
Abstract
Apoptosis plays an important role in delayed neuronal cell death after cerebral ischemia. Activation of Akt/protein kinase B has been recently reported to prevent apoptosis in several cell types. In this article the authors examine whether induction of ischemic tolerance resulting from a sublethal ischemic insult requires Akt activation. Sublethal ischemia gradually and persistently stimulated phosphorylation of Akt-Ser-473 in the hippocampal CA1 region after reperfusion. After lethal ischemia, phosphorylation of Akt-Ser-473 showed no obvious decrease in preconditioned gerbils but a marked decrease in nonconditioned gerbils. Changes in Akt-Ser-473 phosphorylation were correlated with changes in Akt activities, as measured by an in vitro kinase assay. Intracerebral ventricular infusion of wortmannin before preconditioning blocked both the increase in Akt-Ser-473 phosphorylation in a dose-dependent manner and the neuroprotective action of preconditioning. These results suggest that Akt activation is induced by a sublethal ischemic insult in gerbil hippocampus and contributes to neuroprotective ischemic tolerance in CA1 pyramidal neurons.
A sublethal transient global ischemia, also known as preconditioning ischemia, exhibits neuroprotective action against delayed neuronal cell death in the hippocampal CA1 region after a severe ischemic insult in gerbil (Kirino et al., 1991; Kitagawa et al., 1990), rat (Liu et al., 1992), and human (Weih et al., 1999). This phenomenon, known as ischemic tolerance, permits examination of the mechanisms of cell death and neuroprotection after brain ischemia. In neurons, sublethal intracellular Ca2+ elevation through Ca2+ channels (Nakata et al., 1992), α-amino–3-hydroxy–5-methyl-isoxazole–4-propionic acid (AMPA) receptors (Yamaguchi et al., 1999), and/or N-methyl-d-aspartate (NMDA) receptors (Kato et al., 1992; Shamloo and Wieloch, 1999) is involved in induction of ischemic tolerance. In addition, expression of brain-derived neurotrophic factor (Kawahara et al., 1997), heat shock proteins (Aoki et al., 1993), and several transcription factors (Yoneda et al., 1998) is enhanced in CA1 neurons after preconditioning ischemia. Recently, several lines of evidence have been proposed to suggest that apoptosis underlies the molecular mechanism of delayed neuronal cell death after brain ischemia (Choi, 1996). Histologic and biochemical evidence of apoptosis is seen in dying cells after ischemia (MacManus et al., 1993; Nitatori et al., 1995). Several apoptosis-regulatory genes, such as Bax or Bcl-xL, are also induced in vulnerable hippocampal CA1 neurons after brain ischemia (Antonawich et al., 1998; MacManus and Linnik, 1997). In addition, caspaselike enzyme activities are elevated in hippocampal CA1 neurons after reperfusion (Namura et al., 1998), and infusion of caspase-3 inhibitor into the cerebral ventricles rescues cells from delayed neuronal cell death (Chen et al., 1998).
Akt, also known as protein kinase B, is a protein kinase involved in survival signals as a downstream kinase of phosphoinositide 3-kinase (PI3-K) in growth factor-mediated signaling cascades. Phosphorylation of residues Thr-308 and Ser-473 is required for Akt activity (Burgering and Coffer, 1995; Franke et al., 1995; Kohn et al., 1995). Active Akt phosphorylates Bad (Datta et al., 1997; del Peso et al., 1997), caspase-9 (Cardone et al., 1998), and forkhead-related transcription factors (Brunet et al., 1999) and thereby induces antiapoptotic effects. Akt is also known to activate IκB kinase in fibroblasts treated with platelet-derived growth factor (Romashkova and Makarov, 1999). Recently, Ouyang et al. (1999) showed transient activation of Akt phosphorylation in CA1 neurons after global ischemia in rat and suggested that Akt activation delays cell death in these neurons after ischemia. In a previous study, we showed that Akt is also activated by Ca2+ signaling through NMDA receptors (Yano et al., 1998). Specifically, Akt was phosphorylated and activated by Ca2+/calmodulin-dependent protein kinase kinase through NMDA receptor stimulation in a neuroblastoma cell line, NG108–15 cells, which resulted in antiapoptotic effect (Yano et al., 1998). NMDA receptor-dependent stimulation of Akt is also evident in cultured cerebellar granule neurons (Zhang et al., 1998). Given the importance of Ca2+ signaling in ischemic tolerance, we hypothesized that Akt activation plays a fundamental role in ischemic tolerance and thereby protects neurons against apoptosis in global cerebral ischemia.
Here we address whether Akt is activated in sublethal ischemic conditions that produce ischemic tolerance. We also show that inhibition of Akt activity in the hippocampus during preconditioning prevents the neuroprotective action of preconditioning in global cerebral ischemia in the gerbil.
MATERIALS AND METHODS
Ischemia model
All animal experiments were approved by the Committee of Animal Experiments at Kumamoto University School of Medicine. Male Mongolian gerbils (Seac Yoshitomi Ltd., Fukuoka, Japan) weighing 60 to 70 g received transient forebrain ischemia according to procedures reported previously (Hasegawa et al., 2000). Mongolian gerbils do not have a complete circle of Willis, and therefore clamping both carotids produces complete forebrain ischemia. Gerbils were anesthetized with a mixture of 2.5% halothane and nitrous oxide/oxygen (7:3). The bilateral common carotid arteries were surgically exposed and dissected free of surrounding tissues and then occluded quickly with aneurysm clips for a fixed time (2 or 5 minutes). The clips were then removed to restore the blood flow. Occlusion and reperfusion of carotid arteries were verified by direct visual inspection under an operating microscope. The rectal temperature, monitored with a digital thermometer inserted 5 cm into the anus, was maintained at 37†C to 38†C throughout the procedure by placing gerbils under a heating lamp. After they awoke from anesthesia, the gerbils were returned to their cages in a room maintained at a constant temperature of 22†C ± 2†C and allowed to take food and water before the experiments were undertaken.
For experiments of ischemic tolerance, the gerbils were first subjected to 2 minutes of ischemia, then 5 minutes of ischemia 3 days later. They were subsequently decapitated at indicated times after the second ischemia. Control animals were sham-operated and underwent the same experimental procedures, except for occlusion of arteries. Their brains were removed at time points equivalent to ischemic animals.
Western blotting
Animals were killed by decapitation immediately after occlusion or at the indicated times after reperfusion. Brains were removed and rinsed once with cold phosphate-buffered saline. Dissection was done with a scalpel on a chilled plate in phosphate-buffered saline. The hippocampi were removed, cross-sectioned into about six pieces under a binocular microscope, and further cut into subfields CA1, CA3, and the dentate gyrus. Each tissue sample was stored separately at −80†C before use. Frozen tissue was homogenized with a hand homogenizer in 150 μL homogenization buffer consisting of 50 mmol/L N-[2-Hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES) (pH 7.5), 2 mmol/L Ethylenediamine tetraacetic acid (EDTA), 2 mmol/L Ethylene Glycol-bis(β-aminoethyl ether)N,N,N′,N′-tetraacetic acid (EGTA), 0.5% Nonidet P-40, 1 mmol/L sodium orthovanadate, 10 mmol/L sodium pyrophosphate, 50 mmol/L sodium fluoride, 2 mmol/L benzamide, 10 μg/mL leupeptin, 10 μg/mL pepstatin A, 10 μg/mL aprotinin, 5 mmol/L dithiothreitol, and 0.5 mmol/L phenylmethylsulfonyl fluoride, and incubated on ice for 10 minutes, followed by centrifugation at 15,000 × g for 15 minutes. After determination of the protein content of each supernatant fraction with the Bradford solution, 20 μg total protein of each sample was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 10% acrylamide gels. After electrophoresis, proteins were transferred to nitrocellulose membranes. Blotting membranes were incubated with 5% nonfat milk in TBST (10 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 0.05% Tween 20) and probed with a 1:200 dilution of Akt phospho-serine 473 antibody (rabbit antiserum; New England Biolabs, Beverly, MA, U.S.A.) or a 1:1,000 dilution of conventional Akt antibody (sheep antiserum; Upstate Biotech, Lake Placid, NY, U.S.A.) in 5% nonfat milk in TBST at 4†C overnight. Immunoblots were then processed with horseradish-peroxidase–conjugated antirabbit immunoglobulin G (IgG) or antisheep IgG using a chemiluminescence Western blotting detection system kit (Amersham, Arlington Heights, IL, U.S.A.). The blots were exposed to Hyperfilm (Amersham) at room temperature. The images were scanned and analyzed semiquantitatively using the NIH image program.
Akt kinase activity assay
Akt activity was measured with an Akt assay kit (New England Biolabs) according to the manufacturer's protocol with minor modifications. Dissected hippocampi were homogenized using a hand homogenizer in lysis buffer consisting of 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 mg/mL leupeptin, and 1 mmol/L p-amidinophenyl methanesulfonyl fluoride hydrochloride. Extracts containing equivalent amounts of protein were incubated with a slurry of immobilized Akt antibody for 2 hours at 4†C. Immunoprecipitants were washed twice with lysis buffer and twice with kinase buffer containing 25 mmol/L Tris-HCl (pH 7.5), 5 mmol/L β-glycerol phosphate, 2 mmol/L dithiothreitol (DTT), 0.1 mmol/L Na3VO4, and 10 mmol/L MgCl2, and then subjected to an in vitro kinase assay in kinase buffer with 400 μmol/L adenosine triphosphate (ATP) and 1 μg glycogen synthase kinase–3α (GSK–3α) peptide for 30 minutes at 30†C. The reaction was stopped by adding SDS–sample buffer. Phosphorylation at Ser-21 of GSK–3α peptide was analyzed by immunoblotting with anti-phospho-GSK–3α antibody (New England Biolabs) according to the manufacturer's protocol.
Immunohistochemistry
The gerbils were perfused under pentobarbital anesthesia with ice-cold 100 mL phosphate-buffered saline containing 30 mmol/L sodium pyrophosphate, 50 mmol/L NaF, and 50 nmol/L calyculin A, followed by 100 mL 4% paraformaldehyde in 0.1 mol/L phosphate buffer containing 30 mmol/L sodium pyrophosphate, 50 mmol/L NaF, and 50 nmol/L calyculin A. The brains were removed and preserved in the fixation solution overnight. Coronal brain sections (30 μm) were prepared by Vibratome sectioning (Technical Products International, MO, U.S.A.) and washed twice in phosphate-buffered saline for 5 minutes at room temperature, followed by phosphate-buffered saline with 0.5% Triton X-100 for 30 minutes. Nonspecific binding sites were blocked in 3% bovine serum albumin in phosphate-buffered saline (blocking solution) for 1 hour. Brain sections were incubated overnight at 4†C with primary antibodies for both Akt (mouse IgG, Transduction Lab, Lexington, KY, U.S.A.) and phospho-Ser-473 (rabbit IgG, Upstate Biotechnology) in blocking solution, diluted 1:500 and 1:50, respectively, and then washed three times in phosphate-buffered saline for 5 minutes each at room temperature. The sections were labeled for 2 hours at room temperature with fluorescein-labeled antimouse secondary antibodies (1:200) for the anti-Akt antibody (Vector, CA, U.S.A.) and with Texas Red-labeled antirabbit secondaries (Vector) (1:200) for anti-phospho-Ser-473 antibody. For staining of nuclei, sections were incubated with 1 μg/mL propidium iodide for 15 minutes in the dark. After several washes, the sections were mounted on glass slides with coverslips and analyzed with a confocal laser microscope (Fluoview; Olympus, Tokyo, Japan).
Intracerebroventricular administration of wortmannin
Under anesthesia with a mixture of 2.5% halothane and nitrous oxide/oxygen (7:3), gerbils were set on a stereotactic operation frame. A scalp skin incision was made and one burr hole was opened on the right parietal skull at 2 mm lateral and 3 mm posterior to the bregma. A Hamilton syringe was inserted into the brain at a depth of 2.5 mm from the cortex. The point where the needle should be inserted was decided by a preliminary injection of hematoxylin that spread in the ventricles. Intraventricular injection was also confirmed by the observation of cerebrospinal fluid reflux through the burr hole after each injection and of the injection scar on the slices after fixation. Two microliters wortmannin at the indicated concentration or vehicle was continuously infused for 10 minutes. The needle was kept in place for 10 more minutes to allow the drug to diffuse into the ventricle. The preconditioning of 2 minutes of ischemia was given 30 minutes later. Wortmannin was dissolved to 20 μmol/L in phosphate-buffered saline containing 2% dimethylsulfoxide.
Statistical evaluation
Values were expressed as means ± SD. Statistical analysis was performed using a one-way analysis of variance and post hoc Fisher's protected least significant difference test. A level of P < 0.05 was considered statistically significant.
RESULTS
Changes in phosphorylation levels of Akt-Ser-473 after lethal ischemia
We first examined changes in Akt activity after a lethal or sublethal ischemic insult. To evaluate Akt activation, we measured phosphorylation of Akt at Ser-473 with anti-phospho-Ser-473 antibody, which is known to be required for Akt activation. Gerbils were subjected to 5 minutes of global ischemia without or with preconditioning of 2 minutes of ischemia. Seven days after reperfusion, gerbils without preconditioning showed severe neuronal losses in the CA1 region, with a cell survival of 14.9% ± 5.4% (n = 8), whereas the preconditioned gerbils showed neuronal survival of 92.4% ± 5.1% (n = 8). Sham-operated gerbils showed no significant cell death in any area. Under the ischemic conditions with or without preconditioning, immunoblotting analysis was carried out at several time points after reperfusion. Sham-operated hippocampi showed no significant changes in phosphorylation Ser-473 compared with naïve hippocampi (no surgery) in any area (data not shown). Without preconditioning, Ser-473 phosphorylation in the CA1 region was markedly decreased to 17.3% ± 4.6% (P < 0.05, n = 4) of that of sham-operated animals at the end of 5 minutes of ischemia, followed by a transient increase to 196.1% ± 35.7% (P < 0.05, n = 4) at 12 hours after reperfusion. It returned to baseline within 24 hours after ischemia (Fig. 1). No significant change was observed in either CA3 or dentate gyrus (data not shown). In contrast, preconditioning eliminated the dramatic changes in Akt phosphorylation observed after 5 minutes of ischemia, and Akt phosphorylation levels did not significantly change for at least 7 days after reperfusion. Akt protein levels did not change in the CA1 regions of either preconditioned or nonconditioned gerbils until at least 7 days after ischemia.
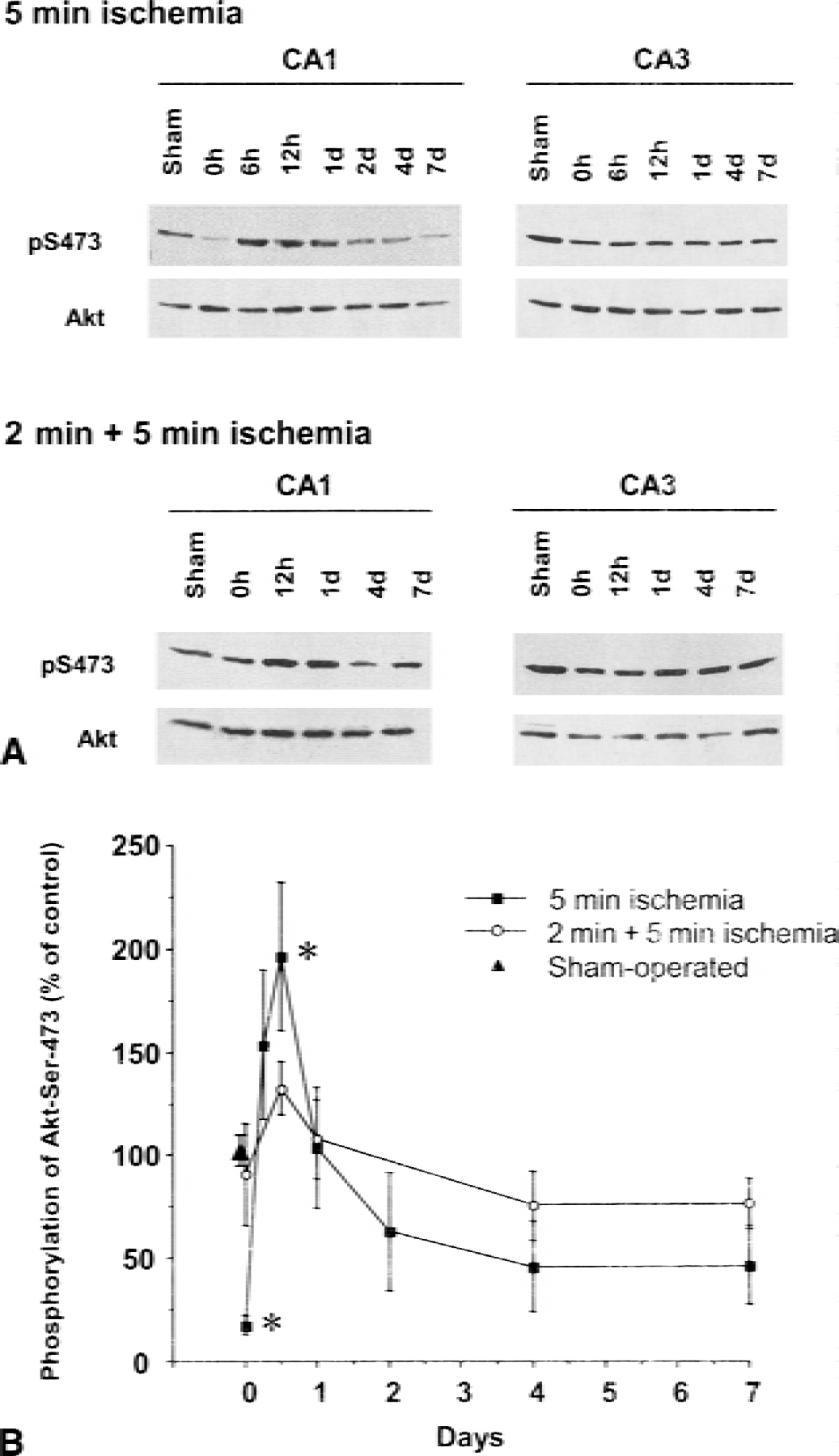
Western blotting analysis of phosphorylation of Ser-473 of Akt after lethal ischemia in the hippocampal CA1 subfields. Extracts were obtained from hippocampal CA1 and CA3 regions at the indicated time points from a sham-operated group, a 5-minute ischemic group, or a group receiving 2 minutes of ischemia followed by 5 minutes of ischemia. (
We next examined levels of Ser-473 phosphorylation after 2 minutes of ischemia. Immunoblotting analysis with anti-phospho-Ser-473 antibody showed gradual increases in Ser-473 phosphorylation after 2 minutes of ischemia (Fig. 2). The level of Ser-473 phosphorylation reached 201.4% ± 9.4% (P < 0.05, n = 4) of sham control on 3 days after reperfusion, when animals were tested by the second ischemia, and the elevated phosphorylation continued at least 7 days. Akt expression levels, however, did not change after 2 minutes of ischemia. In contrast, no significant changes in phosphorylation of Ser-473 were observed in the hippocampal CA3 regions and dentate gyrus after 2 minutes of ischemia.
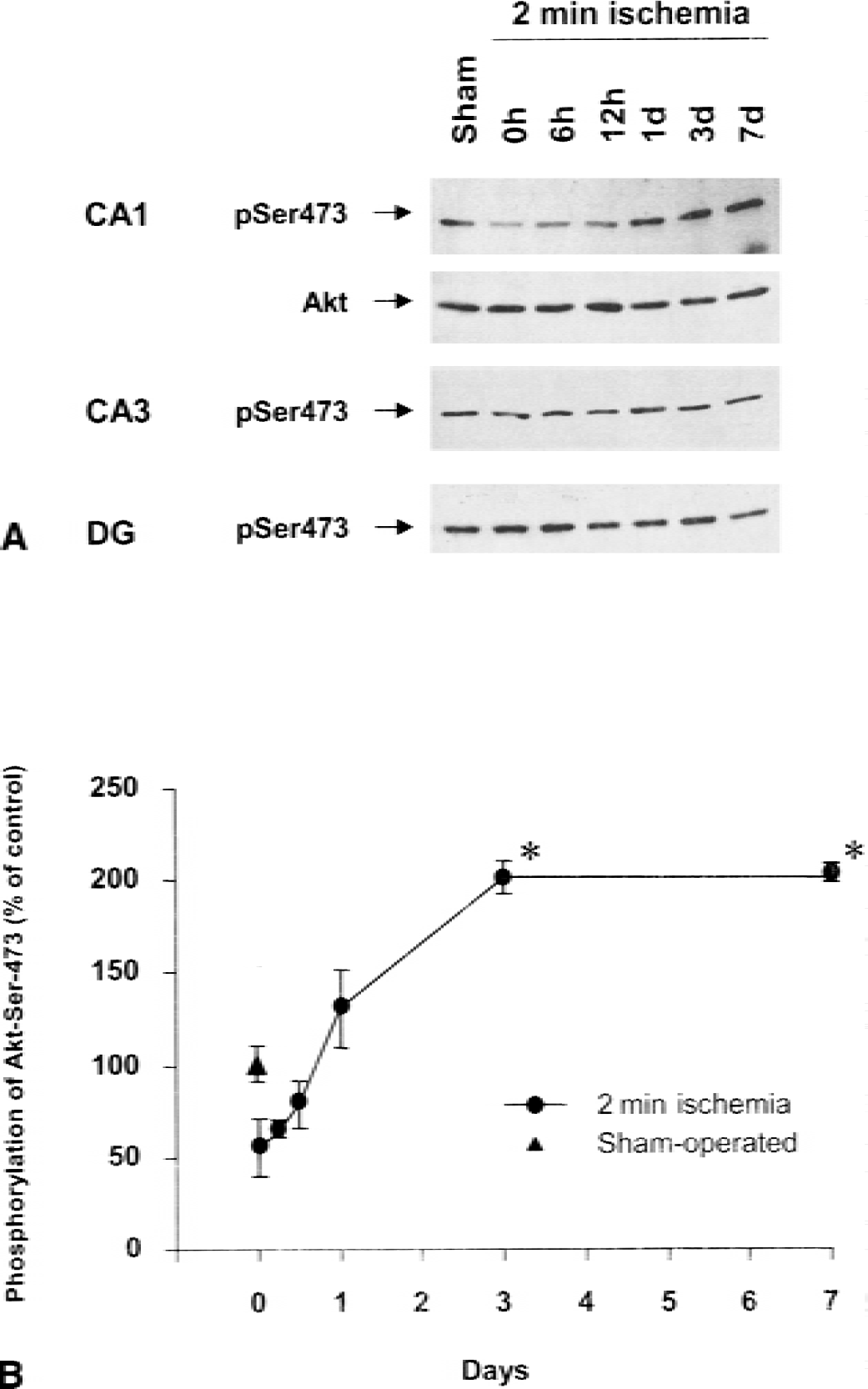
Western blotting analysis of phospho-Ser-473 in hippocampal CA1 regions after preconditioning. Extracts obtained from hippocampal CA1, CA3, or dentate gyrus after sham operation or at the indicated time points after 2 minutes of ischemia were subjected to Western blotting analysis with either anti-Akt antibody or anti-phospho-Ser-473 antibody. (
Changes in Akt activity after ischemia
Because Akt activation requires phosphorylation of residues Thr-308 and Ser-473, potential changes in Akt activity after ischemia were analyzed by a protein kinase assay. After immunoprecipitation of Akt from cell extracts with an anti-Akt antibody, immunocomplexes were subjected to an in vitro kinase assay using GST-GSK3α peptide as substrate. As shown in Fig. 3, phosphorylation of GSK3α peptide was detected after SDS-PAGE by immunoblotting with an anti-phospho-GSK3 antibody. Without anti-Akt antibody, no significant phosphorylation of phospho-GSK3α peptide was observed (data not shown). When phosphorylation of GSK3α peptide was evaluated by densitometry, Akt activity after 5 minutes of ischemia decreased to 42.5% ± 15.0% (P < 0.05, n = 4) compared with that of sham-operated gerbils. In contrast, 2 minutes of ischemia followed by 3 days of reperfusion caused significant elevation to 227.3% ± 17.5% (P < 0.05, n = 4) of that seen in sham-operated animals. In addition, preconditioning of 2 minutes of ischemia blocked the decreases in Akt activity observed after 5 minutes of ischemia; in fact, Akt activity showed a slight but nonsignificant increase after preconditioning. Such changes in Akt activity were similar to the changes in Ser-473 phosphorylation observed in the 5-minute ischemia group, the 2-minute ischemia group, and the 2+5-minute ischemia group, as shown in Figs. 1 and 2.
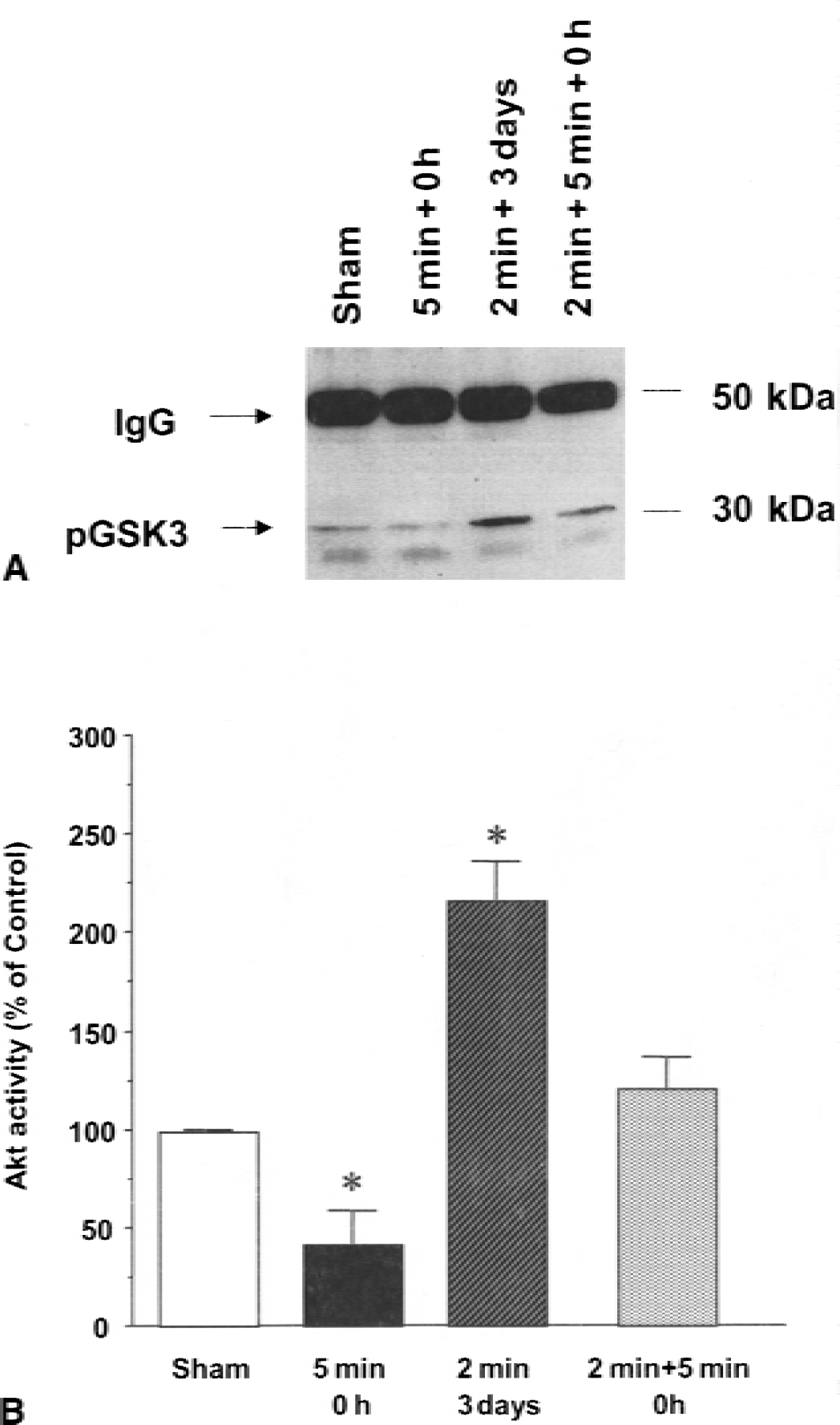
Measurement of Akt activity using GSK3α peptide as substrate in nonconditioned and preconditioned gerbils. Extracts were obtained from hippocampal CA1 regions after sham operation, immediately after 5 minutes of ischemia, 3 days after 2 minutes of ischemia, and immediately after 5 minutes of ischemia with 2 minutes of preconditioning. Immunoprecipitated Akt was subjected to an in vitro kinase assay using GSK3α peptide as substrate. Phosphorylation of GSK peptide was detected by Western blotting analysis using anti-phospho-GSK3α antibody. (
Immunocytochemical localization of Akt and phospho-Akt in CA1 area
Because there are numerous cell types in the hippocampal CA1 region, we investigated the localization of Akt and its activated form by immunocytochemistry. The hippocampal sections from sham-operated or preconditioned animals were stained with antibodies against Akt and phospho-Ser-473 using double-labelled immunofluorescence. The immunofluorescence in control staining without the primary antibodies was negligible (Fig. 4). In sham-operated gerbils, immunoreactivity against Akt was observed in the nucleus, cytoplasm, and apical dendrites of CA1 pyramidal neurons. Intense phospho-Ser-473 immunoreactivity was observed in a dotted pattern in the cytoplasm and weakly in nuclei. When gerbils were subjected to 2 minutes of ischemia (preconditioning) and hippocampal sections were prepared 3 days after reperfusion, phospho-Ser-473 immunoreactivity was markedly increased in nuclei without an increase in Akt immunoreactivity. The dotted staining in the cytoplasm with the phospho-Ser-473 antibody was also observed in the slices from the preconditioned animals. The increased immunoreactivity of phospho-Ser-473 in nuclei was consistent with the results of previous reports. For example, activated Akt was reported to detach from the plasma membrane and translocate to the nucleus in the cultured cells (Andjelkovic et al., 1997; Meir et al., 1997). These results indicate that Akt is predominantly expressed in pyramidal neurons where it is activated, particularly in nuclei, after preconditioning in global cerebral ischemia.
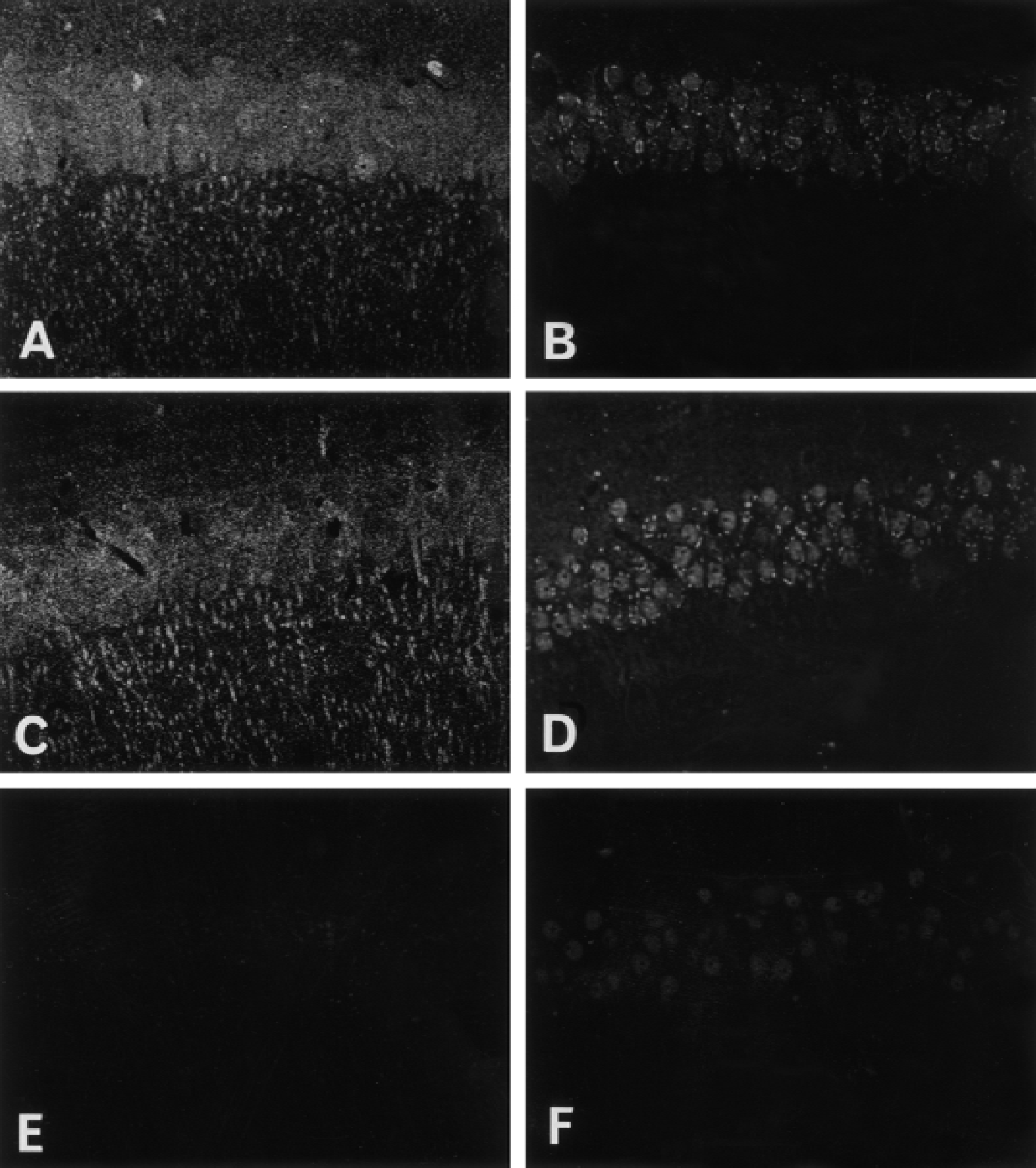
Immunohistochemical analysis of Akt and phospho-Ser-473 expression. Gerbils subjected to sham operation or 2 minutes of ischemia and 3 days after reperfusion were perfusion-fixed with 4% paraformaldehyde containing phosphatase inhibitors. Thirty-micrometer coronal slices were double immunostained with anti-Akt antibody with an FITC-conjugated secondary antibody (
Effects of wortmannin on Akt activity and ischemic tolerance
To investigate the role of Akt activation in induction of ischemic tolerance, we blocked activation of Akt in the hippocampus during the course of tolerance. Gerbils received an infusion into the right cerebral ventricles of wortmannin, a potent inhibitor of PI3-K, 30 minutes before the 2-minute ischemia, and its inhibitory effect was evaluated immediately afterward. Two microliters of either 10 μmol/L or 100 μmol/L wortmannin in phosphate-buffered saline was infused for 10 minutes into the right cerebral ventricle as described above. All animals that received no infusion, vehicle (phosphate-buffered saline) only, or wortmannin survived the experiments. At a high dose (100 μmol/L), wortmannin caused no significant changes in body temperature and did not promote behavioral abnormalities in the animals. Treatment with wortmannin at both concentrations in sham-operated animals produced no neurotoxic effects when cell death was evaluated at 7 days after infusion (data not shown). When hippocampal CA1 regions were analyzed on day 3 after the 2-minute ischemia, increased phosphorylation of Ser-473 was blocked by wortmannin treatment in a dose-dependent manner without changes in Akt protein levels (Fig. 5). Infusion of vehicle did not affect the increase in phosphorylation induced by the 2 minutes of ischemia.
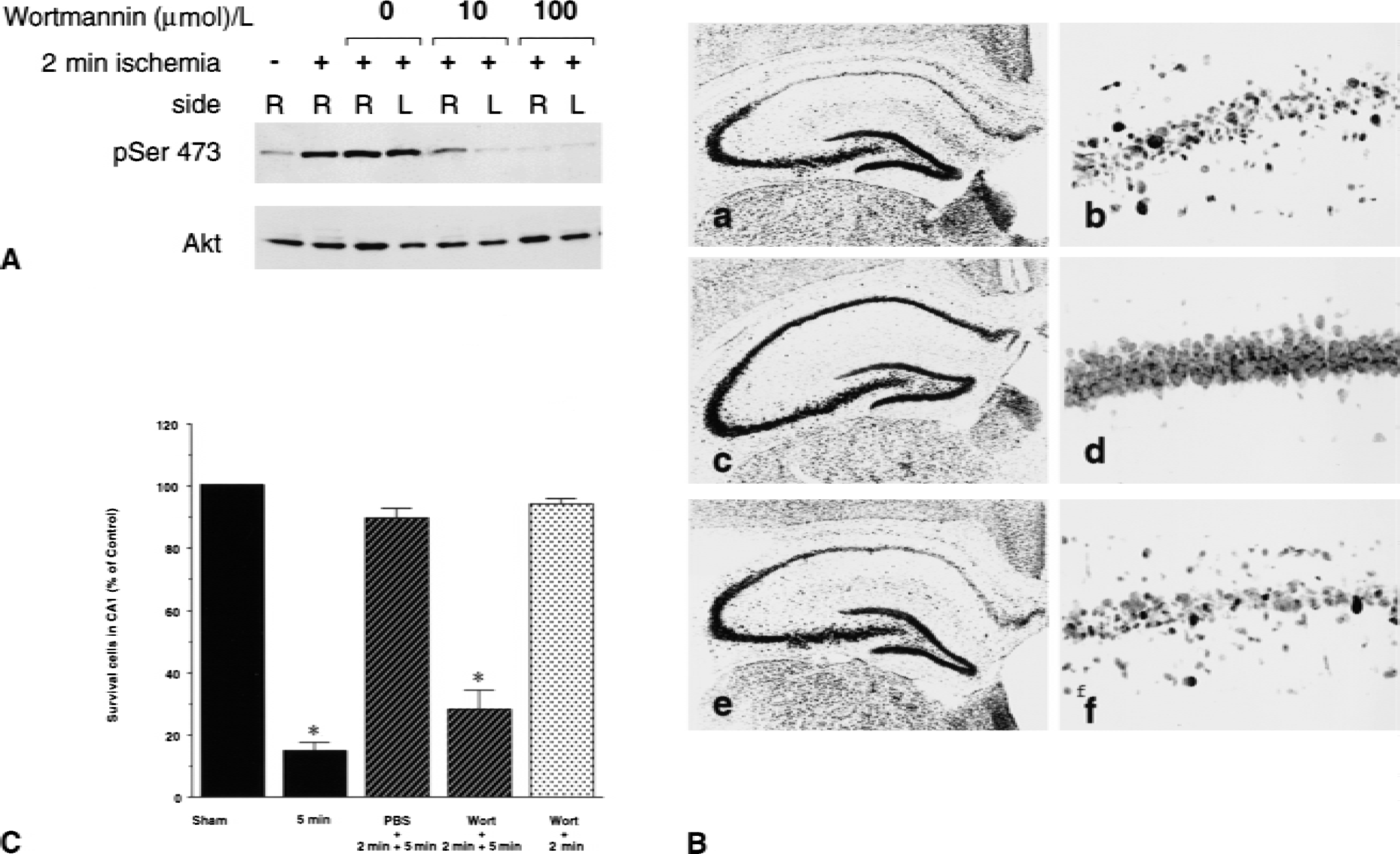
Effect of wortmannin on induction of ischemic tolerance. (
After 5 minutes of ischemia with or without wortmannin treatments before the 2 minutes of preconditioning, delayed neuronal cell death was evaluated on day 7. Animals were perfusion-fixed with ice-cold 4% formaldehyde solution. Surviving neuronal cells, which were evaluated by propidium iodide staining, exhibited large round nuclei in the pyramidal layer. The histologic changes in the CA1 region in gerbils without or with preconditioning or with preconditioning after wortmannin treatment are shown in Fig. 5. Accumulating results of surviving neuronal cells in the CA1 regions are also shown in Fig. 5.
Compared with sham-operated animals, 5 minutes of ischemia induced cell death in approximately 90% of the pyramidal cells, and preconditioning resulted in survival of approximately 90% of the cells in the CA1 region. Infusion of wortmannin before preconditioning significantly blocked the neuroprotective effect of preconditioning. Animals subjected to wortmannin infusion followed by 2 minutes of ischemia without 5 minutes of ischemia did not exhibit neuronal cell death in the CA1 area, indicating that wortmannin at the concentration used did not have a toxic effect on pyramidal neurons.
To evaluate the significance of elevated Akt activity just before the second ischemia, we infused wortmannin into the ventricle on day 3 after the 2-minute ischemia. Phosphorylation of Akt-Ser-473 in the CA1 region 30 minutes after infusion was decreased by infusion of 100 μmol/L wortmannin without changing Akt protein levels, compared with the phosphorylation level observed in gerbils subjected to 2 minutes of ischemia treated with or without infusion (Fig. 6). In histologic outcome studies, however, preconditioned gerbils with or without the posttreatment with wortmannin showed almost complete neuroprotection against lethal ischemia. These results show that a persistent increase in Akt for 3 days before lethal ischemia is relevant for ischemic tolerance induced by sublethal ischemia, and that elimination of the elevated Akt activity at 3 days after the preconditioning does not interfere with the ischemic tolerance.
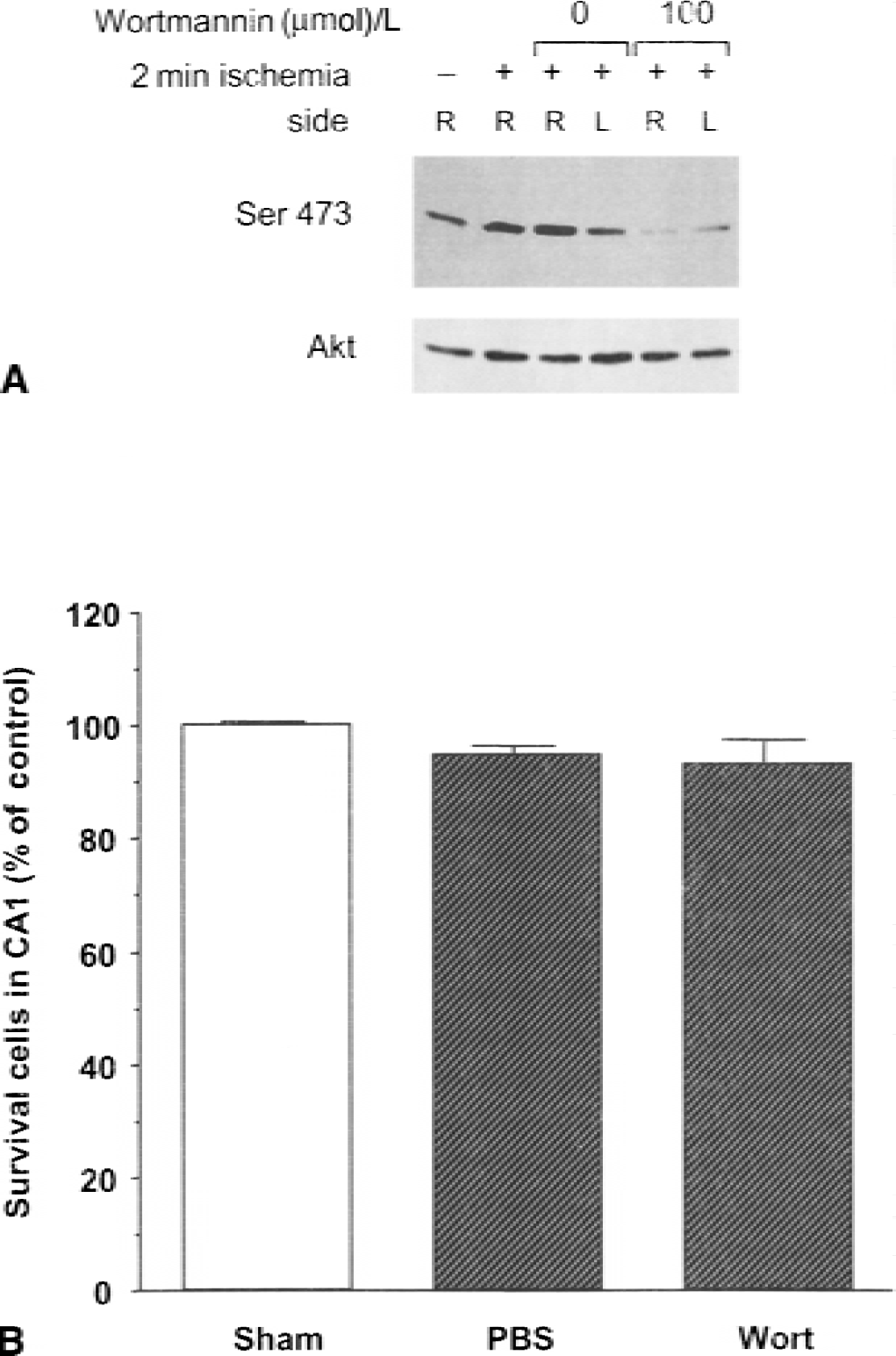
Effect of inhibition of Akt activity immediately before lethal ischemia on ischemic tolerance. (
DISCUSSION
A retrospective case-control study in stroke patients with or without transient ischemic attack before the stroke showed that ischemic tolerance might also occur in the human brain (Weih et al., 1999). Although ischemic tolerance has not received much attention in the field of transient brain ischemia, understanding the molecular mechanisms underlying ischemic tolerance may produce strategies for minimizing neuronal damage in patients with stroke. Using a global cerebral ischemic model in gerbil, we showed here that activation of Akt after sublethal ischemia significantly rescues cells from delayed neuronal cell death after subsequent lethal ischemia in the hippocampal CA1 region. Akt activity after preconditioning was gradually and persistently increased after ischemia, correlating with the development of ischemic tolerance. Interestingly, wortmannin treatments before the 2-minute preconditioning significantly prevented the neuroprotective action of preconditioning, with concomitant elimination of Akt activation. In contrast, wortmannin treatments just before the second ischemia had no effect on the preconditioning-induced neuroprotection. In addition, wortmannin treatment itself did not cause neuronal cell death. These results suggest that the early phase of Akt activation after sublethal ischemia plays an important role in the establishment of ischemic tolerance. When gerbils were subjected to 5 minutes of ischemia without preconditioning, a significant decrease and a subsequent transient increase in the Akt-Ser-473 phosphorylation level were observed. The observation is consistent with the rat ischemic model developed by Ouyang et al. (1999).
How Akt is activated after preconditioning is not known. Preconditioning-induced tolerance was associated with a transient increase in extracellular glutamate concentration (Nakata et al., 1992) and was blocked by treatment with MK801 in gerbil hippocampus (Bond et al., 1999; Kato et al., 1992), showing the role of NMDA receptors. Zhang et al. (1998) reported that stimulation of NMDA receptors rescued cells from apoptosis induced by removal of extracellular K+ in cerebellar granule neurons. The neurotrophic action of NMDA was abolished by a PI3-K inhibitor and was correlated with phosphorylation of insulin receptor substrate–1. We recently showed that sublethal Ca2+ elevation through voltage-dependent Ca2+ channels or NMDA receptors activates Akt through Ca2+/calmodulin-dependent protein kinase kinase in situ (Yano et al, 1998). Ca2+/calmodulin-dependent protein kinase kinase phosphorylated Thr-308 located in the Akt activation loop, with concomitant activation of Akt. In the present study, preconditioning-induced Akt activation was totally abolished by wortmannin, which is reported to be an irreversible inhibitor of PI3-K (Alessi et al., 1996; Kimura et al., 1994), suggesting that the PI3-K pathway is primarily involved in Akt activation in ischemic tolerance. Although a role for Ca2+/calmodulin-dependent protein kinase kinase in Akt activation downstream of PI3-K in ischemic tolerance remains to be investigated, because PI3-K stimulates phospholipase Cγ-mediated calcium signaling (Rameh et al., 1998), this evidence suggests the possibility that sublethal Ca2+ elevation through Ca2+ channels (Nakata et al., 1992) and AMPA or NMDA receptors (Kato et al., 1992; Shamloo and Wieloch, 1999; Yamaguchi et al., 1999) could be involved in Akt activation after sublethal ischemia.
Potential downstream targets of Akt, such as BAD (Datta et al., 1997; Kennedy et al., 1999; Zha et al., 1996) and caspase-9 (Cardone et al., 1998), that are phosphorylated directly by Akt may be responsible for neuroprotection established during ischemic tolerance. However, it is not known whether such phosphorylation events underlie the Akt-induced neuroprotective action during ischemic tolerance, because ischemic tolerance is established at least 24 hours after preconditioning (Kato et al., 1991). In ischemic tolerance, protein synthesis is restored at least 24 hours after preconditioning (Kato et al., 1995). In this context, an attractive hypothesis is that the initial Akt activation after preconditioning accounts for the expression of neuroprotective proteins.
Brain-derived neurotrophic factor (BDNF) is among the candidates responsible for inducing the late phase of Akt activity. BDNF has been shown to mediate NMDA receptor-dependent antiapoptotic signals through Akt-dependent signaling (Bhave et al., 1999). In addition, treatment with BDNF enhances the neuroprotective effect of preconditioning (Kawahara et al., 1997). Other neuroprotective proteins, such as heat shock protein 70 family members and Bcl-2 family members, are also known to be expressed during ischemic tolerance. For example, intense immunoreactivity of Bcl-2 was observed at 30 hours and sustained for at least 4 days after 2 minutes of ischemia in the gerbil CA1 region, whereas increased Bcl-2 immunoreactivity was not seen before 12 hours (Shimazaki et al., 1994). Heat shock protein 70 mRNA and protein were observed in the CA1 area from 6 through 24 hours of reperfusion after 2 minutes of ischemia in gerbils (Kanemitsu et al., 1994). Whether Akt activation regulates expression of Bcl-2 and heat shock proteins is not known. Transcription factors are also attractive targets of Akt, including the cAMP-responsive element binding protein, nuclear factor-κB, and a forkhead-related transcription factor. The forkhead-related transcription factor may be relevant because it is not only directly phosphorylated by Akt but also inhibits expression of proapoptotic proteins such as the Fas ligand (Brunet et al., 1999). cAMP-responsive element binding protein and nuclear factor-κB are also related to antiapoptotic signals in various insults, including cerebral ischemia (Walton and Dragunow, 2000; Yu et al., 1999) and are activated by the Akt signaling cascade in vitro (Du and Montminy, 1998; Romashkova and Makarov, 1999). Our observation that Akt activation after preconditioning was more apparent in nuclei than the cytosol is potentially important. In addition, the increase in Akt activity after preconditioning was specific to hippocampal CA1 pyramidal neurons but not to neurons in the CA3 region and dentate gyrus. Although we cannot conclude that increased immunoreactivity in nuclei is due to nuclear translocation of the protein, Andjelkovic et al. (1997) reported translocation of Akt after its activation.
In conclusion, our results clearly show that Akt is activated by preconditioning in the vulnerable CA1 pyramidal neurons and that such activation plays a neuroprotective role. A speculative view of the function of increased Akt activity in nuclei is that activation could constitute a cellular switch to preserve cell viability against more severe ischemic insults. Although further investigation is required to clarify the molecular targets of Akt underlying its antiapoptotic signals in CA1 pyramidal neurons, the Akt pathway may be an important target for the development of novel therapeutic strategies for protection against ischemic insults.