
Abstract
Activation of terminal caspases such as caspase-3 plays an important role in the execution of neuronal cell death after transient cerebral ischemia. Although the precise mechanism by which terminal caspases are activated in ischemic neurons remains elusive, recent studies have postulated that the mitochondrial cell death-signaling pathway may participate in this process. The bcl-2 family member protein Bax is a potent proapoptotic molecule that, on translocation from cytosol to mitochondria, triggers the activation of terminal caspases by increasing mitochondrial membrane permeability and resulting in the release of apoptosis-promoting factors, including cytochrome c. In the present study, the role of intracellular Bax translocation in ischemic brain injury was investigated in a rat model of transient focal ischemia (30 minutes) and reperfusion (1 to 72 hours). Immunochemical studies revealed that transient ischemia induced a rapid translocation of Bax from cytosol to mitochondria in caudate neurons, with a temporal profile and regional distribution coinciding with the mitochondrial release of cytochrome c and caspase-9. Further, in postischemic caudate putamen in vivo and in isolated brain mitochondria in vitro, the authors found enhanced heterodimerization between Bax and the mitochondrial membrane permeabilization-related proteins adenine nucleotide translocator (ANT) and voltage-dependent anion channel. The ANT inhibitor bongkrekic acid prevented Bax and ANT interactions and inhibited Bax-triggered caspase-9 release from isolated brain mitochondria in vitro. Bongkrekic acid also offered significant neuroprotection against ischemia-induced caspase-3 and caspase-9 activation and cell death in the brain. These results strongly suggest that the Bax-mediated mitochondrial apoptotic signaling pathway may play an important role in ischemic neuronal injury.
Neuronal cell death resulting from cerebral ischemia likely involves both necrotic and apoptotic mechanisms (for reviews see Lipton, 1999; Schulz et al., 1999). Although ischemic neuronal death has classically been considered to be necrotic, recently a growing body of evidence indicates that the apoptosis execution machinery is activated in ischemic neurons and that apoptotic cell death is involved in ischemic injury, especially when the ischemic insult is relatively mild and cellular energy metabolism is not severely compromised. Morphologic and biochemical features of apoptosis have now been reproducibly detected in the ischemic brain, including cell membrane protruding, chromatin condensation, formation of apoptotic bodies, and internucleosomal DNA degradation (Li et al., 1995a, b ; Linnik et al., 1993; MacManus et al., 1993).
The discovery of activation of apoptosis-effector genes in ischemic brain has provided important evidence for the involvement of an apoptotic mechanism in ischemic neuronal death (Chen et al., 1996b, 1998; Krajewski et al., 1995; Namura et al., 1998). These studies identified a group of terminal caspases, particularly caspase-3, as the central executive molecules in ischemic neuronal death. It has been shown that levels of gene expression and protease activity of caspase-3 are increased in injured neurons after ischemia (Chen et al., 1998; Namura et al., 1998; Ni et al., 1998). Caspase-3-mediated proteolytic cleavage of cell death substrates such as poly(ADP-ribose)polymerase and DNA protein kinase has been shown in ischemic brain (Chen et al., 1998; Shackelford et al., 1999). In addition, pharmacologic inhibition of caspase-3 activity significantly decreases infarct size (Hara et al., 1997a, b ; Namura et al., 1998) or attenuates the loss of hippocampal CA1 neurons after transient cerebral ischemia (Chen et al., 1998). These observations strongly suggest a pivotal role of caspase-3 in ischemic neuronal death; however, the precise mechanism through which caspase-3 is activated in ischemic neurons is poorly understood.
The Bcl-2 family proteins are important endogenous regulators of terminal caspases (for review see Adams and Cory, 1998). Recent studies confirm that several members of the Bcl-2 family amplify cell death signals or directly effect apoptosis via the mitochondrial pathway by promoting cytochrome c release and subsequently activating terminal caspases, especially caspase-3 (for reviews see Budihardjo et al., 1999; Kroemer and Reed, 2000). The induction of these death signals is prevented by death suppressors (e.g., Bcl-2, Bcl-x-long) but is promoted by death effectors such as Bax or Bid (for review see Green and Reed, 1998). The mitochon-drial-damaging effect of the potent apoptosis agonist Bax has been extensively studied. On death stimulus, Bax translocates from the cytosol to the mitochondria through a yet-to-be-identified mechanism and induces mitochon-drial membrane permeabilization, presumably by interacting with proteins of the mitochondrial permeability transition pore complex such as voltage-dependent anion channel (VDAC) and adenine nucleotide translocator (ANT) (Jurgensmeier et al., 1998; Martinou, 1999; Marzo et al., 1998; Narita et al., 1998; Rosse et al., 1998; Shimizu et al., 1999). Recently, it has been shown that mitochondrial translocation of Bax constitutes a critical event in the induction of neuronal apoptosis induced by nerve growth factor deprivation or DNA damage (Cregan et al., 1999; Martinou et al., 1999; Putcha et al., 1999; Xiang et al., 1998).
Expression of Bax is induced in selective neurons destined to die after transient cerebral ischemia (Chen et al., 1996b; Gillardon et al., 1996; Krajewski et al., 1995). Thus, Bax could serve as a cell death trigger in ischemic neurons by provoking caspase-3 activation via the mitochondrial pathway. The recent findings that caspase-activating factors such as cytochrome c and caspase-9 are released from mitochondria in ischemic neurons provide strong evidence suggesting that activation of terminal caspases after ischemia may involve a mitochondrial mechanism (Fujimura et al., 1998, 1999; Krajewski et al., 1999; Sugawara et al., 1999). Therefore, in this study, we investigated the intracellular translocation of Bax in the rat model of transient focal cerebral ischemia–reperfusion, testing the hypothesis that Bax-induced mitochondrial changes may contribute to caspase-3 activation in the ischemic brain.
MATERIALS AND METHODS
Animal model of transient focal ischemia
All experiments were performed on male Sprague-Dawley rats each weighing 275 to 300 g (Charles River, Scottsdale, PA, U.S.A.). Transient focal ischemia (30 minutes) was induced in isoflurane-anesthetized rats using intraluminal vascular occlusion of the middle cerebral artery, as described previously (Chen et al., 1996b, 1997). Blood pressure, blood gases, and blood glucose concentration were maintained in the normal range throughout the experiments. Temporalis muscle and rectal temperatures were maintained in the range of 36.5°C to 37.5°C using a heating pad and a temperature-regulated heating lamp. In selective experiments, cortical blood flow was monitored before, during, and after middle cerebral artery occlusion using laser–Doppler flowmetry as previously described (Nagayama et al., 2000). Sham operation was performed in additional animals using the same anesthesia and surgical procedures except that the intraluminal suture was not inserted; these brains served as nonischemic controls.
Mitochondrial and cytosolic protein isolation
Animals were killed with an overdose intraperitoneal injection of 8% chloral hydrate at 1, 3, 6, or 24 hours after 30 minutes of ischemia or 24 hours after sham operation (nine rats per experimental condition). The striatum, which is highly reproducibly affected by middle cerebral artery occlusion in this model, was dissected from the brain. All procedures for protein fractionation were carried out at 4°C, using the method reported previously (Chen et al., 2000). In brief, brain tissues (three brains per sample) were minced and homogenized using a Dounce homogenizer (Vineland, NJ, U.S.A.) in 1 × M-SHE buffer containing 0.21 mol/L mannitol, 0.07 mol/L sucrose, 10 mmol/L Hepes-KOH (pH 7.4), 1 mmol/L ethylenediaminetetraacetic (EDTA), 0.15 mmol/L spermine, and 0.75 mmol/L spermidine. The following freshly prepared protease inhibitors were then added: 1 μg/mL each of leupeptin, aprotinin, and pepstatin; 1 mmol/L phenylmethylsulfonyl acid (PMSF); and 1 mmol/L dithiothreitol (DTT). After lysis for 30 minutes on ice, unbroken cells and nuclei were pelleted at 1,200 × g. The supernatant, containing the mitochondria, was centrifuged at 10,000 × g for 15 minutes to pellet the mitochondria, and the resulting supernatant (cytosolic fraction) was removed while taking care to avoid the pellet. To isolate mitochondrial protein further, the pellet was resuspended in a solution containing 3% Ficoll 400, 0.12 mol/L mannitol, 0.03 mol/L sucrose, and 25 μmol/L EDTA (pH 7.4), and gently layered twice in 6% Ficoll 400 solution to produce a discontinuous density gradient. After centrifugation at 10,400 × g for 25 minutes, the sediment was resuspended in a lysis buffer containing 10 mmol/L Hepes (pH 7.4), 142.5 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-traacetic acid (EGTA), 0.5% Nonidet P-40, 0.5 mmol/L PMSF, 10 μg/mL aprotinin, and 1 μg/mL each of leupeptin, chymostatin, antipain, and pepstatin (Narita et al., 1998), followed by sonication. The lysate was centrifuged at 130,000x g for 1 hour, concentrated to 5- to 10-mg/mL protein, and stored at −80°C in aliquots.
Western blot analysis
For the detection of Bax and cytochrome c in subcellular fractions, mitochondrial or cytosolic proteins were denatured in sodium dodecyl sulfate (SDS)-loading buffer (100 mmol/L Tris-HCl, 200 mmol/L dithiothreitol, 4% SDS, 0.2% bromophenol blue, and 20% glycerol) at 100°C for 6 minutes and then separated on 12% SDS-polyacrylamide gels (40 μg per lane). Immunoblotting was performed as described previously (Chen et al., 1996b), using a chemiluminescent detection system (Clontech, Palo Alto, CA, U.S.A.). The working dilutions for Bax polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and cytochrome c monoclonal (Pharmingen, San Diego, CA, U.S.A.) antibodies in the present study were 1:500 and 1:3,000, respectively. The recombinant Bax protein (Yan et al., 2000) and rat heart cytochrome c (Sigma, St. Louis, MO, U.S.A.) were used in preabsorption experiments to confirm the specificity of the immunoreactivity (Chen et al., 1998). To control for equal sample loading in each subcellular fraction, immunoblotting was performed using antibodies against the mitochondrial marker cytochrome c oxidase (Pharmingen) and the cytosolic marker α-tubulin (Sigma). Immunoreactivity for Bax and cytochrome c on each individual lane of the blots was semiquantified by a gel densitometric-scanning program using the Microcomputer Imaging Device (MCID, St. Catherine's, Ontario, Canada) image analysis system (Chen et al., 1998).
Immunoprecipitation
To examine protein-protein interaction in mitochondria, immunoprecipitation was performed using the Santa Cruz Biotechnology protocol with slight modifications. Mitochondria lysates (150 μg protein per sample) were incubated with 1 μg of the primary antibody (Bax), and the mixture was incubated for 2 hours at 4°C. The samples were then transferred to Eppendorf tubes containing 20 μL Protein A/G PLUS-Agarose beads (Sigma) and incubated overnight at 4°C. The beads were collected by centrifugation and washed four times with the lysis buffer containing 10 mmol/L Hepes (pH 7.4), 142.5 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L EGTA, 0.5% Nonidet P-40, 0.5 mmol/L PMSF, 10 μg/mL aprotinin, and 1 μg/mL each of leupeptin, chymostatin, antipain, and pepstatin. After the final wash, the beads were resuspended in 40 μL sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, boiled for 5 minutes, and centrifuged. The supernatant was then subjected to electrophoresis and subsequent immunoblotting using the antibody of interest, including VDAC (Calbiochem, San Diego, CA, U.S.A.), ANT (a gift from Dr. H.H. Schmid, University of Minnesota, Austin), Bcl-2 (Dako, Carpinteria, CA, U.S.A.), and Bcl-w (StressGene, Victoria, British Columbia, Canada).
Immunohistochemistry and double-labeling
Animals were anesthetized with 8% chloral hydrate at 1, 3, 6, 24, or 72 hours after 30 minutes of ischemia or 24 hours after sham operation (n = 4 per time point). Their brains were rapidly removed, frozen in 2-methylbutane at −30°C, covered with mounting medium and stored at −80°C. Coronal sections (15-μm thick) were cut on a cryostat at −20°C and collected on precleaned Probe-On-Slides (Fisher Scientific, Pittsburgh, PA, U.S.A.). Sections at the level of midcaudate (anteroposterior, +0.2 mm from the bregma) were selected and processed for immunohistochemical staining. The sections were fixed with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS; pH 7.4) for 15 minutes followed by three washes in PBS. After the sections were permeabilized with 1% Triton X-100 for 30 minutes followed by three PBS washes, they were pre-blocked using 2% normal goat serum in PBS for 20 minutes. Sections were then incubated for 2 hours at 37°C in the primary antibody diluted (1:250 for Bax; 1:1,000 for cytochrome c) in PBS (pH 7.4), containing 2% goat serum, 0.2% Triton X-100, 0.5% bovine serum albumin, and 0.2% glycine. Sections were washed in PBS four times for 10 minutes each and then incubated for 1.5 hours at room temperature at a 1:2,500 dilution with goat antirabbit Cy3.18 (for Bax) or goat anti-mouse Cy2 (for cytochrome c) (Jackson ImmunoResearch Inc., West Grove, PA, U.S.A.). All steps were performed in the dark. Sections were then washed in PBS four times for 15 minutes each, mounted in gelvatol, and coverslipped. A Zeiss light microscope (Carl Zeiss, Oberkochen, Germany) equipped for epifluorescent illumination was used for observation with excitation/emission wavelengths of 550/565 nm (red) and 495/515 nm (green-yellow) for Bax and cytochrome c, respectively. To detect nonspecific immunostaining, alternate sections were incubated with an antibody that was preabsorbed with the specific antigen before immunohistochemistry (Chen et al., 1996b).
To determine the colocalization of Bax with the mitochondrial marker cytochrome c oxidase or with cytosolic cytochrome c, double-label immunohistochemistry was performed on sections obtained at 6 and 24 hours after ischemia. Sections were first processed for Bax immunohistochemical staining as described above followed by four PBS washes, and then incubated in the anticytochrome c antibody (1:1,000) or anticytochrome c oxidase antibody (1:3,000) for 2 hours at 37°C, followed by PBS washes and incubation with the secondary antibody (goat antimouse Cy2 immunoconjugate). The remaining procedures were the same as described above.
In vitro effect of ANT inhibition on isolated mitochondria
Bongkrekic acid (BA) is a potent inhibitory ligand of the mitochondrial ANT (Henderson and Lardy, 1970; Klingenberg and Appel, 1980), which inhibits apoptosis induced by various death-inducing stimuli (Marchetti et al., 1996a, b ). To determine the effect of BA on Bax-induced mitochondrial changes, the in vitro effect of BA on isolated brain mitochondria was studied. Mitochondria were purified from normal nonischemic rat brain as previously described (Yan et al., 2000). Mitochondria were suspended at 10 mg protein/mL in the MSB buffer containing 400 mmol/L mannitol, 50 mmol/L Tris-HCl (pH 7.2), 5 mg/mL bovine serum albumin, and 10 mmol/L KH2PO4. For the cytochrome c release assay, mitochondria (1 mg protein/mL) were incubated with recombinant Bax (50 μg/mL) in the absence or presence of BA (Biomol Research Laboratories, Plymouth Meeting, PA, U.S.A.) in the MSB buffer for 30 minutes. The Bax fusion protein is a truncated human Bax that lacks the 21-amino acid hydrophobic C-terminus but retains the function (Yan et al., 2000; Yin et al., 1994). After the incubation, mitochondria were pelleted by centrifugation. The resulting supernatants were analyzed by immunoblotting with the monoclonal antibody against cytochrome c. The mitochondria pellets were subjected to coimmunoprecipitation with the Bax antibody followed by immunoblotting with the antibody against ANT.
In vivo drug administration
To determine the potential protective effect of inhibiting ANT against ischemic cell death, BA in vivo infusion studies were performed. BA was dissolved at a concentration of 2 mg/mL in mock cerebrospinal fluid (CSF) and further diluted 1:3 or 1:10 in mock CSF. In vivo infusion of BA was performed using a 10-μL Hamilton syringe (Hamilton, Reno, NV, U.S.A.) through a preimplanted 21-gauge cannula in the left ventricle as previously described (Chen et al., 1998). Thirty minutes before and 2 hours after ischemia, each animal received two ventricular infusions of 2.5 μL each over a 5-minute time period. The resulting doses for BA infusion treatment were 0.56 μg × 2, 1.67 μg × 2, and 5 μg × 2 per animal. The doses of BA used in this study were suggested by the results of others' in vitro and in vivo studies showing the dose ranges of this drug in inhibiting apoptosis (Hirsch et al., 1998; Marchetti et al., 1996b; Zamzami et al., 1996). Infusion of the same volumes of mock CSF served as control for BA treatment. All treatments were assigned to animals in a randomized and blind manner. Temporalis and rectal temperatures were monitored in all animals before, during, and up to 2 hours after ischemia. For histologic outcome experiments, rats were killed at 72 hours after ischemia (n = 6 per group). The brains were rapidly removed and cryostat coronal sections were cut continuously throughout the caudate putamen. Sections were taken every 0.4 mm from the anterior limit to the posterior limit of the caudate putamen (total eight or nine sections per brain) and stained with cresyl violet. The infarct volume in the caudate was determined using the method previously described (Chen et al., 1996b). Adjacent brain sections were processed for the in situ detection of DNA fragmentation using the terminal deoxynucleotidyl-transferase-mediated dUTP-biotin nick end labeling (TUNEL) method (Chen et al., 1998).
For biochemical experiments, rats received vehicle or drug infusion using the optimal dosage of BA determined in the histologic experiments. Animals were killed at 6 or 24 hours after ischemia (n = 3 per condition). Caudate putamen was dissected and processed for measurement of caspaselike activities.
Measurement of caspaselike activities
Measurement of caspase-3- and caspase-9-like protease activity in brain cell extracts was performed as described previously (Chen et al., 1998) with slight modification. The animals were anesthetized using 8% chloral hydrate and decapitated at 6 or 24 hours after ischemia with or without drug treatment, or at 24 hours after sham operation (n = 4 per experimental condition). Brains were quickly removed and tissues were dissected from the caudate putamen of the two hemispheres separately. Protein extracts were prepared on ice by Dounce homogenization of tissues in a lysis buffer containing 25 mmol/L N-[2-Hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES), 5 mmol/L EDTA, 1 mmol/L EGTA, 5 mmol/L MgCl2, 5 mmol/L DTT, 0.5 mmol/L PMSF, and 10 μg/mL each of pepstatin, leupeptin, and aprotinin. Cell lysate was centrifuged at 14,000 g for 30 minutes at 4°C, and the supernatant was used for the enzymatic assay. One hundred micrograms of the protein extracts was incubated for 1 hour at 37°C with the reaction buffer containing 25 mmol/L HEPES (pH 7.5), 10% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylam-monio]-1-propane sulfonate, 5 mmol/L DTT, and 5 mmol/L EDTA in a total volume of 150 μL. The reaction mixture also contained 25 μmol/L fluorogenic peptide cleavage substrate (Biomol, Plymouth Meeting, PA, U.S.A.). Two different substrates were used for the assays (Medical & Biological Laboratories, Watertown, MA, U.S.A.): Ac-DEVD-AFC for caspase-3-like activity and Ac-LEHD-AFC for caspase-9-like activity. One unit of protease activity corresponds to the caspaselike activity that cleaves 1 pmol AFC per minute at 30°C at saturating substrate concentrations. To detect nonspecific protease activity, in parallel experiments the protein extracts were incubated in the reaction buffer with inhibitors for caspase-3 (DEVD-CHO) and caspase-9 (IEHD-CHO) at room temperature for 30 minutes before the addition of assay substrates.
Data analysis
All data are presented as mean ± SD. Comparisons of caspase activities, Bax protein expression, cytochrome c release, infarct size, cell survival, or DNA-damaged cells were made after ischemia with or without drug treatment versus sham controls. Comparisons were made using analysis of variance and post hoc Fisher's t-tests. A level of P < 0.05 was considered statistically significant.
RESULTS
Mitochondrial translocation of Bax after transient cerebral ischemia
In this study, we examined Bax protein expression in normal and ischemic brains, focusing on the caudate putamen, a region reproducibly injured by transient focal ischemia (Chen et al., 1996a, 1997). In particular, the changes in subcellular distribution of Bax after ischemia were investigated using Western blot analysis after subcellular protein fractionation and using double-label fluorescence immunohistochemistry.
Representative Western blots are presented in Fig. 1, which shows mitochondrial accumulation of Bax after ischemia. In nonischemic caudate putamen, basal Bax immunoreactivity was detectable in the cytosol and to a lesser extent in the mitochondria. Bax immunoreactivity was increased 1.8- to 3.7-fold in the mitochondrial fraction from 3 to 24 hours after 30 minutes of middle cerebral artery occlusion. In contrast, Bax immunoreactivity was not significantly changed in the cytosolic fraction until 24 hours after ischemia, when a 2.8-fold increase was detected.
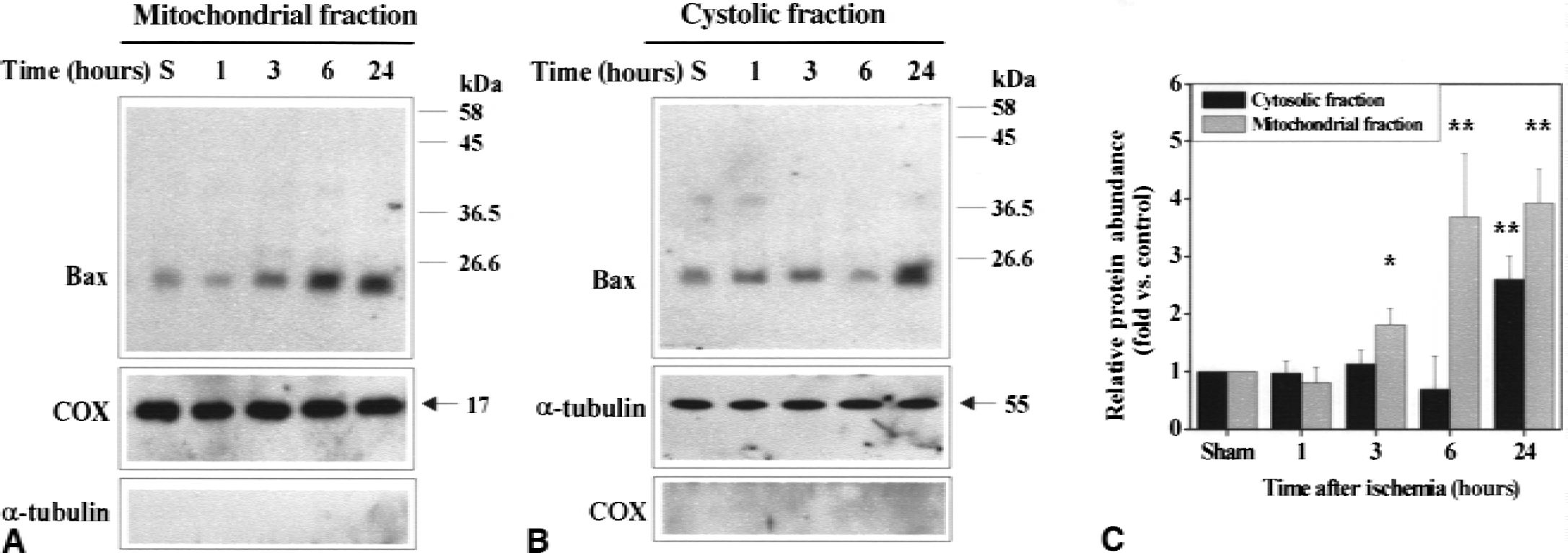
Immunohistochemical identification of Bax was performed on brain sections using indirect immunofluorescence (Fig. 2). Compared with the nonischemic brain, Bax immunoreactivity was markedly increased in the ischemic caudate putamen at 24 hours after ischemia. Most cells that showed increased Bax immunoreactivity were distributed in the medial caudate putamen, a region corresponding to the inner border zone of the infarction. Figure 2 also shows Bax immunoreactivity in the medial caudate putamen under higher magnification (×400). In nonischemic caudate putamen, Bax staining was faint and diffusive in the cytosol and generally difficult to distinguish from the background. Three hours after 30 minutes of ischemia, increases in Bax immunoreactivity began to be detectable in many medium-sized neurons. The increased immunoreactivity of Bax had a punctate appearance, suggesting a membrane-bound rather than a soluble protein. This punctate pattern of Bax immunoreactivity was most prominent in the media caudate putamen at 6 hours after ischemia. At 24 or 72 hours after ischemia, increased Bax immunoreactivity in neurons showed a mixed diffusive and punctate pattern, consistent with the immunoblotting results showing that Bax was also increased in the cytosolic fraction at 24 hours after ischemia.
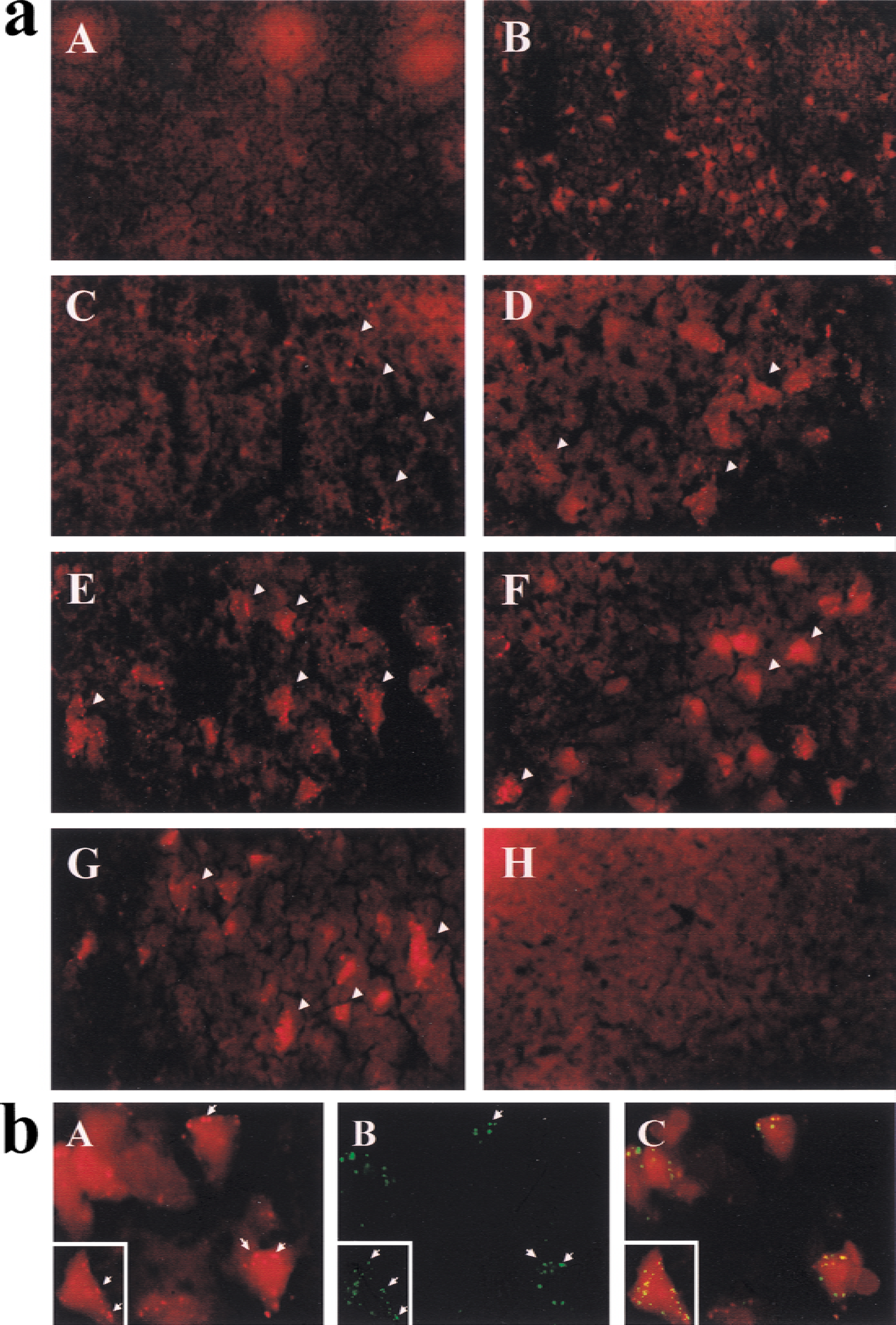
Subcellular localization of Bax immunoreactivity in neurons was further studied using double-label immunohistochemistry with the mitochondrial membrane-bound enzyme cytochrome c oxidase. The punctate membrane-bound features of Bax immunoreactivity in ischemic neurons are now shown to be those of mitochondrial membrane localization (Fig. 2).
Correlation of Bax translocation and cytochrome c and caspase-9 release after ischemia
One major putative consequence of Bax translocation to mitochondria is the release of cytochrome c. To determine whether Bax translocation is associated with cytochrome c release in the ischemic brain, we examined the temporal profile of cytochrome c release in the caudate putamen after 30 minutes of focal ischemia. Western blots revealed that cytochrome c immunoreactivity was detectable in the cytosolic fraction from 3 to 24 hours after ischemia, with the highest concentration seen at the 24-hour time point (Fig. 3). In contrast, cytochrome c immunoreactivity was decreased in the mitochondrial fraction at 6 and 24 hours after ischemia, consistent with the partial loss of cytochrome c from these organelles. Concomitantly, the procaspase-9 immunoreactivity was also decreased in the mitochondrial fraction (Fig. 3), suggesting that caspase-9 was released from the mitochondria after ischemia.
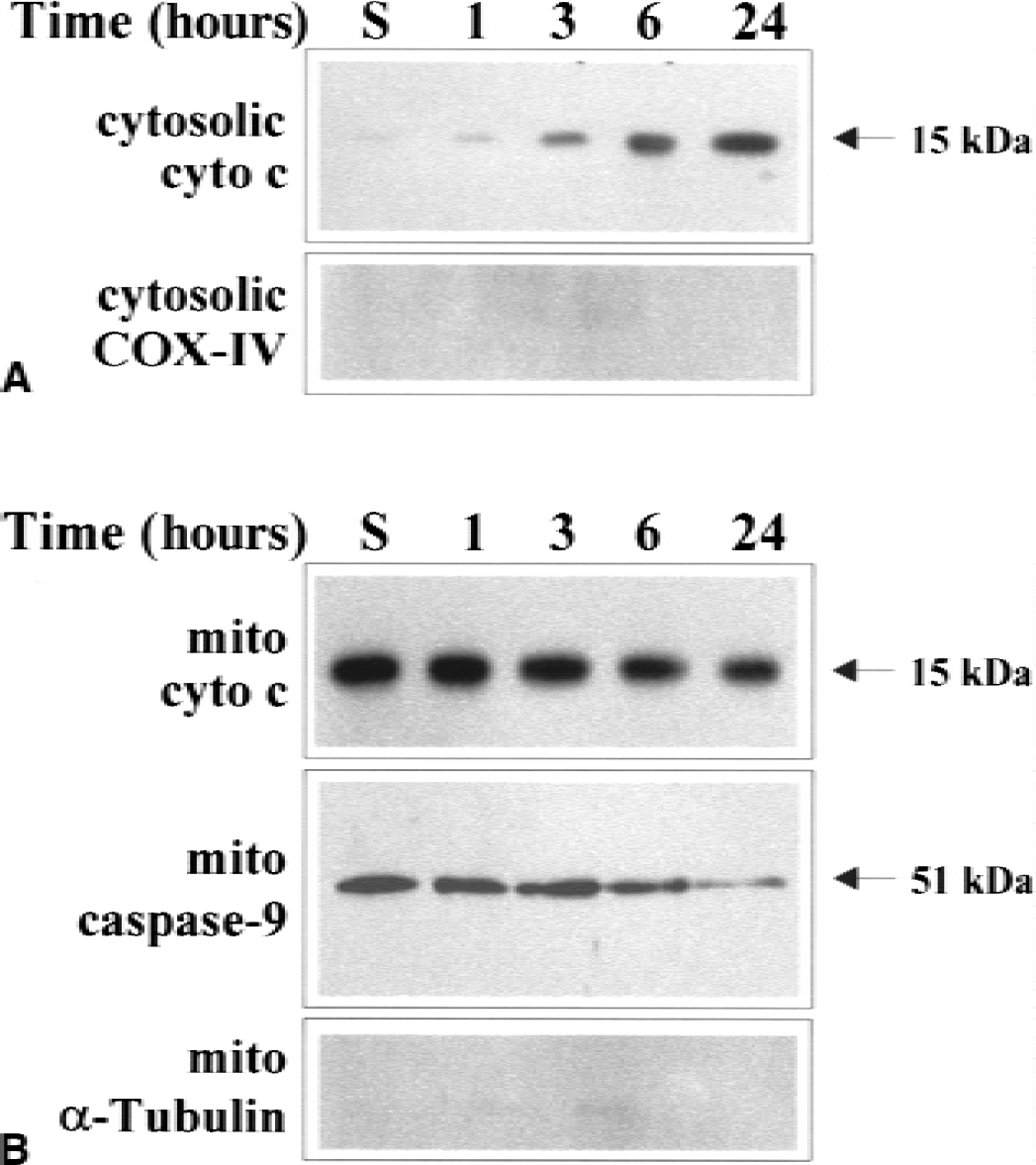
Representative Western blots show the temporal profiles of release of cytochrome c and caspase-9 from the mitochondria after ischemia. Cytosolic and mitochondrial protein was purified from the caudate putamen at 1, 3, 6, and 24 hours after ischemia or 24 hours after sham operation and subjected to Western blot analysis. Cytochrome c immunoreactivity was increased in the cytosol 3 to 24 hours after ischemia
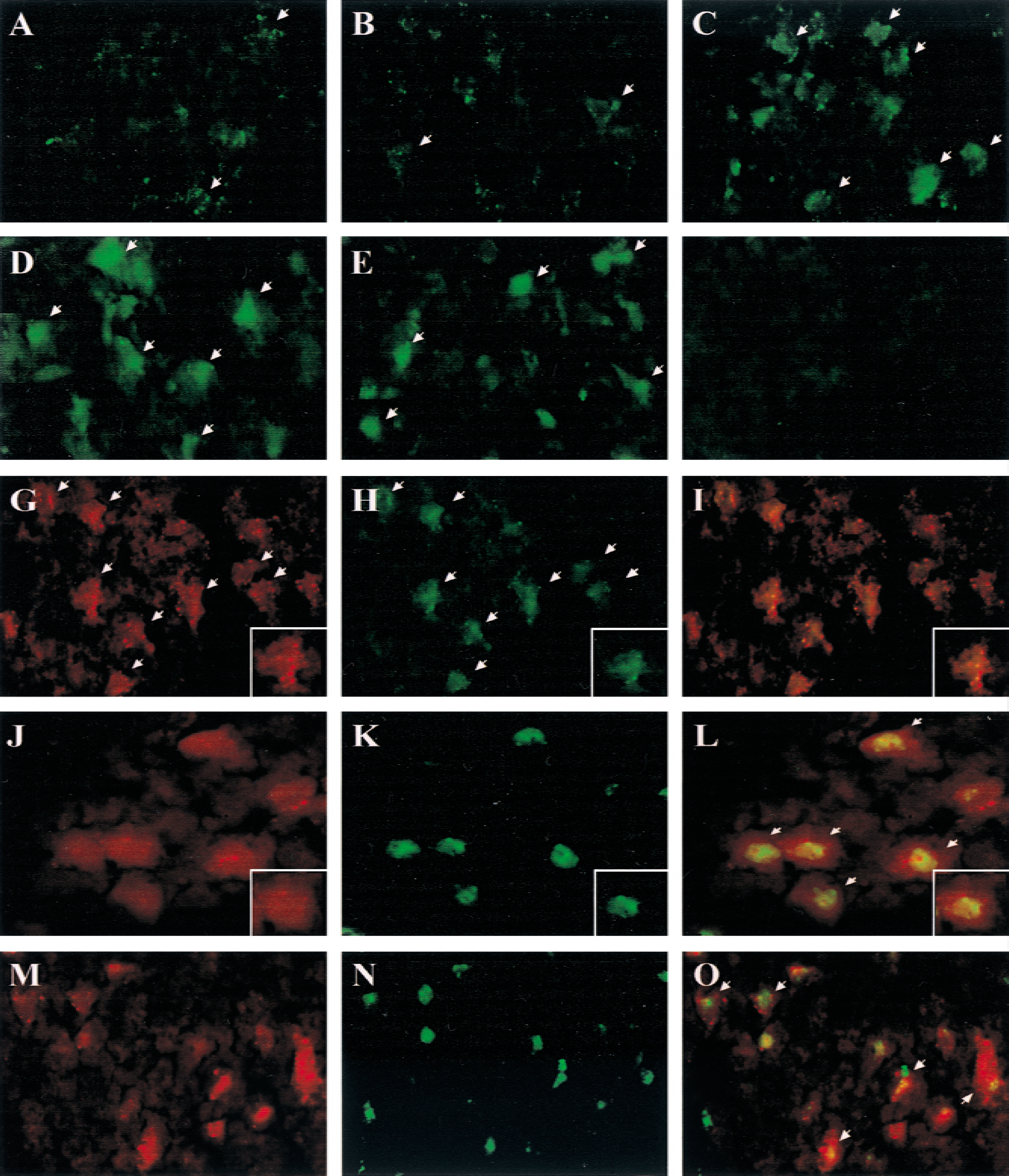
Figure 4 shows representative immunofluorescent images of cytochrome c in medial caudate putamen neurons after ischemia. In normal nonischemic caudate putamen, cytochrome c immunoreactivity was present in a punctate pattern in scattered cells, suggesting that the antibody had little access to the cytochrome c antigen residing in the mitochondrial intermembrane matrix and resulted in limited detection of cytochrome c immunoreactivity. Beginning at 3 hours, and more prominently at 6 to 24 hours after ischemia, cytosolic diffusive staining of cytochrome c was detected in a large number of caudate putamen neurons. At the earlier time points (3 to 6 hours), cells showing cytosolic staining of cytochrome c were mainly distributed in the medial caudate putamen, the same region where mitochondrial translocation of Bax was found to be abundant. At the later time point (24 hours), however, cytochrome c-immunoreactive cells were also found in the dorsal-lateral caudate putamen region corresponding to the infarct core (not shown). Cytosolic staining of cytochrome c was not found in the contralateral hemisphere at any time point.
Correlation between Bax intracellular translocation, cytochrome c release, and cell death in caudate neurons after ischemia.
Double-label immunofluorescence was performed to colocalize Bax with cytochrome c immunoreactivity. Cytosolic cytochrome c immunoreactivity overlapped with mitochondria-bound Bax in many medial caudate putamen neurons at 6 (Fig. 4) and 24 hours (not shown) after ischemia. In all six ischemic brains examined (n = 3 per time point), more than 80% of cytosolic cytochrome c-positive neurons in the ischemic medial caudate putamen showed punctate Bax immunoreactivity.
Double-label immunofluorescence also revealed that the cells showing increased Bax immunoreactivity were mainly neurons, because Bax immunoreactivity was predominantly distributed in cells that were stained positively with the neuronal nuclear marker NeuN (Fig. 4).
Studied in three brains obtained at 72 hours after ischemia, colocalization of Bax immunoreactivity and in situ DNA fragmentation (TUNEL positive) was detected in a large number of neurons in the ischemic medial caudate putamen (Fig. 4). Many DNA-fragmented neurons showed punctate Bax immunoreactivity.
Increased protein dimerization between Bax and mitochondrial proteins after ischemia
It has been suggested that Bax increases mitochondrial membrane permeability and triggers cytochrome c release by interacting with and posttranslationally modifying several mitochondrial membrane proteins that form the permeability transition pore complex (PTPC) (Jurgensmeier et al., 1998; Marzo et al., 1998; Narita et al., 1998; Pastorino et al., 1998; Rosse et al., 1998; Shimizu et al., 1999; Wolter et al., 1997). To test the hypothesis that ischemic injury may involve enhanced interaction between Bax and mitochondrial proteins, immunoprecipitation was performed using the monoclonal anti-Bax antibody and then probing the Bax immunoprecipitates with antibodies against VDAC and ANT, respectively. Coimmunoprecipitation of Bax with VDAC and ANT was detected in samples prepared from the caudate putamen of rat brains (Fig. 5). Compared with the nonischemic controls, the contents of VDAC and ANT in the Bax immunoprecipitates were increased up to 3.4-fold and 6.1-fold, respectively, 6 to 24 hours after ischemia. The increased formation of Bax/ANT and Bax/VDAC dimerization in ischemic brains was not accompanied by changes in the protein levels of ANT or VDAC in the mitochondrial fraction, indicating that Bax translocation to the mitochondria was directly responsible for this protein-protein interaction.
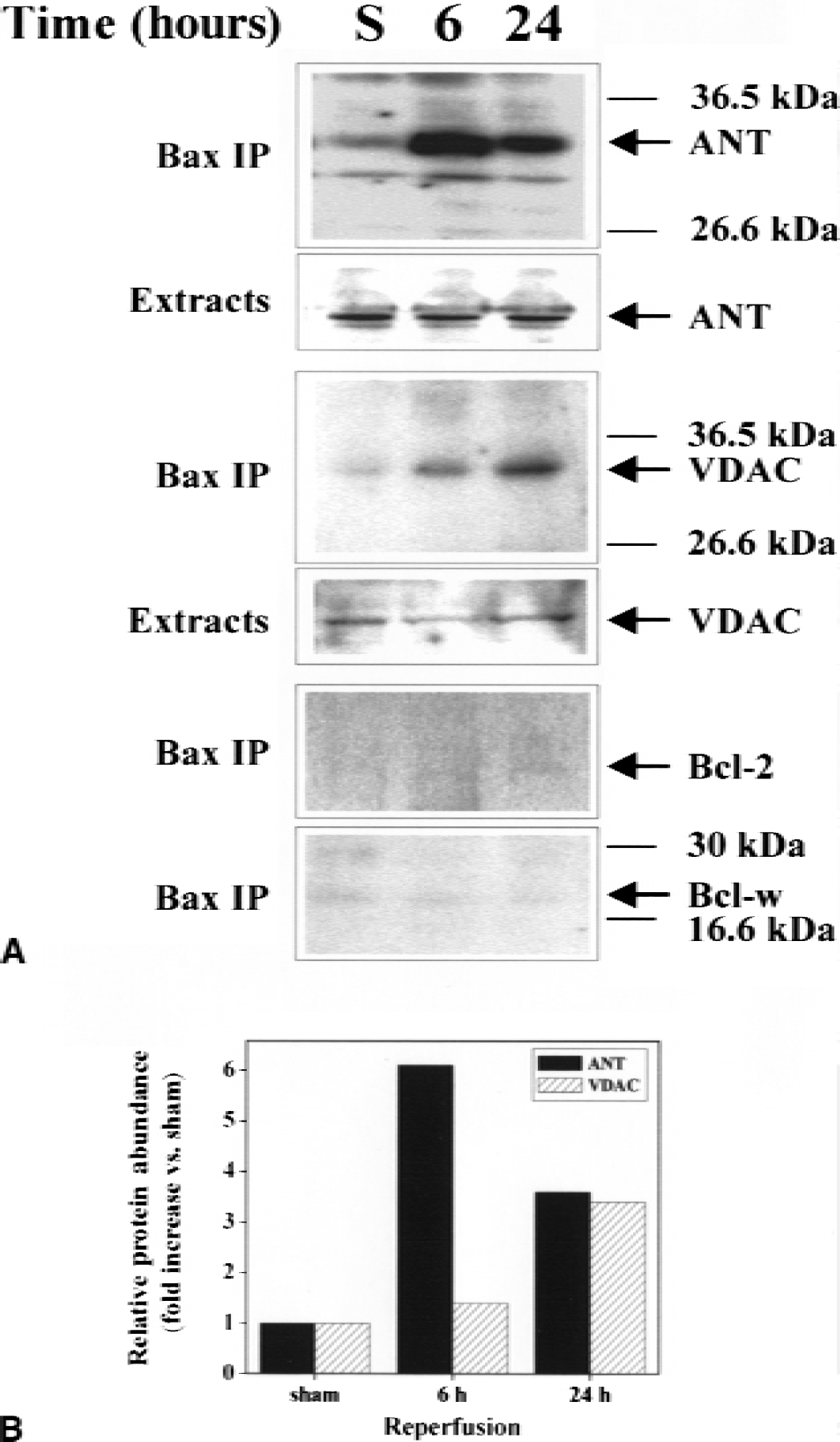
The possible interactions between Bax and the antiapoptotic members of Bcl-2 family, including Bcl-2 and Bcl-w, were also investigated in nonischemic and ischemic samples. No changes in coimmunoprecipitation of Bax with Bcl-2 or Bcl-w were detected in any samples tested.
Effects of the ANT inhibitor
Bongkrekic acid inhibits apoptosis by binding to and inhibiting the major PTPC component ANT (Furlong et al., 1998; Marchetti et al., 1996a, b ; Zamzami et al., 1996). To determine the effect of BA on Bax-induced mitochondrial changes, isolated brain mitochondria were incubated with the recombinant Bax protein (Yan et al., 2000) for 30 minutes in the absence or presence of BA. In the absence of BA, incubation with recombinant Bax resulted in redistribution of cytochrome c and pro-form of caspase-9 (procapase-9) from mitochondria to the supernatant. In the presence of BA, however, the release of procaspase-9 induced by Bax was inhibited in a dose-dependent manner with the maximal effect of BA at 50 to 100 μmol/L (Fig. 6). However, BA had no inhibitory effect on Bax-triggered cytochrome c release. Immunoprecipitation and Western blot analysis revealed that incubation of recombinant Bax with the isolated mitochondria resulted in increased formation of Bax/ANT and Bax/VDAC dimerization. The addition of BA in the reaction mixture prevented the formation of Bax/ANT dimerization but had no effect on the formation of Bax/VDAC complex.
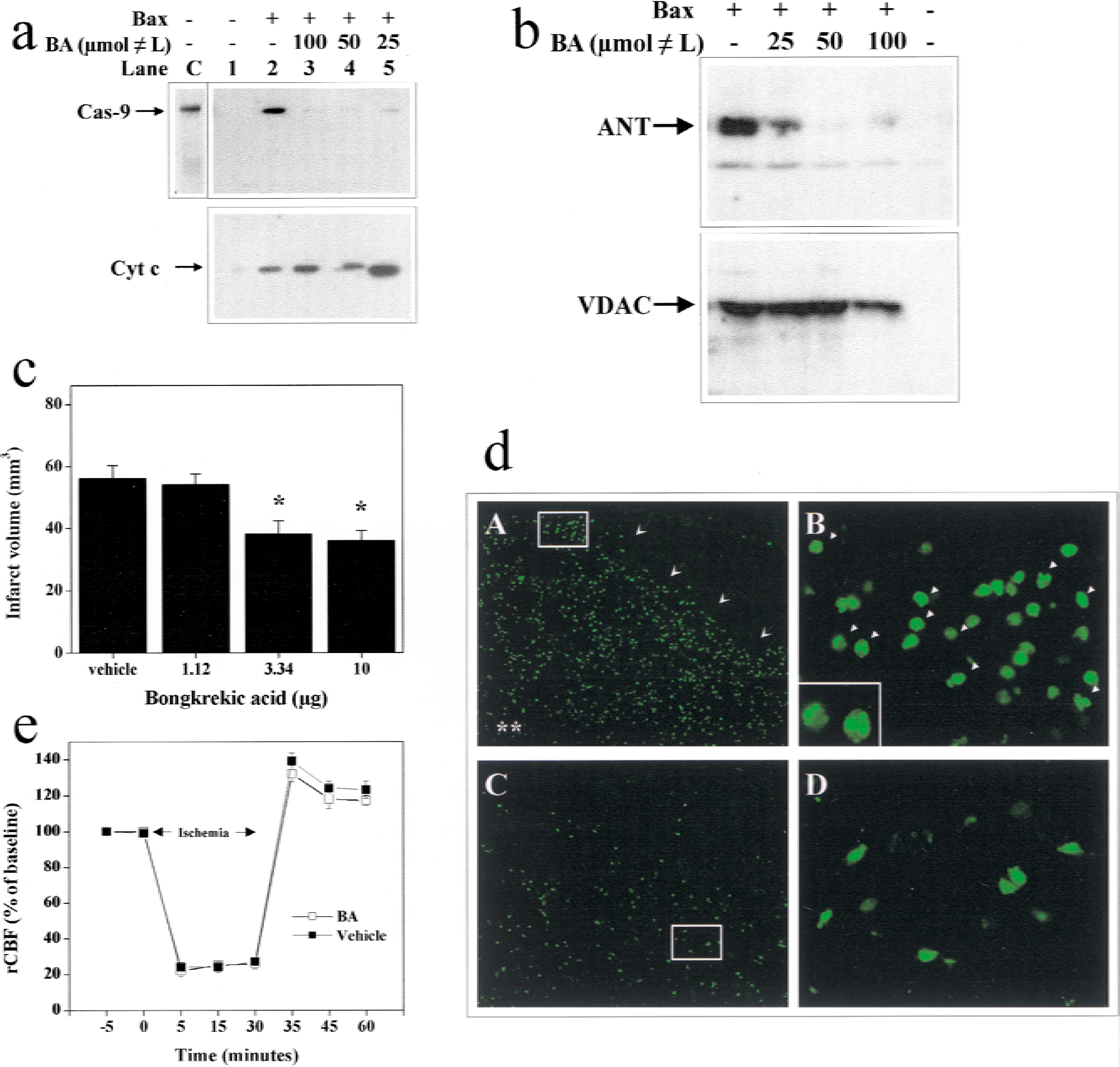
Effects of the adenine nucleotide translocator (ANT) inhibitor bongkrekic acid (BA) on mitochondrial damage and ischemic cell death.
The effect of BA on ischemic cell death was determined in the in vivo infusion experiments. Middle cerebral artery occlusion for 30 minutes produced ipsilateral cerebral infarcts averaging 55 mm3 in volume in the caudate putamen after 72 hours. Pretreatment with BA 30 minutes before and 2 hours after ischemia reduced infarct size in a dose-dependent manner, reaching maximal protection by 40%. Cerebral salvage occurred primarily in the medial caudate putamen, leading to significantly decreased cell death, as demonstrated by the reduced amount of TUNEL staining. In contrast, BA had no measurable protection in the dorsal-lateral caudate putamen, which represents the ischemic core (not shown). Protection by BA was not accompanied by alterations in regional cerebral blood flow (rCBF), as determined using laser–Doppler flowmetry up to 30 minutes after ischemia. Physiologic variables, including temporalis muscle and body temperature, were not significantly different between BA- and vehicle-treated animals (data not shown).
As determined at 6 and 24 hours after ischemia, BA significantly reduced caspase-9- and caspase-3-like activity in caudate putamen cell extracts (Fig. 7).
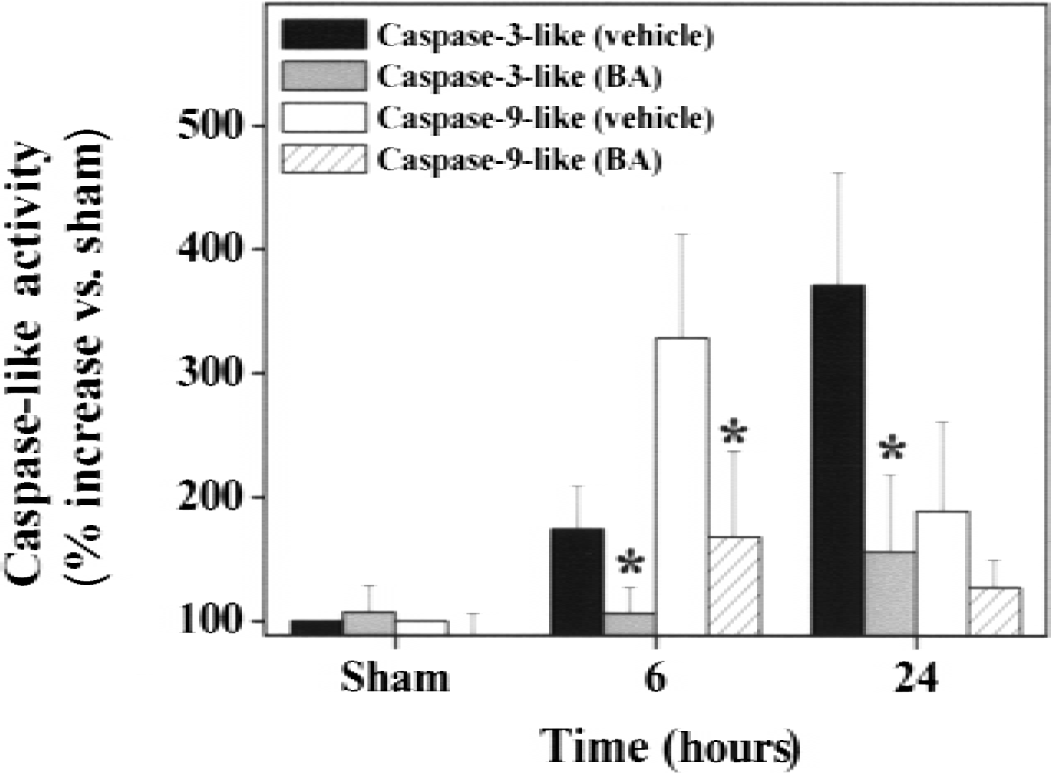
Effects of bongkrekic acid (BA) on terminal caspase activities in the brain after ischemia. Cell extracts were prepared from the caudate putamen at 6 and 24 hours after 30 minutes of focal ischemia or 24 hours after sham operation and then subjected to measurements for caspase-9- and caspase-3-like protease activity using specific substrates. Protease activities are expressed as percentage increases versus sham controls. Treatment with BA significantly decreases caspase-9- and caspase-3-like activities. Data are mean ± SD, n = 4 per group. *P < 0.05 versus vehicle-treated brains (analysis of variance and post hoc Fisher's t-tests).
DISCUSSION
The mitochondrion is the key intracellular site for sensing various death-inducing stimuli and transmitting them to the executional process of apoptosis. Release of soluble intermembrane proteins such as cytochrome c and apoptosis-inducing factor from mitochondria into cytosol is a crucial step in the mitoch ondrial apoptotic-signaling pathway (for review see Kroemer and Reed, 2000). In the cytosol, cytochrome c forms a complex with Apaf-1, a homolog of the Caenorhabditis elegans protein ced-4, which then binds to and activates caspase-9. This triggers the activation of terminal caspases such as caspase-3, which in turn lead to cell death (Kluck et al., 1997; Kroemer and Reed, 2000; Liu et al., 1996; Yang et al., 1997; Zou et al., 1997). Although the downstream cascade for the execution of apoptosis has now become clear, the mechanism by which the mitochondrial-signaling pathway is activated during apoptosis is complex and appears to depend on the types of death stimuli and the cell types receiving such stimuli. It has been postulated that mitochondrial translocation of the cytosolic proapoptotic protein Bax is an important trigger for cytochrome c release and caspase activation during apoptosis in many cell types, including neuronal cells (Cregan et al., 1999; Putcha et al., 1999; Xiang et al., 1998).
Our data show that Bax translocation from cytosol to mitochondria constitutes an early event in ischemic brain injury. Although the up-regulation of overall Bax protein expression did not occur until 6 to 24 hours after transient focal ischemia, an increased accumulation of Bax in the mitochondrial membranes was readily detectable at 3 hours after ischemia and thereafter. This translocation of Bax was shown immunohistochemically at the cellular level and confirmed in purified mitochondrial extracts by Western blot analysis. The early increases (3 to 6 hours after ischemia) in mitochondrial Bax occurred without the predicted decreases in cytosolic levels of Bax. These results suggest that the de novo synthesis of Bax may contribute the increased Bax translocation after ischemia. Medial caudate putamen, a region that developed abundant apoptotic cell death in this ischemic model, showed prominent changes in Bax after ischemia. The regional distribution and temporal profile of Bax translocation after ischemia coincided with those of cytochrome c and procaspase-9 release, but both events occurred before the induction of caspase-3 activity (>6 hours) and cell death (24 hours) in this model. Thus, these results are consistent with the postulated role of Bax in triggering the mitochondria-dependent apoptotic pathway in ischemic injury.
Two mechanisms by which Bax targets mitochondria and promotes cell death have been hypothesized (for review see Adams and Cory, 1998). Bax can form heterodimerization with antiapoptotic family members such as Bcl-2 and Bcl-x-l, therefore blocking their antiapoptotic effects (Oltvai et al., 1993; Yin et al., 1994). Deletion analysis has indicated that the BH3 domain of Bax is required for it to bind to Bcl-2 or Bcl-x-l and promote cell death. However, the functional significance of such protein–protein interactions between Bax and antiapoptotic Bcl-2 family proteins in triggering apoptosis has been questioned: the results of recent studies clearly point out that Bax can trigger cell death through mechanisms independent of the BH3 domain and independent of its interaction with Bcl-2 and Bcl-x-l (Eskes et al., 1998; Jurgensmeier et al., 1998). Therefore, it has been suggested that the second mechanism by which Bax triggers apoptosis is the enhancement of mitochondrial outer membrane permeability, which in turn leads to the release of cytochrome c and other apoptosis-promoting molecules (Jurgensmeier et al., 1998; Pastorino et al., 1998; Rosse et al., 1998; Wolter et al., 1997). In vitro cell-free studies showed that Bax can form pH- and voltage-dependent ion-conducting channels in planar lipid bilayer membranes, and this channel-forming activity in mitochondrial membrane is likely involved in the regulation of mitochondrial membrane permeability (Antonsson et al., 1997; Schlesinger et al., 1997).
The direct effect of Bax on cytochrome c release has been extensively studied using isolated mitochondria, including those of neuronal origin. Addition of submicromolar amounts of recombinant Bax to isolated mitochondria results in rapid depletion of cytochrome c from the intermembrane space of the mitochondria (Eskes et al., 1998; Marzo et al., 1998; Yan et al., 2000). This effect of Bax is mediated via the conformational changes in the PTPC proteins, especially ANT and VDAC (Marzo et al., 1998; Shimizu et al., 1999, 2000). The formation of heterodimerization between Bax and ANT and VDAC is directly responsible for the conformational changes in PTPC and increased membrane permeability (Beutner et al., 1998; Marzo et al., 1998; Shimizu et al., 1999). In strong support of the role of changes in PTPC proteins in Bax-induced cell death, it has been shown that ectopic expression of Bax-induced cell death occurs in wild-type but not in ANT- or VDAC-deficient cells (Marzo et al., 1998; Shimizu et al., 1999).
Using coimmunoprecipitation, we obtained strong evidence of enhanced Bax/ANT and Bax/VDAC interactions in brain mitochondria after ischemia. Consistent with the deduced role of Bax/ANT heterodimerization in triggering the mitochondrial apoptotic pathway, we found that the ANT inhibitor BA interrupted the formation of Bax/ANT complex and inhibited Bax-induced release of procaspase-9 from the isolated brain mitochondria. Concomitantly, intraventricular infusion of BA significantly inhibited caspase-9- and caspase-3-like activity and attenuated ischemia-induced cell death in the caudate putamen in vivo. These results show, for the first time, that inhibiting ANT activity offers neuroprotection against ischemic cell death. Hence, enhanced mitochondrial ANT activity may play an important role in mediating ischemic injury; further, the formation of Bax/ANT heterodimerization likely contributes to postischemic changes in ANT. Based on the in vitro and in vivo observations, we suggest that BA achieves the antiapoptotic effect in neurons, at least in part, by inhibiting Bax-triggered, mitochondria-dependent activation of terminal caspases. We further speculate that Bax-triggered caspase activation in the ischemic brain may involve the release of both procaspase-9 and cytochrome c, but blocking the release of either factor may be sufficient to prevent caspase activation.
As shown here, BA was capable of blocking Bax-induced procaspase-9 release and attenuating cell death but had no effect on cytochrome c release from brain mitochondria. Thus, it is possible that the presence of cytochrome c in the cytosol may not necessarily result in caspase activation unless other cofactors such as caspase-9 reach certain concentrations in the cytosol as well. This speculation is strikingly relevant to the recently reported finding that in cultures of cerebrocortical neurons, Bongkrekic acid (BA) prevented N-methyl-d-aspartate (NMDA) receptor-mediated caspase-3 activation and apoptosis without affecting cytochrome c release (Budd et al., 2000). Taken together, these results and ours suggest that, in addition to preventing cytochrome c release, mitochondrion-level modulations that prevent caspase-9 release may be a potential therapeutic strategy for neuronal injury resulting from ischemia or related insults.
In summary, our results show that transient focal cerebral ischemia induced overexpression and intracellular translocation of the proapoptotic protein Bax from cytosol to mitochondria in the brain, with a temporal profile and regional distribution coinciding with that of cytochrome c and caspase-9 release. Translocated Bax formed a heterodimeric complex with the mitochondrial proteins ANT and VDAC in postischemic brain in vivo and in isolated brain mitochondria in vitro and triggered the release of cytochrome c and caspase-9 from the isolated mitochondria. The ANT inhibitor BA, which interrupted Bax and ANT interactions and inhibited Bax-triggered caspase-9 release in vitro, showed significant protection against ischemia-induced caspase activation and cell death in the brain. These results strongly suggest that the Bax-mediated mitochondrial apoptotic signaling pathway may play an important role in ischemic neuronal injury.
Footnotes
Acknowledgments:
The authors thank Carol Culver for excellent editorial assistance and Pat Strickler for secretarial support.