
Abstract
The serine-threonine kinase, Akt, prevents apoptosis by phosphorylation at serine-473 in several cell systems. After phosphorylation, activated Akt inactivates other apoptogenic factors, such as Bad or caspase-9, thereby inhibiting cell death. The present study examined phosphorylation of Akt at serine-473 and DNA fragmentation after transient focal cerebral ischemia in mice subjected to 60 minutes of focal cerebral ischemia by intraluminal blockade of the middle cerebral artery. Phospho-Akt was analyzed by immunohistochemistry and Western blot analysis. The DNA fragmentation was evaluated by terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL). Immunohistochemistry showed the expression of phospho-Akt was markedly increased in the middle cerebral artery territory cortex at 4 hours of reperfusion compared with the control, whereas it was decreased by 24 hours. Western blot analysis showed a significant increase of phospho-Akt 4 hours after focal cerebral ischemia in the cortex, whereas phospho-Akt was decreased in the ischemic core. Double staining with phospho-Akt and TUNEL showed different cellular distributions of phospho-Akt and TUNEL-positive staining. Phosphorylation of Akt was prevented after focal cerebral ischemia by LY294002, a phosphatidylinositol 3-kinase inhibitor, which facilitated subsequent DNA fragmentation. These results suggest that phosphorylation of Akt may be involved in determining cell survival or cell death after transient focal cerebral ischemia.
INTRODUCTION
The serine-threonine kinase, Akt, which is also referred to as protein kinase B and RAC, is the cellular homologue of the viral oncogene v-Akt. This protein kinase plays a critical role in controlling the balance between survival and apoptosis (Burgering and Coffer, 1995; Franke et al., 1995, 1997). Akt is activated by insulin and various growth and survival factors, and functions in a wortmannin-sensitive pathway involving phosphatidylinositol 3-kinase (PI3-kinase) (Burgering and Coffer, 1995; Franke et al., 1995). Akt contains an amino-terminal pleckstrin homology domain, which binds phosphorylated lipids at the membrane in response to activation of PI3-kinases. Akt is activated by phospholipid binding and activation-loop phosphorylation at threonine-308 by phosphatidylinositol(3,4,5)-trisphosphate-deinase 1 (Alessi et al., 1996), and also within the C-terminus at serine-473 by integrin-linked kinase (Delcommenne et al., 1998). After phosphorylation, Akt promotes cell survival and prevents apoptosis by inactivating several targets, including Bad (Datta et al., 1997; del Peso et al., 1997), glycogen synthase kinase-3 (Cross et al., 1995; Srivastava and Pandey, 1998), forkhead transcription factors (Brunet et al. 1999; Kops and Burgering, 1999), or caspase-9 (Cardone et al., 1998). According to recent reports, phospho-Akt also appears to negatively regulate the mitogen-activated protein kinase and extracellular-signal-responsive kinase kinase (MEK) and extracellular-signal-responsive kinase (ERK) pathway, inactivating Raf at serine-259 (Rommel et al., 1999; Zimmermann and Moelling, 1999).
Changes in phospho-Akt levels were reported after global cerebral ischemia (Ouyang et al., 1999; Namura et al., 2000; Yano et al., 2001) and focal cerebral ischemia (FCI) (Janelidze et al., 2001; Yoshimoto et al., 2001). These reports showed a temporal increase of phospho-Akt at serine-473 (Ouyang et al., 1999; Namura et al., 2000; Janelidze et al., 2001; Yano et al., 2001); however, it is not completely clear how phosphorylation of Akt plays a role in the downstream events after ischemia. It has been reported that cytochrome c is released from mitochondria to the cytosol after Akt activation in a global ischemia model (Ouyang et al., 1999). Because Bcl-2 and Bcl-XL proteins are negatively regulated by Bad (Yang et al., 1995; Zha et al., 1996), it is conceivable that Akt activation attenuates Bcl-2 suppression by Bad, and then cytochrome c release from mitochondria is prevented at an early stage after global ischemia. However, this study does not prove the direct interaction between Akt activation and cytochrome c release. To address the direct effect of Akt in cell survival, a drug study using a PI3-kinase inhibitor would be useful. A report showed that wortmannin, a PI3-kinase inhibitor, did not block neuroprotection mediated by brain-derived neurotrophic factor after hypoxic-ischemic injury, indicating that the Akt pathway did not play a critical role in brain-derived neurotrophic factor-mediated neuroprotection (Han and Holtzman, 2000). Wortmannin inhibited the neuroprotective effect by preconditioning in a gerbil global-ischemia model, suggesting that the neuroprotective action of preconditioning was mediated by Akt activation (Yano et al., 2001), whereas the opposite result—that preconditioning did not increase in phospho-Akt level, indicating Akt does not play a major role in the neuroprotective action of preconditioning—is also reported (Namura et al., 2000). However, the mechanism of cell survival mediated by phosphorylation of Akt after FCI is still unclear.
In the present study, we investigated the possibility that intracerebroventricular injection of LY294002, another PI3-kinase inhibitor, could reduce phosphorylation of Akt and increase subsequent DNA damage after transient FCI. We also examined the regional distribution of phospho-Akt by immunohistochemistry.
MATERIALS AND METHODS
Focal cerebral ischemia
All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by Stanford's Administrative Panel on Laboratory Animal Care. Adult male CD-1 mice (35–40 g) were subjected to transient FCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture, as previously described (Yang et al., 1994).
The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of the femoral artery allowed the monitoring of blood pressure and arterial blood gases. Samples for analysis were taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. Blood gases were analyzed by a pH and blood gas analyzer (Chiron Diagnostics Ltd., Essex, U.K.). After a midline skin incision, the left external carotid artery was exposed and its branches were electrocoagulated. An 11-mm 5–0 surgical monofilament nylon suture that was blunted at the end was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minutes of MCA occlusion, blood flow was restored by withdrawing the nylon suture.
Drug injection
To investigate the role of the PI3-kinase pathway after FCI, a PI3-kinase inhibitor was injected. The inhibitor, LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1benzopyran-4-one), was purchased from Cell Signaling (Beverly, MA, U.S.A.) and dissolved in dimethyl sulfoxide and phosphate-buffered saline (PBS). The scalp was incised on the midline and the skull was exposed. LY294002 (5 or 50 nmol in 25% dimethyl sulfoxide in PBS) and the vehicle (25% dimethyl sulfoxide in PBS) were injected intracerebroventricularly (2 μL, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). LY294002 and the vehicle were injected 1 hour before MCA occlusion.
Western blot analysis
Whole-cell protein extraction was performed. Samples were obtained from the caudate putamen (ischemic core) and the MCA territory cortex on the ischemic sides and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 4, and 24 hours of reperfusion and homogenized in seven volumes of cold suspension buffer (20 mmol/L HEPES-potassium hydroxide [pH 7.5], 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L ethyleneglycoltetracetic acid, 0.7% protease and phosphatase inhibitor cocktails [Sigma, St. Louis, MO, U.S.A.]). The homogenate was centrifuged at 10000 g for 20 minutes at 4°C and the supernatant was used for the analysis. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, U.S.A.) to the supernatant, equal amounts of the samples were loaded per lane. The primary antibodies were 1:1000 dilution of rabbit polyclonal antibody against phospho-Akt (serine-473) and Akt (Cell Signaling) or 1:10000 dilution of anti–β-actin monoclonal antibody (Sigma). Western blot analyses were performed with horseradish peroxidase-conjugated antirabbit immunoglobulin G (Cell Signaling) or antimouse immunoglobulin G (Chemicon International, Temecula, CA, U.S.A.) using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, England). The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, U.S.A.) and the results were quantified using Multi-Analyst software (Bio-Rad).
Immunohistochemistry of phospho-Akt and neuron-specific nuclear protein
Anesthetized animals were perfused with 200 mL 10 U/mL heparin in 0.9% saline and 4% paraformaldehyde in 0.1 mol/L PBS (pH 7.4) after 1, 4, and 24 hours of reperfusion, and in normal controls (n = 3). Brains were removed, postfixed for 12 hours in 4% paraformaldehyde, sectioned at 50 μm with a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with rabbit polyclonal antiphospho-Akt (serine-473) antibody IHC Specific (for immunohistochemistry; Cell Signaling) at a dilution of 1:200. Immunohistochemistry was performed using the avidin-biotin technique and the nuclei were counterstained with methyl green solution for 10 minutes. As a negative control, alternative sections were incubated without primary antibodies. To confirm the specificity of the antibody, immunohistochemistry with the antiphospho-Akt antibody preabsorbed with the phospho-Akt serine-473 peptide (Cell Signaling) was also performed.
To evaluate neuronal expression of phospho-Akt, we performed double immunofluorescent staining for phospho-Akt and neuron-specific nuclear protein (NeuN). The sections fixed by 4% paraformaldehyde were immunostained with the phospho-Akt antibody as described previously with biotinylated goat antirabbit immunoglobulin G (Vector Laboratories, Burlingame, CA, U.S.A.) followed by Fluorescein Avidin DCS (Vector Laboratories). The sections were incubated with blocking solution and reacted with mouse monoclonal anti-NeuN antibody (Chemicon) at a dilution of 1:200, followed by Texas Red conjugated donkey antimouse immunoglobulin G antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) at a dilution of 1:250. Subsequently, the slides were covered with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories). Fluorescence of fluorescein was observed at excitation of 495 nm and emission of more than 515 nm, and fluorescence of Texas Red was observed at excitation of 510 nm and emission of more than 580 nm. Fluorescence of DAPI was also observed at excitation of 360 nm and emission of more than 460 nm.
Immunofluorescent double labeling with phospho-Akt immunohistochemistry and TUNEL
To clarify the spatial relationship between phospho-Akt expression and DNA fragmentation, we performed double staining for phospho-Akt and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) using a fluorescent method. The sections fixed by 4% paraformaldehyde were immunostained with the phospho-Akt antibody as described above, with biotinylated goat antirabbit immunoglobulin G (Vector Laboratories, Burlingame, CA, U.S.A.) followed by Fluorescein Avidin DCS (Vector Laboratories). Then, the sections were incubated with NeuroPore (Trevigen, Gaithersburg, MD, U.S.A.) for 30 minutes. The sections were placed in 1X terminal deoxynucleotidyl transferase buffer (Life Technologies, Gaithersburg, MD) for 30 minutes, followed by reaction with terminal deoxynucleotidyl transferase enzyme (Life Technologies) and biotinylated 16-dUTP (Boehringer Mannheim, Indianapolis, IN, U.S.A.) at 37°C for 90 minutes. The sections were washed two times in saline-sodium citrate (150 mmol/L sodium chloride, 15 mmol/L sodium citrate, pH 7.4) for 15 minutes, followed by washing in PBS two times for 15 minutes. Texas Red Avidin DCS (Vector Laboratories) was applied to the sections for 30 minutes. Subsequently, the slides were covered with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories). Fluorescence was assessed as described previously.
In situ labeling of DNA fragmentation
To estimate DNA damage, cell counting was performed after TUNEL staining. The sections were treated as described previously. After incubation with biotinylated 16-dUTP, the sections were incubated with avidin-biotin horseradish peroxidase solution (ABC kit; Vector Laboratories) for 30 minutes, then washed for 15 minutes with 0.175 mol/L sodium acetate. Staining was observed using 0.025% diaminobenzidine and 0.075% hydrogen peroxide in PBS with 0.4 mg/mL nickel sulfate. The sections were rinsed with water and stained with methyl green for 10 minutes.
Quantification and statistical analysis
To analyze the amount of the phospho-Akt and β-actin, we used optical density as the unit. The data are expressed as mean ± SD. To quantify the DNA-fragmented cells after ischemia, the number of TUNEL-positive cells in the caudate putamen and the ischemic cortex was counted at 24 hours of reperfusion, in a high-powered field (400X) and expressed as number/mm2. Comparisons among multiple groups were performed using an analysis of variance (Fisher protected least-significant difference test), whereas comparisons between two groups were achieved using the t-test. P < 0.05 was considered statistically significant.
RESULTS
Physiological data and cerebral infarction
Physiological parameters showed no significant differences in mean arterial blood pressure and arterial blood gas analysis between each time point. The preischemic physiological values (mean ± SD) were as follows (n = 4): mean arterial blood pressure, 90 ± 9.1 mm Hg; arterial oxygen tension, 155 ± 25.1 mm Hg; arterial carbon dioxide tension, 32 ± 3.4 mm Hg; pH, 7.3 ± 0.05. There was no deviation from these values during the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hour after reperfusion, and extended to the entire MCA territory at 4 hours by cresyl violet staining (data not shown). The time-dependent increase of infarction in the mouse brain using intraluminal suture blockade was consistent with previous reports that used the same focal-stroke model in mice (Murakami et al., 1998).
Western blot analysis of phospho-Akt and total Akt expression after transient MCA occlusion
As shown in Fig. 1, the bands of phospho-Akt (serine-473) and Akt were observed at 60 kDa in the whole-cell fraction from mice brains. In both the caudate putamen and the MCA territory cortex, phospho-Akt was constitutively expressed in the nonischemic control brain. In the caudate putamen, phospho-Akt was diminished after ischemia and the decrease was significant at 24 hours (Fig. 1B, P < 0.01). In the cortex, phospho-Akt was significantly increased 4 hours after reperfusion (P < 0.05), whereas it was decreased by 24 hours (Fig. 1D). Akt showed no significant increase or decrease after reperfusion in either the caudate putamen (Fig. 1A) or the cortex (Fig. 1C). These data suggest that phosphorylation of Akt is temporarily accelerated in the cortex after transient FCI.
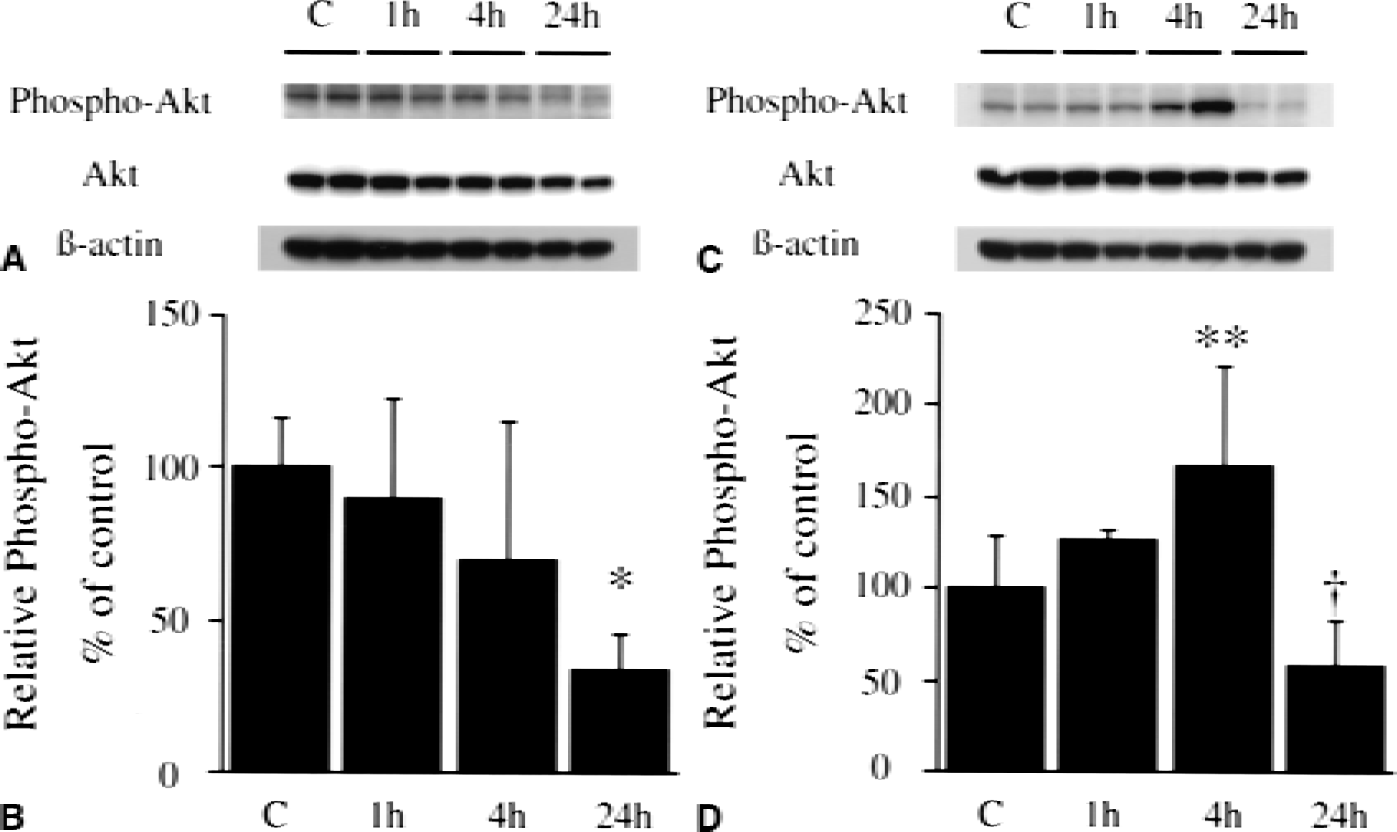
Western blot analysis of phospho-Akt (serine-473) and Akt in the caudate putamen (
Phospho-Akt immunohistochemistry after transient FCI
Phospho-Akt (serine-473) was constitutively expressed in the entire region of the normal mouse brain, including the caudate putamen (Fig. 2A) and the cortex (Fig. 2B). After 1 hour of reperfusion, the expression of phospho-Akt was reduced in the caudate putamen (ischemic core). This reduction was sustained until 24 hours of reperfusion (Fig. 2C, 4 hours; Fig. 2E, 24 hours). However, phospho-Akt expression was increased at 4 hours of reperfusion in the MCA territory cortex (Fig. 2D). After 24 hours of reperfusion, immunoreactivity of phospho-Akt was diminished compared with the control in the cortex (Fig. 2F). Immunostaining of the cortex without a primary antibody (Fig. 2G) or after a preabsorption procedure (Fig. 2H) showed no immunoreactivity, suggesting the specificity of the antibody to the antigen. Phospho-Akt expression in the contralateral hemisphere after reperfusion was not different from a healthy brain (data not shown).
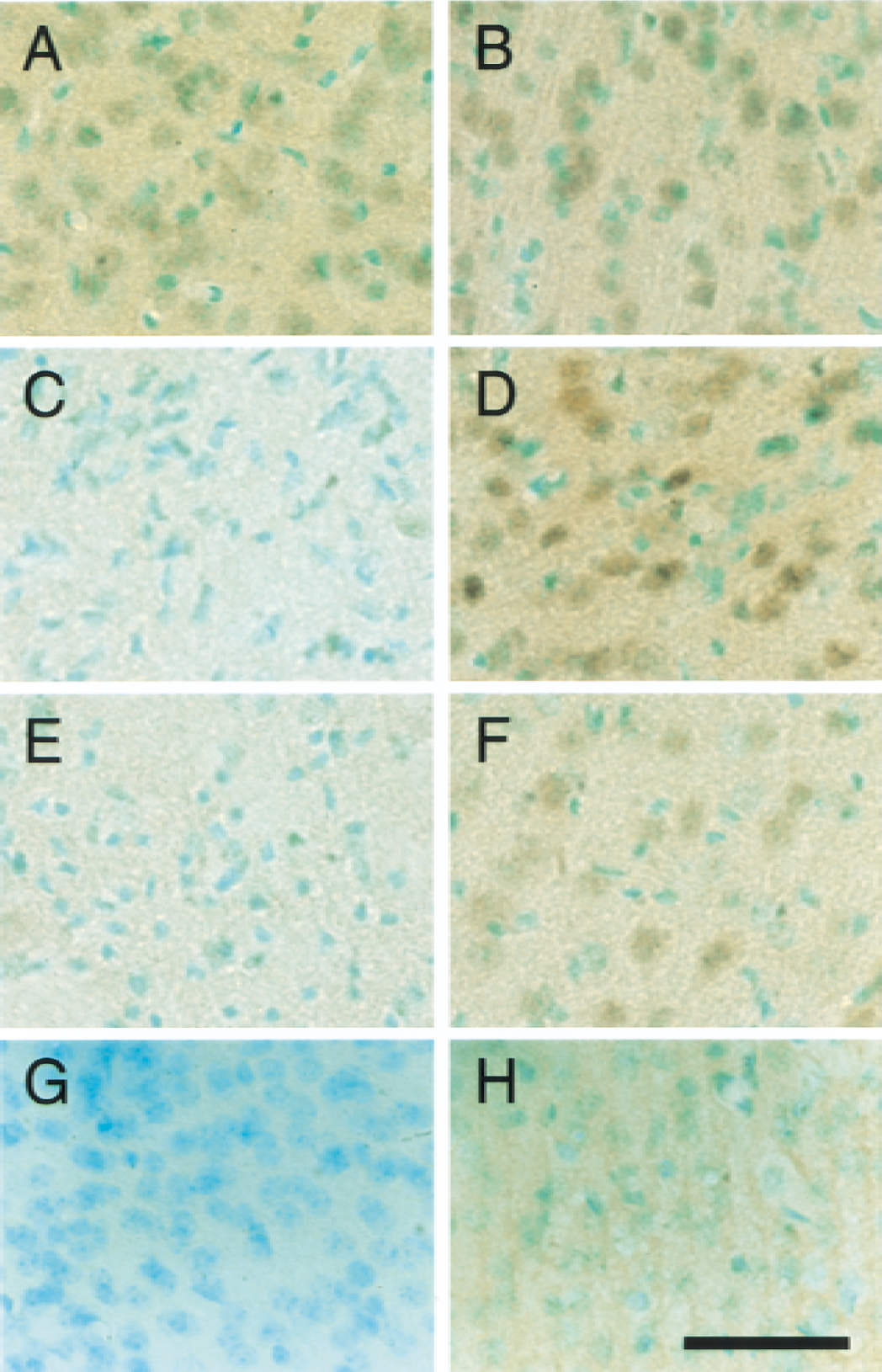
Phospho-Akt (serine-473) immunostaining with methyl-green counterstaining in coronal brain sections from the caudate putamen (
Neuronal expression of phospho-Akt after transient FCI
Double immunofluorescence for phospho-Akt (serine-473) (Fig. 3A) and NeuN (Fig. 3B) demonstrated that phospho-Akt–positive cells in the ischemic cortex colocalized mainly with neurons 4 hours after transient FCI (Fig. 3C, overlapped image). This result suggests neuronal expression of phospho-Akt in the cortex after FCI.
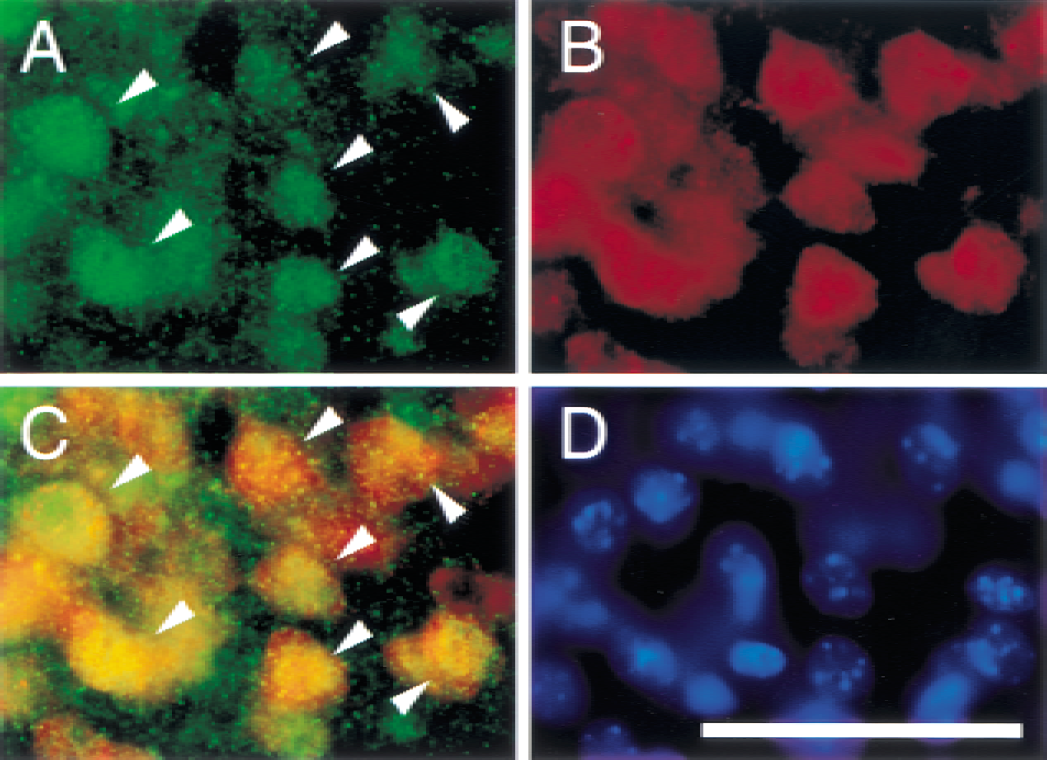
Representative photomicrographs show immunofluorescent staining for phospho-Akt (serine-473) and NeuN after transient FCI. At 4 hours of reperfusion, phospho-Akt-positive cells were observed in the ischemic cortex (
Double labeling with phospho-Akt expression and DNA fragmentation detected by TUNEL staining after transient FCI
After 4 hours of reperfusion, phospho-Akt (serine-473) was strongly detected in the cortex (Fig. 4C), whereas it was hardly seen in the caudate putamen (Fig. 4A). No TUNEL-positive cells were observed in the caudate putamen (Fig. 4B) or in the cortex (Fig. 4D) 4 hours after reperfusion. Twenty-four hours after FCI, a significant number of TUNEL-positive cells were seen in the caudate putamen (Fig. 4F) or in the ischemic cortex (Fig. 4H) with shrunken, condensed nuclei and apoptotic bodies. In the outer area adjacent to the ischemic cortex, phospho-Akt–positive cells were still observed 24 hours after reperfusion (Fig. 4I); however, no colocalization was observed with TUNEL-positive cells (Fig. 4J). These data suggest that the cellular population of phospho-Akt is totally different from that of DNA fragmentation.
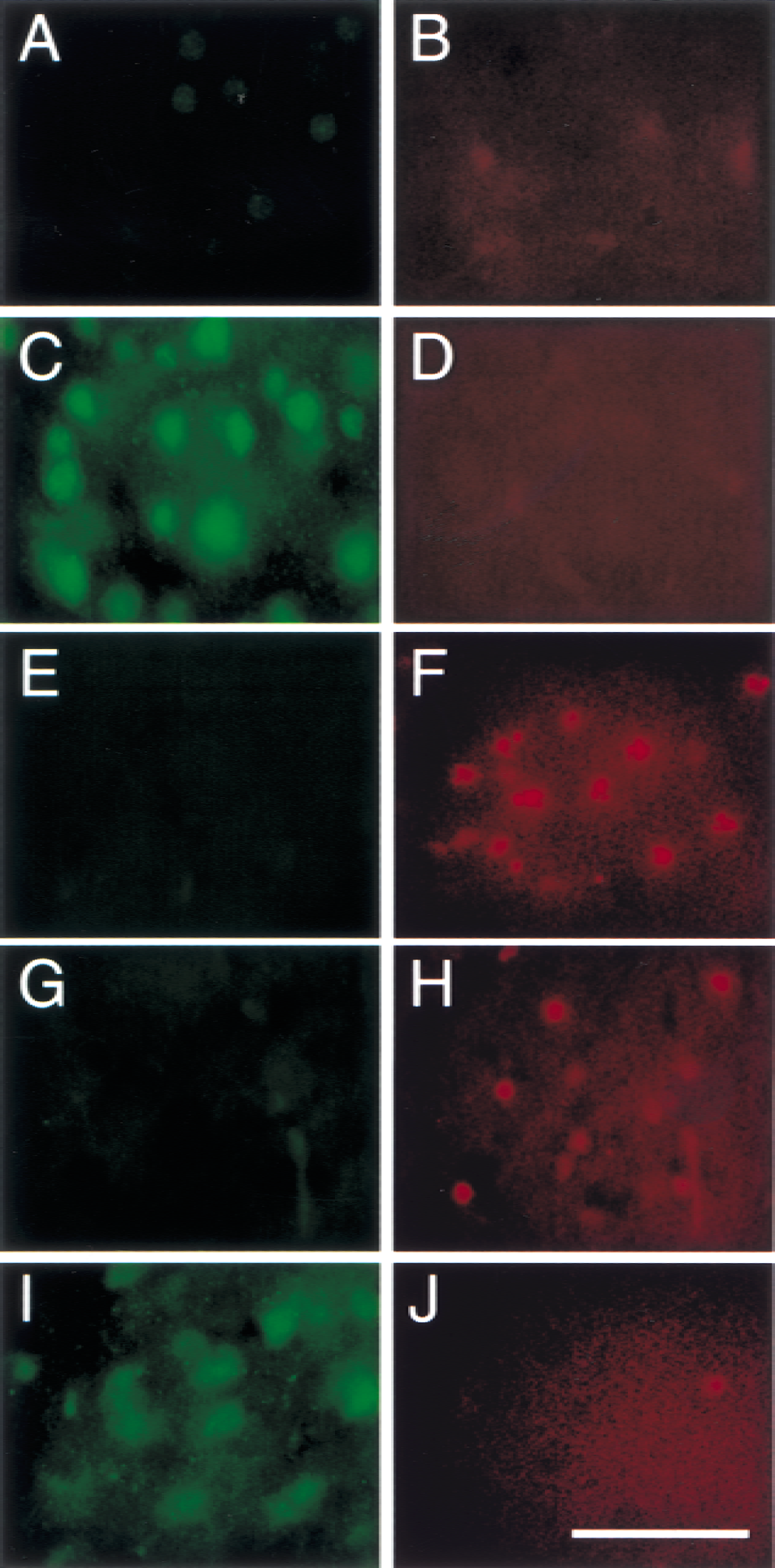
Representative photomicrographs show immunofluorescent staining for phospho-Akt (serine-473) and TUNEL after transient FCI. At 4 hours of reperfusion, phospho-Akt was strongly detected in the cortex (
PI3-kinase inhibitor blocks phosphorylation of Akt and accelerates DNA fragmentation after transient FCI
After 4 hours of reperfusion, phospho-Akt (serine-473) was decreased in the 50 nmol LY294002-injected animals compared with the vehicle-injected animals (Fig. 5A, caudate putamen; Fig. 5B, ischemic cortex). The inhibition was incomplete in the 5 nmol LY294002-injected animals (data not shown). Statistical analysis showed a significant difference between the two groups in the ischemic cortex (Fig. 5D, P < 0.005, n = 4). To evaluate apoptotic cell death, TUNEL staining was performed 24 hours after transient FCI in both the LY294002-injected animals and the vehicle-injected animals (Fig. 6). TUNEL-positive cells were observed in the entire MCA territory in both the vehicle-treated group (Fig. 6A) and the LY294002-treated group (Fig. 6B) with shrunken, darkly stained nuclei and apoptotic bodies (arrows); some nuclei showed fragmentation (arrowheads). Quantitative analyses showed a significant increase of TUNEL-positive cells in the LY294002-treated group compared with the vehicle-treated group in the ischemic cortex (Fig. 6D, P < 0.05, n = 4; vehicle, 0.860 ± 0.478 × 103cells/mm2/ LY294002, 1.548 ± 0.106 × 103cells/mm2), but not in the caudate putamen (Fig. 5C, n = 4; vehicle, 1.104 ± 0.068 × 103cells/mm2/ LY294002, 1.072 ± 0.091 × 103cells/mm2).
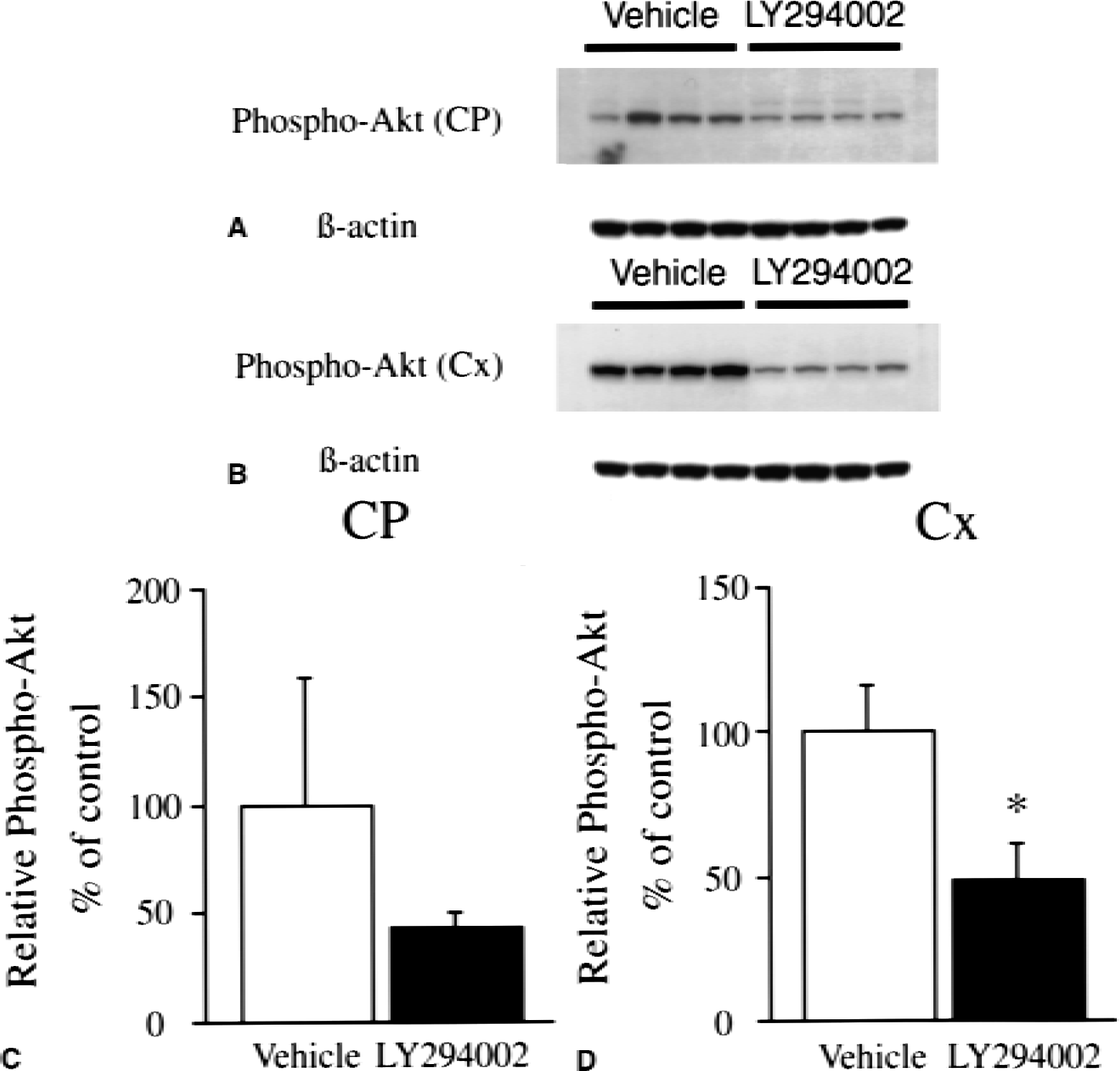
Western blot analysis of phospho-Akt (serine-473) in the caudate putamen (
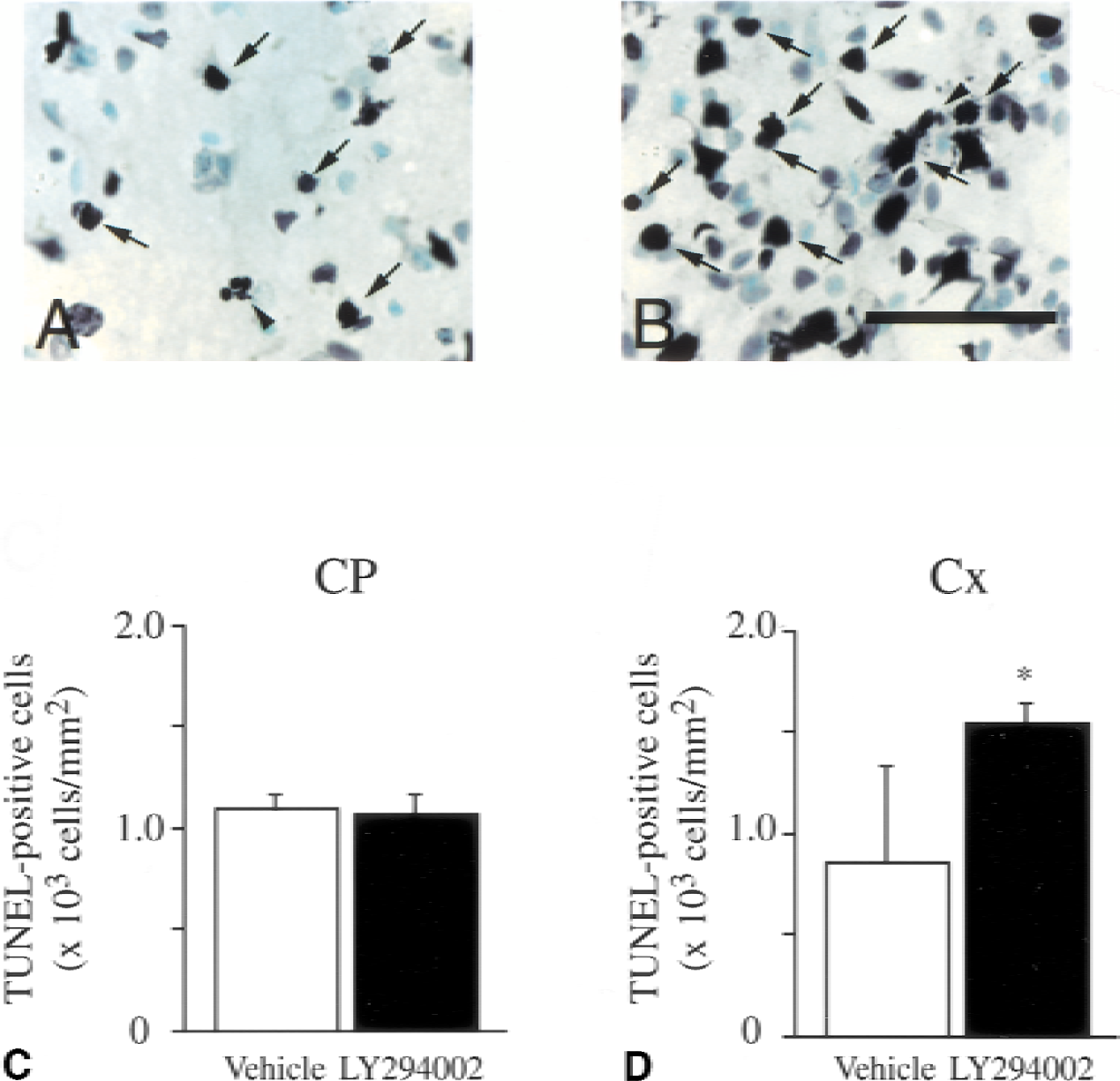
Representative photomicrographs show TUNEL-positive cells in the ischemic cortex (Cx) of the vehicle-injected (
DISCUSSION
Phosphorylation of Akt after ischemia
Akt activation is known as one of the principal factors to prevent apoptosis. Several studies have revealed phosphorylation of Akt against various stimuli both in vitro (Chalecka-Franaszek and Chuang, 1999; Namikawa et al., 2000; Zhou et al., 2000) and in vivo (Ouyang et al., 1999; Namura et al., 2000; Janelidze et al., 2001; Yano et al., 2001). Although Akt is phosphorylated at serine-473 and at threonine-308, the exact role of phosphorylation at each site is still unclear.
In the present study, we have revealed several points. First, phosphorylation of Akt was temporally accelerated at serine-473 in the ischemic cortex after transient FCI, but not in the ischemic core (Figs. 1 and 2). A study has shown that phosphorylation of Akt at serine-473 was enhanced after FCI (Janelidze et al., 2001); however, we are the first to mention regional distribution of phospho-Akt after FCI. In the ischemic core, phospho-Akt was simply decreased after ischemia. It is conceivable that the cellular damage in the ischemic core was too lethal to activate the Akt cell-survival pathway. However, the cortical lesion might have activated Akt because the regional damage was relatively moderate. The regional difference in Akt activation has been seen in the global ischemia model (Yano et al., 2001), and the difference might reflect the degree of cellular damage. The authors demonstrated that in the CA1 region of the hippocampus, phospho-Akt was dephosphorylated right after ischemia and rephosphorylated above the control level for 6 hours after 5 minutes of global ischemia, whereas no significant change was observed in the CA3 region and the dentate gyrus (Yano et al., 2001). They also demonstrated that the difference in ischemic severity could make a difference in the phospho-Akt level in the CA1 region —lethal damage reduced phospho-Akt in the CA1 region whereas sublethal damage increased phospho-Akt 7 days after global ischemia. These results are supported by another report showing that in the lethal stress of 15 minutes of global ischemia, alterations in phospho-Akt levels were also observed in the dentate gyrus (Ouyang et al., 1999), suggesting that a severe insult could phosphorylate Akt in the dentate gyrus after global ischemia. Thus, phosphorylation of Akt might largely depend on the severity of the stress, which might cause regional differences in phospho-Akt after FCI.
Second, phospho-Akt–positive cells colocalized with NeuN-positive cells in the cortex after FCI (Fig. 3). This result suggests that phospho-Akt might be associated with neuronal cell survival after FCI. Moreover, phospho-Akt–positive cells did not colocalize with DNA damage 24 hours after FCI (Fig. 4). Immunofluorescent staining showed that phospho-Akt–positive cells were observed in the outer area adjacent to the ischemic cortex at 24 hours of reperfusion. However, TUNEL-positive cells were observed in the ischemic core and the ischemic cortex. Because phospho-Akt is thought to be involved in cell survival, it is supposed that only moderate damage could encourage phosphorylation of Akt, which contributed to neuronal cell survival and less subsequent DNA damage.
Third, the PI3-kinase inhibitor prevented phosphorylation of Akt after FCI (Fig. 5), followed by increased DNA damage (Fig. 6). These results show that the PI3-kinase pathway had a role in reducing apoptosis after transient FCI, at least in part. Regarding infarct volume, cresyl violet staining suggested a possibility that LY294002 injection might enlarge the infarct size, though we did not assess this finding statistically (data not shown). Previous studies revealed that the PI3-kinase inhibitor could reduce Akt activity in vitro (Crowder and Freeman, 1998; Chalecka-Franaszek and Chuang, 1999). In the global ischemia model, histologic damage was assessed by propidium-iodide staining after administration of wortmannin (Yano et al., 2001). In the current study, we demonstrated that LY294002 injection increased DNA damage in the ischemic cortex but not in the ischemic core, suggesting that Akt-mediated cell survival was critical in the moderately damaged lesion.
Downstream of Akt
Akt is activated after phosphorylation, thereby preventing apoptosis phosphorylating of the downstream factors. Bad is one of the downstream factors of the Akt pathway, and a proapoptotic member of the Bcl-2 family that can displace Bax from binding to Bcl-2 and Bcl-xL, resulting in cell death (Yang et al., 1995; Zha et al., 1996). Survival factors such as IL-3 can inhibit the apoptotic activity of Bad by activating intracellular signaling pathways that result in the phosphorylation of Bad at serine-112 and serine-136 (Zha et al., 1996). Phosphorylation at these sites results in the binding of Bad to 14-3-3 proteins and the inhibition of Bad binding to Bcl-xL and Bcl-2 (Zha et al., 1996). Akt has been shown to promote cell survival by its ability to phosphorylate Bad at serine-136 (Datta et al., 1997; del Peso et al., 1997). Although it has not been shown how Bad works after FCI, a recent study showed that Bax, which could be displaced by Bad and which induces apoptosis, translocated from the cytosol to mitochondria after transient FCI (Cao et al., 2001). Bax translocation can trigger cytochrome c release from mitochondria, and there is evidence that cytochrome c is released from mitochondria to the cytosol after FCI (Fujimura et al., 1998). As a downstream event of cytochrome c release, caspase-9 (Noshita et al., 2001) and caspase-3 (Namura et al., 1998) were activated after FCI, followed by DNA fragmentation. Because Akt inhibits activation of caspase-9 and caspase-3 after cytochrome c release in vitro (Zhou et al., 2000), it is possible that Akt activation after FCI might be crucial for these downstream elements in apoptosis.
Besides activation after cytochrome c release, caspase-9 is directly inactivated by phospho-Akt. A report showed that phospho-Akt inactivated caspase-9 by phosphorylating at serine-196 (Cardone et al., 1998), suggesting that Akt contributed to cell survival by inhibiting caspase-9. There are some other downstream elements of the Akt cell survival, and investigating of these elements would help clarify the role of Akt after FCI.
The MEK/ERK pathway plays a critical role in the regulation of cell growth and differentiation (Crews et al., 1992; Alessi et al., 1994; Rosen et al., 1994) and has a relation to apoptosis. Evidence shows that intracerebroventricular injection of the MEK1 inhibitor could reduce infarct size after FCI (Alessandrini et al., 1999), suggesting that the activation of this pathway is crucial for cerebral infarction. A recent study showed that phospho-Akt also appears to negatively regulate the MEK/ERK pathway, inactivating Raf at serine-259 (Rommel et al., 1999; Zimmermann and Moelling, 1999). In that study, inhibition of Akt seemed to prevent phosphorylation of Raf at serine-259, which resulted in acceleration of Raf and ERK activation (Zimmermann and Moelling, 1999). However, another study showed that intracerebroventricular injection of wortmannin, a PI3-kinase inhibitor, did not prevent phosphorylation of ERK caused by injection of brain-derived neurotrophic factor (Han and Holtzman, 2000), suggesting that Akt inhibition might not be effective in the ERK pathway. The investigation of how Akt interacts with the ERK pathway could help to clarify the mechanism of apoptosis after FCI.
CONCLUSION
Our results have shown that phosphorylation of Akt was temporally accelerated at serine-473 in the ischemic cortex after transient FCI, and that phospho-Akt–positive cells did not colocalize with DNA damage. After inhibiting the PI3-kinase pathway, subsequent DNA damage was increased. These findings suggest that the PI3-kinase pathway might mediate cell survival after transient FCI.
Footnotes
Acknowledgments:
The authors thank C. Christensen for editorial assistance, and L. Reola, B. Calagui, and G. Omar for technical assistance.