracellular concentrations may actually reflect changes in AD- or SD-like depolarization. Sick et al (1999) showed that hypothermia did not change the onset time of the extracellular potassium level ([K+]o) shift and the concentration of [K+]o in the ischemic core. Furthermore, hypothermia did not affect the duration and frequency of SD-like depolarization and [K+]o in the penumbra, although hypothermia promoted [K+]o recovery after reperfusion. In contrast, Chen et al (1993) showed that hypothermia reduced the number of SD-like depolarizations; differences between the core and the penumbra were not addressed in this study. Moreover, intracellular Ca2+ levels play a critical role in neurotoxicity. A few reports have convincingly showed that hypothermia merely delays Ca2+ mobilization without altering its concentration from normothermic levels (Kristian et al, 1992; Mitani et al, 1991). Again, these studies were performed in the context of global ischemia. Whether hypothermia has similar effects in focal ischemia is not known.
Hypothermia delays onset time of AD in global ischemia or inhibits frequency of SD in focal ischemia
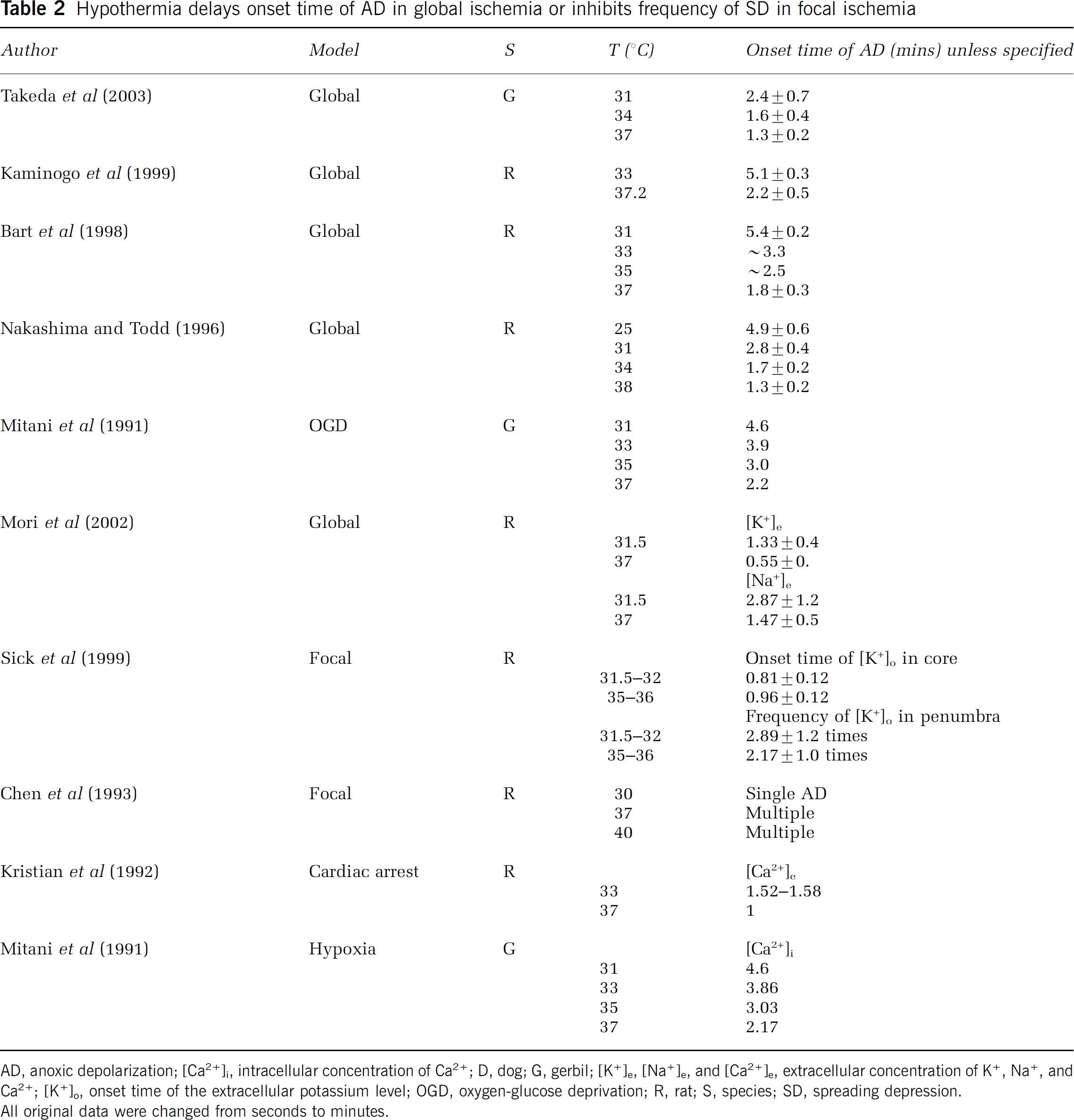
AD, anoxic depolarization; [Ca2+]i, intracellular concentration of Ca2+; D, dog; G, gerbil; [K+]e,[Na+]e, and [Ca2+]e, extracellular concentration of K+, Na+, and Ca2+; [K+]o, onset time of the extracellular potassium level; OGD, oxygen-glucose deprivation; R, rat; S, species; SD, spreading depression. All original data were changed from seconds to minutes.
Glutamate Release
Unlike the consistent effects of hypothermia on anoxic depolarization in global ischemia, the effects of hypothermia on extracellular glutamate concentration ([Glu]e) are controversial (Table 3). It has been reported that mild hypothermia completely blocks (Busto et al, 1989b; Li et al, 1999; Martinez-Tica and Zornow, 2000; Patel et al, 1994), partially blocks (Mitani and Kataoka, 1991; Winfree et al, 1996; Yamamoto et al, 1999), merely delays (Asai et al, 2000, 1998), or does not inhibit an increase in [Glu]e after cerebral ischemia (Berger et al, 1998; Zhao et al, 1997). Furthermore, the effect of hypothermia on [Glu]e depends on the brain area, ischemic severity, and animal age (Berger et al, 2002; Boris-Moller and Wieloch, 1998; Lo et al, 1993; Van Hemelrijck et al, 2003).
The effects of hypothermia on glutamate release
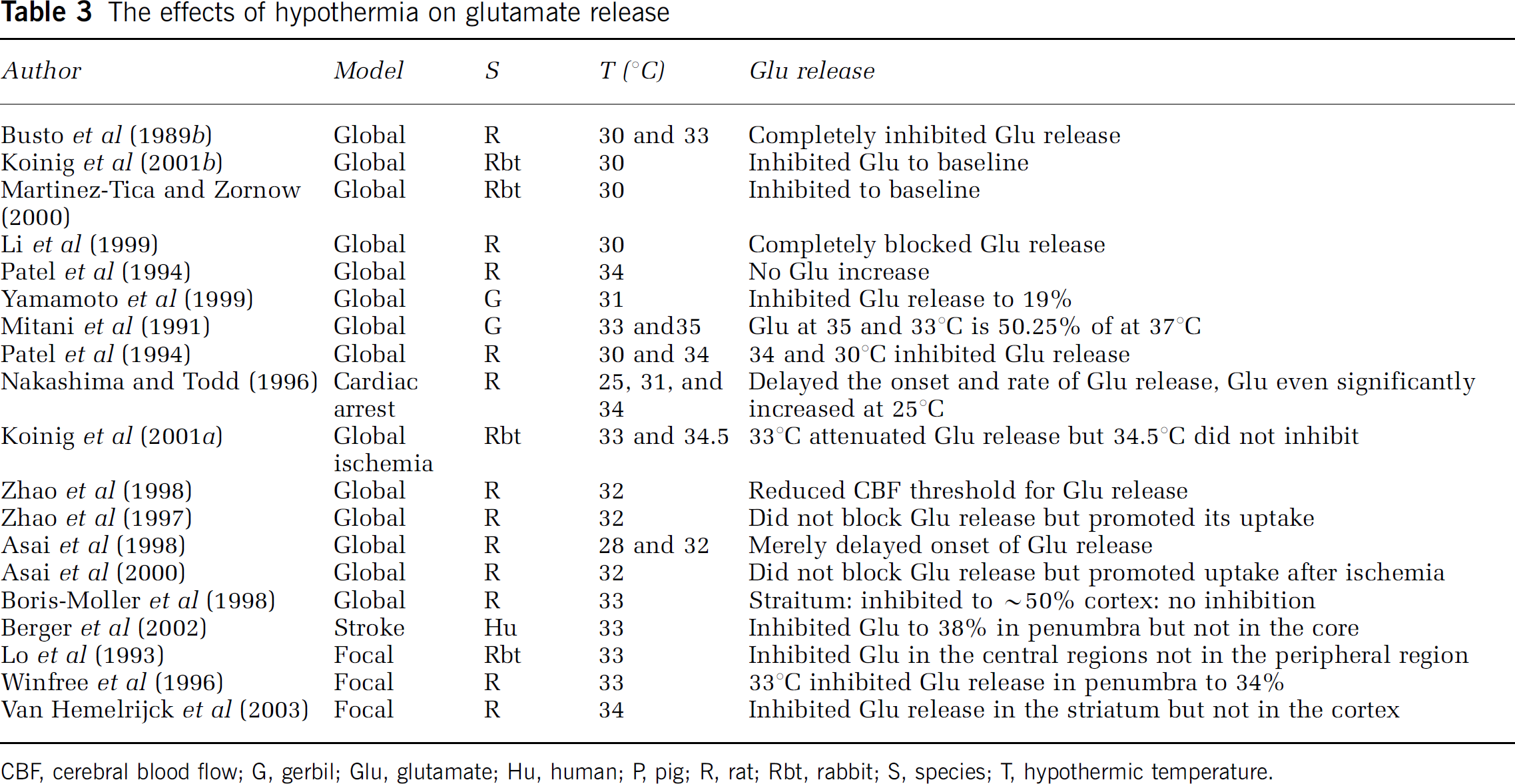
CBF, cerebral blood flow; G, gerbil; Glu, glutamate; Hu, human; P, pig; R, rat; Rbt, rabbit; S, species; T, hypothermic temperature.
It is not clear why there is such variation across studies concerning [Glu]e. It may reflect species variation, ischemia models, and differences in systems used for monitoring glutamate. We postulate that the different methodology used for detecting glutamate release may be largely responsible for such variations as seen in CBF detection. As we discussed, various studies have shown strikingly consistent results about the effect of hypothermia on AD in global ischemia, probably because the way of detecting AD using direct current potential electrodes is simple and straightforward. However, for glutamate release detection, samples are usually collected using microdialysis probes and are then measured by high-performance liquid chromatography (Li et al, 1999; Phillis et al, 2000; Rossi et al, 2000; Zhao et al, 1999). Extracellular glutamate enters the probe through a semipermeable membrane and is washed out by a continuous flow of solution. The type of probe, the speed of flow, the sample-collecting period, and the high-performance liquid chromatography detection systems could each affect the results.
The mechanisms of glutamate release after cerebral ischemia are not completely understood. [Glu]e reflects a balance of release and uptake. Glutamate starts to release almost at the same time that AD occurs. After reperfusion and ATP recovery, released glutamate is taken back into the intracellular space when AD starts to recover.
As occurrence of AD reflects ATP depletion that leads to glutamate release, AD usually synchronizes glutamate release under both normothermia and hypothermia after stroke (Asai et al, 2000, 1998; Obrenovitch and Urenjak, 1997; Zhao et al, 1997). Thereafter, released glutamate stimulates N-methyl-
In summary, intraischemic mild hypothermia merely delays ATP depletion, the occurrence of AD, glutamate release, and Ca2+ mobilization during severe cerebral ischemia. This delay is not sufficient to explain the strong protection of mild or moderate hypothermia (Bart et al, 1998; Takeda et al, 2003), but it might contribute to a reduction in ischemic damage, especially when combined with the myriad other beneficial effects. Nevertheless, in the case of mild to moderate hypothermia, neuroprotection cannot be solely attributed to its effect on intraischemic events; its effect on subsequent cascades also plays a critical role, which will be discussed in the following sections.
Hypothermia Does Not Simply Block Cell Signaling Pathways of Apoptosis or Necrosis
Although hypothermia generally blocks protein synthesis in the absence of an insult (Sayegh et al, 1992), it actually improves protein synthesis compared with normothermia after ischemia (Wu et al, 1995; Yamashita et al, 1991). Sparing of tissue may contribute to the improved protein synthesis; however, hypothermia selectively upregulates some proteins after ischemia. For example, expression of the mineralocorticoid receptor increased under hypothermia after stroke, which may promote neuronal survival in the hippocampal dentate gyrus (Macleod et al, 2003). Hypothermia also enhanced the glucose-regulated protein-78, of which overexpression via adenovirus gene therapy rescued hippocampal neurons (Aoki et al, 2001). In addition, mild hypothermia improved the protein synthesis rate after 12 h of recovery in hippocampal slices from immature or mature fetuses subjected to oxygen-glucose deprivation from 10 to 40 mins, and improved protein synthesis rate in slices from adult animals exposed to 20 to 30 mins, but not 40 mins, oxygen-glucose deprivation. The neuroprotective effect of mild hypothermia on protein synthesis depends on the animal's age and the severity of the ischemia (Berger et al, 1998). We summarize such specific effects of hypothermia in Table 4.
Hypothermia specifically regulates protein levels or mRNA of genes or BBB
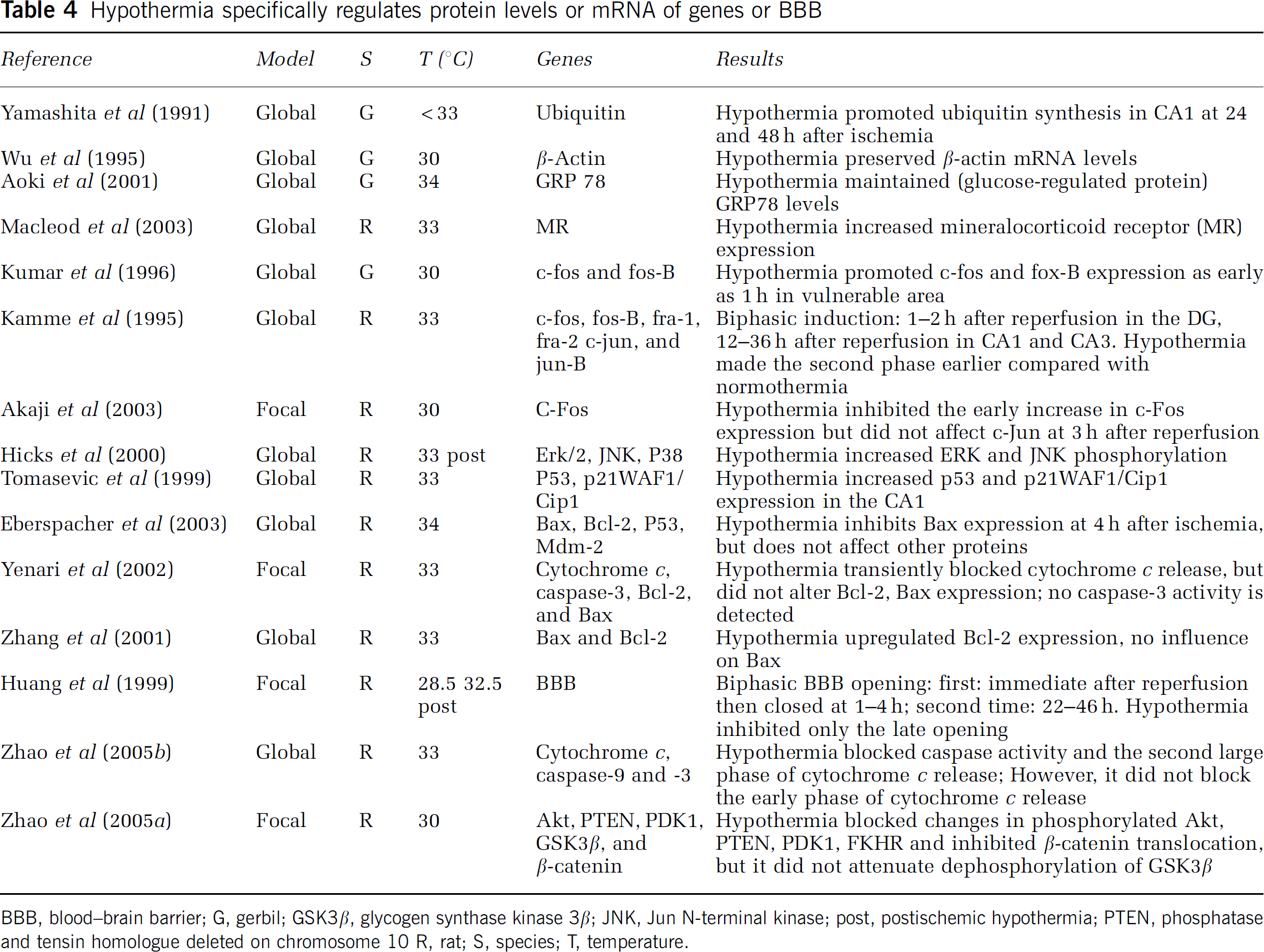
BBB, blood-brain barrier; G, gerbil; GSK3β, glycogen synthase kinase 3β; JNK, Jun N-terminal kinase; post, postischemic hypothermia; PTEN, phosphatase and tensin homologue deleted on chromosome 10 R, rat; S, species; T, temperature.
The Jun N-Terminal Kinase/Early Genes and the Erk Pathway
Jun N-terminal kinase (JNK) and Erk1/2 are two well-characterized members of the mitogen-activated protein kinase family; they are implicated in both apoptosis and neuroprotection after stroke, thus their exact roles remain elusive. In addition, JNK functions by regulating the early genes, c-jun and jun-B, which are upregulated immediately after cerebral ischemia (Akins et al, 1996). As with JNK and Erk1/2, it is not known whether such early gene expression is beneficial or detrimental. Hicks et al (2000) showed that phosphorylated/activated JNK increased at 6, 12, and 24 h after cardiac arrest in rats, and that mild hypothermia further increased phosphorylated JNK at 24 h, but not at 6 and 12 h compared with normothermia; moreover, phosphorylated JNK also increased in the ischemia-resistant regions of the dentate gyrus and of the CA3. The hypothermia-induced increase in phosphorylated JNK coincides with an increase in downstream early gene expression of JNK (Kamme et al, 1995). Kamme et al (1995) showed that the early gene expression patterns of c-jun and jun-B, as well as c-fos and fos-b, is biphasic after global ischemia: the early phase occurred at 1 to 2 h after reperfusion in the dentate gyrus and the late phase appeared at 12 to 36 h in CA-1 and −3. Mild hypothermia accelerated the occurrence of the second phase of gene expression earlier in CA1. Furthermore, Kumar et al (1996) showed that both c-fos and fos-B were induced in the hippocampal dentate gyrus as early as 1 h after global ischemia, and expression extended to vulnerable areas at 4 to 6 h. Hypothermia accelerated their expression to 1 h in the vulnerable regions and temperature did not change expression of the two genes at 6 h, 1 and 2 days. Although another experiment showed that hypothermia decreased expression of c-fos, this occurred in the penumbra of a focal ischemia, and hypothermia did not inhibit c-jun expression at 3 h after cerebral ischemia (Akaji et al, 2003). The conclusion that upregulation of immediate early genes is detrimental is at odds with the fact that these genes are generally induced in hypothermia (Akins et al, 1996).
As with JNK, the function of the kinase Erk1/2 after ischemia is not understood. Whereas some studies state that Erk1/2 expression contributes to ischemic damage (Sugawara et al, 2002), others showed that Erk1/2 activity promoted neuronal survival after ischemia (Kilic et al, 2005). Consistent with the latter, Hicks et al (2000) showed that hypothermia promotes Erk1/2 expression after global ischemia, and Erk1/2 overexpression correlates with selective upregulation of brain-derived neurotrophic factor, an upstream element of the Erk1/2 pathway (D'Cruz et al, 2002).
Hypothermia Unexpectedly Enhances the p53 Pathway
Striking findings have emerged concerning the tumor suppressor p53, a protein that causes cell death. p53 expression after stroke has been associated with neuronal damage, and p53 induction is generally believed to contribute to apoptosis after cerebral ischemia (Halterman and Federoff, 1999). Therefore, induction of p53 is used as a marker for neuronal death, but how it causes neuronal death is not clear. Surprisingly, Tomasevic et al (1999) found that p53 induction might be neuroprotective through their research in hypothermia. They investigated the expression of both p53 and one of its effector genes, p21WAF/Cip1, which is upregulated in p53-dependent cell cycle arrest and apoptosis. Both p53 and p21WAF/Cip1 mRNA are upregulated in the resistant area of the hippocampus after transient global ischemia in normothermic and hypothermic rat brains. p53 protein levels are increased in CA3 neurons after ischemia in normothermic rats. But hypothermia increases p53 expression in the protected CA1 neurons, where it translocates into the nuclei, suggesting that its induction is not a marker for neuronal death (Tomasevic et al, 1999). Actually, p53 and p21WAF/Cip1 are also involved in DNA repair, and lack of their activity leads to failure of repair (Tomasevic et al, 1999). Therefore, the authors argue that p53 induction correlates with neuronal survival, which is supported by a recent report using p53 knockout mice (Maeda et al, 2001). Maeda et al induced a 1 h transient focal ischemia in wild type (p53 + /+), heterozygous (p53 + / −), and homozygous (p53−/−) mice for p53 deficiency. Infarct size increased inversely to the p53 gene dosage, suggesting that p53 prevents rather than worsens ischemic damage. Taken together, hypothermia may enhance p53 expression promoting repair after stroke (Tomasevic et al, 1999).
Caspase and Cytochrome c-Mediated Apoptotic Pathways
Hypothermia blocks apoptosis after experimental stroke. Mild hypothermia inhibits Fas and caspase-3 expression after 1 h of focal ischemia (Phanithi et al, 2000), it also inhibits Bax overexpression 4 h after 30 mins of incomplete cerebral ischemia but does not affect Bcl-2, p53, and Mdm-2 expression (Eberspacher et al, 2003). In addition, Yenari et al (2002) showed that hypothermia transiently attenuated cytochrome c release but did not alter Bcl-2 and Bax expression in a focal ischemia model. Caspase activity was not observed, however, which suggests that ischemic damage is caspase-independent (Yenari et al, 2002). In contrast, they found that hypothermia specifically increased Bcl-2 protein expression after global ischemia (Zhang et al, 2001).
Recently, we observed a biphasic cytochrome c release after global ischemia: a small peak of cytochrome c release occurred at 5 h, and a larger peak occurred at 48 h after ischemia onset (Zhao et al, 2005b). Moreover, caspase-9 and −3 activity significantly increased at 12 and 24 h after the first phase of cytochrome c release, suggesting that the small amount of cytochrome c might be responsible for the subsequent caspase activity. To our surprise, mild hypothermia did not block the first phase of cytochrome c release, but it did significantly block caspase activity and the second phase of cytochrome c release. These results suggest that low levels of cytochrome c release might be reversible after global ischemia, and may define a time window for intervention for stroke treatment.
Hypothermia Does Not Indiscriminately Upregulate all Signals in the Akt/protein kinase B Pathway
The PI3K/Akt kinase pathways promote neuron survival after ischemia (reviewed by Zhao et al, 2006; Figure 1). Akt activity is believed to be regulated by phosphorylation at Ser-473 and Thr-308 via some upstream molecules, such as phosphoinositide-dependent protein kinase-1 and phosphatase and tensin homologue deleted on chromosome 10 (PTEN). Whereas activated phosphoinositide-dependent protein kinase-1 phosphorylates Akt, activated PTEN dephosphorylates Akt through degrading phosphatidyliositol-3,4,5-bisphosphate to phosphatidyliositol-4,5-bisphosphate. Activated Akt then blocks caspase/cytochrome c-mediated apoptosis by phosphorylating Akt substrates, such as forkhead homologue in rhabdomyosarcoma and glycogen synthase kinase 3β (GSK3β).
The level of phosphorylated Akt at Ser-473 (P-Akt) has been used to represent Akt activity, and most reports indicate that P-Akt is transiently increased within hours after stroke, but then decreased after 24 h or longer (Zhao et al, 2006). Protection by some agents such as corticosteroids, brain-derived neurotrophic factor, preconditioning, and copper—zinc superoxide dismutase overexpression maintains P-Akt after stroke (Zhao et al, 2006).
We recently showed that PI3K/Akt pathway indeed plays critical roles in neuroprotection by moderate hypothermia (Zhao et al, 2005a). Phosphorylated Akt levels are transiently increased after stroke, but phosphorylation levels of PTEN, phosphoinositide-dependent protein kinase-1, GSK3β, and forkhead homologue in rhabdomyosarcoma are decreased (Zhao et al, 2005a). Intriguingly, in vitro Akt kinase assays showed that the true Akt activity decreased when P-Akt levels reached a peak, suggesting that P-Akt does not represent the true Akt activity. Consistent with this observation, the true Akt activity and phosphorylation levels of PTEN, phosphoinositide-dependent protein kinase-1, GSK3β, and forkhead homologue in rhabdomyosarcoma are decreased at early time points in the penumbra after ischemia, and preceded degradation of MAP-2 and cytochrome c release. Thus, disruption of the Akt pathway might mediate ischemic damage (Zhao et al, 2005a).
Hypothermia serves as a useful tool for understanding the roles of the Akt pathway in neuroprotection after stroke. To our surprise, hypothermia blocks the increases in P-Akt after stroke, but it maintains the true Akt activity, further suggesting that P-Akt does not represent true Akt activity (Zhao et al, 2005a). A functional role for this hypothermia-maintained activity is supported by the finding that the PI3K/Akt inhibitor, LY294002, enlarged infarct size in hypothermic animals, suggesting that the PI3K/Akt pathways play critical roles in neuroprotection (Zhao et al, 2005a).
Further evidence for the role of Akt activity in hypothermic protection comes from the study of a negative regulator of the Akt pathway, PTEN. The activity of PTEN is downregulated by phosphorylation at Ser-380 (P-PTEN) (Torres and Pulido, 2001; Vazquez et al, 2000). Dephosphorylated (activated) PTEN results in dephosphorylation of phosphatidyliositol-3,4,5-bisphosphate to phosphatidyliositol-4,5-bisphosphate and prevents recruitment of Akt to the membrane for phosphorylation (Zhao et al, 2006). Our data indicate that hypothermia blocks ischemic damage by attenuating a decrease in P-PTEN after stroke onset. In fact, the level of P-PTEN is markedly and consistently preserved by hypothermia at various time points from 0.5 to 48 h after stroke, compared with P-Akt, phosphorylated phosphoinositide-dependent protein kinase-1, P-GSK3β, and phosphorylated forkhead homologue in rhabdomyosarcoma. Therefore, hypothermia may exert its neuroprotective action partly through its effects on P-PTEN.
However, hypothermia does not indiscriminately attenuate every aspect of the Akt pathway. Glycogen synthase kinase 3β, a kinase downstream of Akt, contributes to ischemic neurotoxicity (Kelly et al, 2004). Glycogen synthase kinase 3β activity is downregulated when phosphorylated at Ser-9 by Akt. Although hypothermia maintains many steps in the Akt pathway after stroke, it does not elevate GSK3β (Ser-9) phosphorylation (Zhao et al, 2005a). It is a puzzle that the protein level of the active form of nonphosphorylated GSK3β was increased after hypothermic stroke, which is supposed to induce cell death, but no ischemic damage was detected under hypothermia. We therefore explored if hypothermia affects the downstream target of GSK3β, β-catenin, a critical transcription factor. Glycogen synthase kinase 3β phosphorylase β-catenin leading to its degradation and apoptosis (Zhao et al, 2005a). Despite protein levels of phosphorylated β-catenin being unknown, we have found that β-catenin translocates into nuclei in the penumbra after stroke, and that hypothermia blocks such translocation. This result suggests that hypothermia may block ischemic damage by blocking events downstream of GSK3β (Zhao et al, 2005a).
In sum, hypothermia provides multiple protective effects against damaging cascades after stroke. However, contrary to the popular belief that hypothermia simply blocks all apoptotic or necrotic pathways, it actually specifically upregulates or downregulates expression of certain beneficial or detrimental genes. Nevertheless, one must be cautious not to over extrapolate these findings to draw conclusions concerning the roles of such genes in pro- or anti ischemic damage, since hypothermia may provide overall protection in several pathways, but does not always promote expression of all beneficial genes nor does it inhibit every detrimental event.
Clinical Translation and Future Directions
Hypothermic studies done in the laboratory have led to clinical investigations for cerebral ischemia. Four recent landmark prospective randomized-controlled studies showed that induced mild to moderate hypothermia improved neurologic function in patients suffering cardiac arrest from ventricular fibrillation (Hypothermia after Cardiac Arrest Study Group, 2002; Bernard et al, 2002) and reduced risk of death or disability in neonates hypoxic—ischemic encephalopathy (Gluckman et al, 2005; Shankaran et al, 2005). In the case of acute stroke treatment, temperature on admission correlates with neurologic outcome: hyperthermia is associated with poor prognosis (den Hertog et al, 2007; Krieger et al, 2005). Therefore, hyperthermia prevention for acute stroke patients is warranted (Ginsberg and Busto, 1998) by using antipyretics to inhibit hyperthermia (e.g., den Hertog et al, 2007).
Several preliminary clinical trials have been completed to test the protection of induced mild to moderate hypothermia against focal stroke (den Hertog et al, 2007; Krieger et al, 2005) and three trials are still ongoing (http://www.strokecenter.org). In these studies, cooling methods from surface cooling to endovascular cooling have been used. Mild to moderate hypothermia was instituted from 6 h to 1 day after stroke onset, and target temperatures were achieved from 1 to 6 h after cooling application (den Hertog et al, 2007; Krieger et al, 2005). These studies have confirmed the feasibility and safety of hypothermia application, but no definite beneficial effect was reported.
Although we cannot directly extrapolate settings in the laboratory into clinical trials, a comparison might provide insights for future clinical trials. In the laboratory, hypothermia must be initiated at very early hours after reperfusion in transient focal ischemia to achieve neuroprotection, and it has minimal protective effects in permanent ischemic models. However, in most clinical studies, mild to moderate hypothermia was initiated as late as 5 to 6 h after focal stroke, and it took one to several hours to reach target temperature (den Hertog et al, 2007; Krieger et al, 2005). In addition, patients might not have reperfusion or reperfusion occurred in a very late stage. Although laboratory studies have provided strong rational for clinical application of hypothermia for acute stroke treatment, we must emphasize that the protective effect of hypothermia is limited even in the laboratory. In the laboratory, a number of crucial variables need to be considered, including the onset time of hypothermia, its depth, and whether the models used include reperfusion. Compared with experimental stroke in the laboratory, it is not surprising that beneficial effects were not found in the past clinical trials. Therefore, entrance criteria for clinical trials must be carefully re-considered. For patients with focal stroke, early reperfusion and rapid hypothermia initiation should be used to achieve protection.
Understanding the protective mechanisms involved in hypothermia provides insights for clinical trials. To date, the intraischemic hypothermia studies suggest that the effects of hypothermia on acute events during ischemia are not the sole decisive events underlying its neuroprotection. Overemphasizing the effects of hypothermia on ATP depletion, anoxic depolarization, glutamate release, and Ca2+ mobilization during ischemia may be misleading. Instead, studies of the effects of hypothermia on the expression of various genes after reperfusion have enhanced our understanding of the mechanisms of ischemic neuronal damage. Hypothermia not only blocks multiple damaging cascades after stroke, it also selectively upregulates some protective genes. In fact, hypothermia provides a unique opportunity for us to understand the function of some genes that have puzzled us.
Unfortunately, despite the great potential clinical relevance of postischemic hypothermia, few have studied its protective mechanisms; future projects will concentrate on cellular and molecular mechanisms of postischemic hypothermia. In addition, how gender, age, and rewarming factor into protection by hypothermia need to be studied in the laboratory. Finally, the literature shows that the protective effects of hypothermia, especially when applied after stroke, are highly variable; additional well-designed experiments are needed to resolve these outstanding issues.
Footnotes
Acknowledgements
We thank Dr David Schaal, Dr Bruce Schaar, and Ms Felicia F Beppu for manuscript assistance and Ms Elizabeth Hoyte for figure preparation.