
Abstract
Previous histopathologic results have suggested that one mechanism whereby hyperglycemia (HG) leads to exaggerated ischemic damage involves fragmentation of DNA. DNA fragmentation in normoglycemia (NG) and HG rats subjected to 30 minutes of forebrain ischemia was studied by terminal deoxynucleotidyl transferase mediated DNA nick-labeling (TUNEL) staining, by pulse-field gel electrophoresis (PFGE), and by ligation-mediated polymerase chain reaction (LM-PCR). High molecular weight DNA fragments were detected by PFGE, whereas low molecular weight DNA fragments were detected using LM-PCR techniques. The LM-PCR procedure was performed on DNA from test samples with blunt (without Klenow polymerase) and 3′-recessed ends (with Klenow polymerase). In addition, cytochrome c release and caspase-3 activation were studied by immunocytochemistry. Results show that HG causes cytochrome c release, activates caspase-3, and exacerbates DNA fragments induced by ischemia. Thus, in HG rats, but not in control or NGs, TUNEL-stained cells were found in the cingulate cortex, neocortex, thalamus, and dorsolateral crest of the striatum, where neuronal death was observed by conventional histopathology, and where both cytosolic cytochrome c and active caspase-3 were detected by confocal microscopy. In the neocortex, both blunt-ended and stagger-ended fragments were detected in HG, but not in NG rats. Electron microscopy (EM) analysis was performed in the cingulate cortex, where numerous TUNEL-positive neurons were observed. Although DNA fragmentation was detected by TUNEL staining and electrophoresis techniques, EM analysis failed to indicate apoptotic cell death. It is concluded that HG triggers a cell death pathway and exacerbates DNA fragmentation induced by ischemia.
Transient forebrain ischemia leads to a delayed type of brain damage that usually affects only the neuronal population. Preischemic hyperglycemia (HG) aggravates such damage, typically causing a more rapid evolution of the damage, pan-necrotic lesions (“infarction”), and postischemic seizures. Hyperglycemia seems to exert its adverse effect by causing a further decrease of intra-and extracellular pH during and immediately after the ischemic transient (Siesjö et al., 1996; Li and Siesjö, 1997).
The mechanisms of HG-exaggerated damage have not been defined. One possibility is that the exaggerated acidosis triggers DNA damage. Two types of results support this contention. First, previous EM data published by the authors' laboratory, pertaining to 30 minutes of forebrain ischemia in the rat, show that, in HG animals, recirculation is accompanied by signs of extensive rapidly occurring chromatin condensation (Kalimo et al., 1981). Second, intracellular acidification triggers apoptosis by directly or indirectly activating ICE-like protease (Furlong et al., 1997) and caspase (Matsuyama et al., 2000), or by activating a DNase II (Barry and Eastman, 1993). In the current study, 30-minute transient forebrain ischemia was used to test whether preischemic HG causes earlier appearing and more extensive DNA fragmentation than in normoglycemia (NG) animals as assessed by biochemical techniques. Furthermore, the authors studied whether the DNA fragmentation is correlated to cytochrome c release and caspase-3 activation. Finally, the authors explored if extensive accumulation of TUNEL-positive neurons observed in the cingulate cortex in HG rats have an apoptotic morphology, as assessed by EM criteria.
MATERIALS AND METHODS
Cerebral ischemia
All animal use procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Ethics Committee.
Fifty-three overnight-fasted male Wistar rats (Simonsen, MA, U.S.A.), weighing 290 to 330 g, were used in the current experiments. Of 53 rats, 12 were used for conventional histopathologic and TUNEL staining, 12 for immunocytochemistry, 24 for PFGE and LM-PCR, and 5 for EM. Anesthesia was introduced with 3.5% halothane in a mixture of N2O:O2 (70:30) and maintained at 1% to 1.5% halothane during surgery. Ventilation was adjusted to give an arterial Paco2 35 to 40 mm Hg and Pao2 approximately 100 mm Hg. Surgical procedures are the same as previously described by the authors (Li et al., 1995). Heparin (30 IU/kg) was given intravenously before the first blood sampling. Electroencephalogram was monitored throughout the experimental period. Body and head temperatures were maintained close to 37°C by a combination of a homeothermic blanket control unit and lamp heating. Transient forebrain ischemia (2-vessel occlusion plus hypotension) lasting 30 minutes was performed as described by Smith et al. (1984). Hyperglycemia was achieved by infusion of a 25% glucose solution (1.5 mL) for 30 minutes before ischemia to yield a plasma glucose concentration ≈ 20 mmol/L. Fasted rats infused with the same amount of 0.9% saline served as NG control (plasma glucose concentration ≈ 5 mmol/L).
Evaluation of brain damage
After 60 minutes of recirculation after 30 minutes of transient forebrain ischemia, rats were transcardially perfusion-fixed with 4% paraformaldehyde in PBS (pH 7.4). Brains were processed for dehydration and embedding. Paraffin-embedded sections (5 μm) were stained with a combination of acid fuchsin and celestine blue. Damage was evaluated in the neocortex, hippocampus, thalamus, and striatum under light microscopy. Two types of neurons were defined as dead. The first type was bright red-stained acidophilic neurons with a shrunken triangular shape and dense purple nucleus, which was usually observed in neocortical structures and hippocampal areas. The second type was neurons with shrunken nucleus and clear perineuronal sponginess, which was frequently observed in the thalamus in HG animals.
TUNEL staining
Brain sections adjacent to those for histopathologic evaluation were used for TUNEL staining. In situ detection of DNA fragmentation on brain sections after ischemia or sham operation was performed using the methods reported by Gavrieli et al. (1992) and Wijsman et al. (1993). Sections were deparaffinized in xylene and rehydrated by a passage through a decreasing ethanol series (100%, 95%, 90%, 80% for 3 minutes each), followed by 2 washes in PBS (pH 7.4) at 2 minutes per wash. Sections were treated with proteinase K 20 μg/mL in TE buffer (10 mmol/L Tris, 5 mmol/L EDTA, pH 7.4) for 15 minutes at room temperature to increase cell permeability and then were immersed in 3% hydrogen peroxide in PBS at room temperature for 5 minutes to remove endogenous peroxidase. Slides were incubated with a labeling buffer containing TdT and divalent cation (Mn2+) inside a humidity chamber at 37°C for 1 to 2 hours. The reaction was stopped by transferring the sections to a termination buffer (300 mmol/L sodium chloride, 30 mmol/L sodium citrate) for 5 minutes. After 2 washes in PBS, sections were incubated with Strep-HRP for 10 minutes at room temperature and then transferred to diaminobenzidine tetrahydrochloride (DAB) working solution for staining. Sections were counterstained with methyl green. After two washes in PBS, pH 7.4, sections were dehydrated in an ascending ethanol series. After immersion in xylene, the sections were coverslipped in Permount. For control studies, the sections were treated the same way except that DNase I replaced TdT.
Immunocytochemistry for cytochrome c and caspase-3
Animals were perfused with ice-cold 4% paraphormaldehyde in PBS. Their brains were removed and postfixed overnight in the same fixative solution before being cut coronally at 50 μm with a vibratome. Brain sections were washed twice in PBS and then in PBS containing 0.2% triton X-100 (TX 100) for 30 minutes. Nonspecific binding sites were blocked in 3% bovine serum albumin (BSA) in PBS/0.2% TX 100 for 30 minutes. Brain sections were incubated with primary antibody for cyt c (monoclonal antibody; Biosource, Camarillo, CA, U.S.A.), diluted 1:400 in PBS/0.1% TX 100 and 1% BSA, overnight at 4°C, then washed in PBS containing 0.1% TX 100, 3 times for 10 minutes. Sections were labeled for 2 hours with fluorescein isothiocyanate-labeled anti-mouse secondary antibody (1:200) and propidium iodide to stain nuclei.
Cryostat section of 12 μm at the bregma level of −3.8 mm was used for detection of caspase-3. Biotin-DEVD-CHO [N-biotin-Asp-Glu-Val-Asp-CHO (aldehyde)] (Biomol, Plymouth Meeting, PA, U.S.A.), which binds only to activated caspase-3, was used to determine caspase-3 activity. Brain sections were incubated with 1 μmol/L Biotin-DEVD-CHO in HD solution (10 mmol/L Hepes, 1 mmol/L dithiothreitol) buffered with PBS, pH 7.4, at 37°C for 1 hour. Sections were washed with PBS twice and then conjugated with streptavidin CY3 (1:500 dilution in PBS) for 30 minutes. Sections were washed again with PBS twice, fixed in 4% paraformaldehyde for 10 minutes. Sections were mounted on glass slides, coverslipped, and analyzed on a BioRad MRC 1024 laser-scanning confocal microscope (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
Pulse-field gel electrophoresis
After 30 and 60 minutes of recirculation, animals were decapitated under deep halothane anesthesia and the brains were quickly removed. Neocortex and hippocampus were carefully dissected on ice and kept deep frozen at −80°C. PFGE, as described by Walker et al. (1993), was performed to detect high molecular weight fragments of 50 kbp.
Deeply frozen neocortical and hippocampal samples (40 to 50 mg) were suspended in 80 μL TE buffer (10 mmol/L Tris, 125 mmol/L EDTA, pH 9.0) with the aid of a 1 mL pipette tip. The suspension was immediately mixed with an equal volume of 1.5% low melting-point agarose (Incert Agarose; FMC Bioproducts, Rockland, ME, U.S.A.), held at 37°C. The mixture was cast into a 1-mL syringe and set at 4°C for 5 minutes. Plugs then were immersed in lysis buffer (10 mmol/L Tris, 1 mmol/L EDTA, and 10%N-lauryl sarcosine, pH 8.0) containing proteinase K at 20 mg/mL (Gibco BRL, Burlington, ON, Canada), and incubated with rotation at 37°C for 18 to 20 hours. The next day, the plugs were rinsed in TE and incubated with 10 mg/mL RNase A (Sigma, Burlington) in TE buffer at 4°C for 1 hour. The plugs were then rinsed twice in TE at 4°C for 30 minutes each and pulled back into a 1-mL syringe and stored at 4°C. A slice of 2-mm plug was loaded onto a well in 0.8% agarose (FMC) gel in TBE electrophoresis buffer (0.089 mol/L Tris, 0.089 mol/L Borate, and 2.5 mmol/L EDTA, pH 8.3) and sealed in place with 1% low melting-point agarose (FMC). The gel was subsequently run on a Q-life Autobase page system (Q-Life, Kinston, ON, Canada) in TBE buffer at 14°C. After staining with ethidium bromide (1 μg/mL) for 30 minutes, the gel was destained in distilled water and photographed with Type 55 film (Polaroid, Fisher Biotech, Pittsburgh, PA, U.S.A.) under UV transillumination.
Ligation-mediated polymerase chain reaction
Low molecular weight DNA fragments were detected using LM-PCR techniques as described by Staley et al. 1997, see also MacManus et al., 1999). Briefly, a brain tissue sample (100 mg) was homogenized in TE buffer (pH 9.0), with 40 μg proteinase K. The homogenate was centrifuged at 3000 g for 3 minutes, the pellet was then resuspended in 10 mmol/L MgCl2/0.35% nonidet P-40 and centrifuged again at 3000 g for 3 minutes. The resulting pellet was resuspended in solution containing 50 mmol/L Tris, 2 mmol/L MgCl2, and 2 mmol/L CaCl2. For each 100-μL volume, 55 μL 5 m NaCl, 25 μL 0.5 m EDTA, 25 μL 10% sodium dodecyl sulfate, and 10 μL 20 mg/mL proteinase K were added. The sample was incubated at 55°C for 3 hours, then extracted with phenol:chloroform:isoamyl alcohol (25:24:1), and precipitated with 5 mol/L NaCl and ethanol at −20°C overnight. The next morning, after centrifugation (14000 g × 10 minutes) and washing (70% ethanol), DNA was resuspended in TE buffer. The sample was then incubated with RNase A (10 mg/mL) at 37°C for 1 to 2 hours. DNA was extracted with phenol:chloroform, chloroform, and ether, and then was precipitated with 0.3 mol/L NaOAc and 2 volumes of ethanol overnight. After centrifugation, washing, and resuspension in TE (pH 8.0), the optical density was read at 260 nm. In a separate set of samples, 2 μg DNA was treated with 5 units of Klenow polymerase (Gibco BRL) with the 4 deoxyribonucleotides at pH 8.0 for 15 minutes at 30°C, followed by inactivation at 75°C for 10 minutes.
The LM-PCR procedure was performed on DNA from both the test samples with blunt (without Klenow polymerase) and 3′-recessed-ends (with Klenow polymerase). Two oligonucleotides (24 bp: 5′-AGCACTCTCGAGCCTCTCACCGCA-3′ and 12 bp: 5′-TGCGGTGAGAGG-3′) were annealed and ligated to 200 ng target DNA using 1 μL 20 mmol/L adenosine triphosphate, 1 μL 20 mmol/L DTT, and 4 U T4 DNA ligase (New England BioLabs, Mississauga, ON, Canada). The polymerase chain reaction (PCR) was performed with 2.5 ng target DNA with 65 pmol of the 24 bp oligonucleotide and 3 U Taq polymerase (Bibco-BRL), 2 mmol/L MgCl2 with 0.324 mmol/L deoxyribonucleotides in 67 mmol/L Tris, 16 mmol/L (NH4)2SO4, 10 mmol/L β-mercaptoethanol, and 0.1 mg/mL BSA in a 50-μL volume. The sample was amplified for 28 cycles. An aliquot of the samples was run on a 1.8% Synergel (Diversified Biotech, Boston, MA, U.S.A.) in TAE electrophoresis buffer (40 mmol/L Tris, 1 mmol/L EDTA, pH 8.0 with acetic acid). Gels with DNA-size markers were run for 16 to 18 hours at 20 V at room temperature, stained with ethidium bromide, and subsequently photographed with Type 55 film (Polaroid) under UV transillumination.
Electron microscopy studies
Rat brains collected from sham and 60-minute recovery after 30 minutes of ischemia in both NG and HG rats were perfused with 2.5% glutaraldehyde. Coronal brain sections (200-μm-thick) at the level of bregma −3.8 mm were postfixed with 4% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). Sections then were soaked in 1% osmium tetroxide in 0.1 mol/L cacodylate buffer for 2 hours, rinsed in distilled water, and stained with 1% aqueous uranyl acetate overnight. Tissue sections then were dehydrated in an ascending series of ethanol to 100% followed by dry acetone, and embedded in Durcopan ACM. Ultrathin sections were counterstained with lead citrate before examination by EM.
Statistics
Parametric pathologic data were analyzed with analysis of variance followed by Scheffe's test. Student's t-test was used for TUNEL data.
RESULTS
Physiologic variables
Blood glucose concentrations and physiologic parameters were measured 5 minutes before induction of ischemia. As expected, blood glucose concentrations were significantly greater in HG than in NG animals (19.8 ± 1.8 versus 4.3 ± 0.4, mean ± SD).
Paco2 was 39.6 ± 6.3 mm Hg in HG and 37.7 ± 4.2 mm Hg in NG rats. Pao2 was 114 ± 14 versus 112 ± 14 mm Hg and pH was 7.39 ± 0.03 versus 7.42 ± 0.04 units in HG versus NG animals.
Brain and body temperatures were maintained at 37.2°C ± 0.2°C in both HG and NG animals. Mean arterial blood pressure was 110 mm Hg. There were no significant differences in physiologic parameters between NG and HG groups.
Morphologic changes under light microscopy
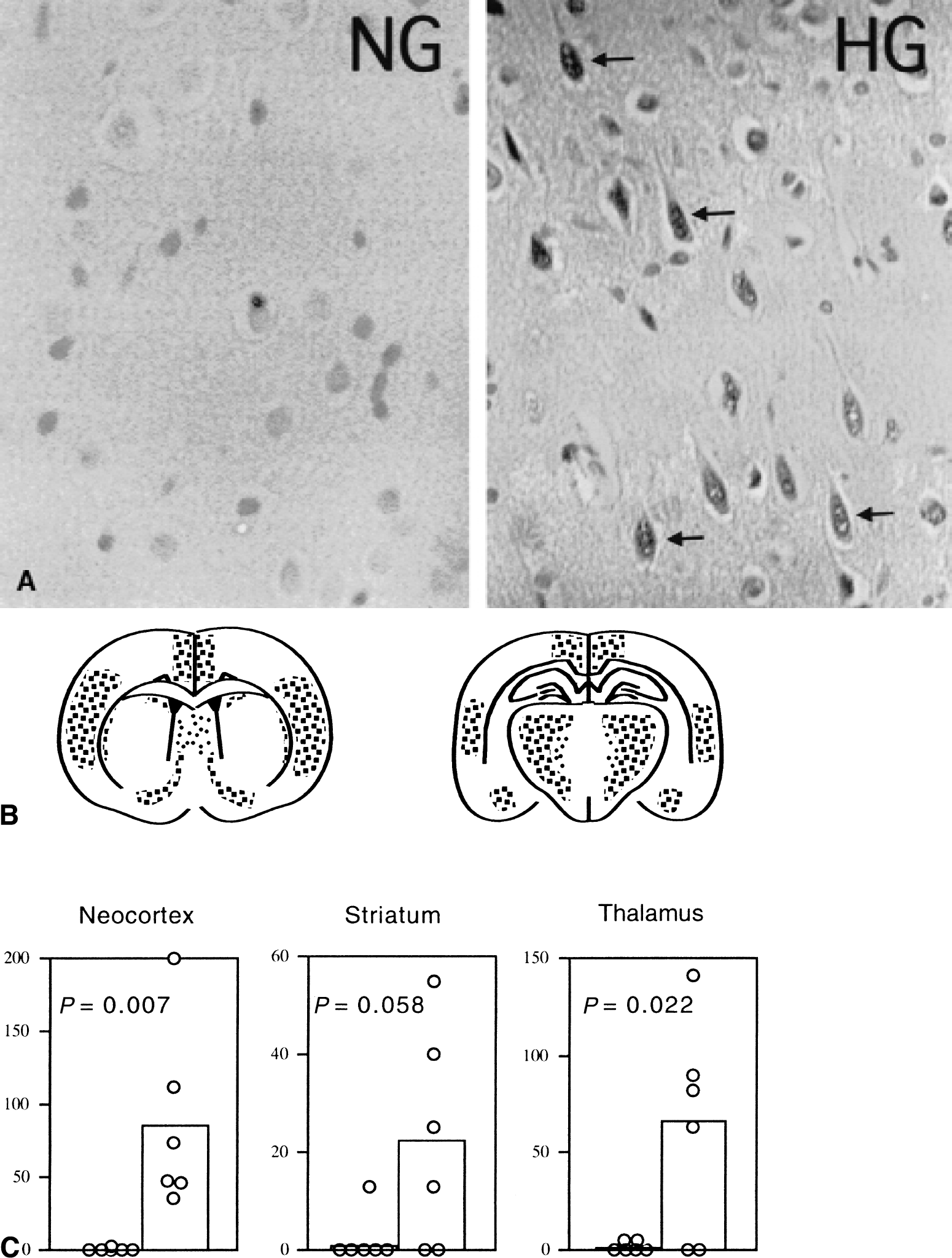
In NG animals, neurons were intact in all brain regions examined after 60 minutes of recirculation, except that many condensed, dark purple-stained basophilic neurons were presented in the neocortex. In HG animals, some of the neurons in the neocortex, thalamus, and striatum were dead, whereas those in the hippocampal CA1, CA3, and dentate gyrus regions were virtually spared at such an early reperfusion stage. Neurons in the neocortex and striatum appeared as a mixture of basophilic and acidophilic neurons, distributed evenly over the whole cortex and dorsolateral crest of striatum. Damage in the thalamus presented as an increased sponginess. TUNEL staining was performed in the adjacent sections to look for evidence of nuclear DNA breakdown. Consistent with the pathologic findings, TUNEL-positive cells could barely be found in NG rats but were abundant in HG animals (Fig. 1A). TUNEL-positive cells were mainly distributed in the cingulate cortex, parietal and auditory cortex layers 2 to 5, thalamus, and dorsolateral crest of the striatum (Fig. 1B). Many of the TUNEL-positive cells contained punctate chromatin. A summary of these data is presented in Fig. 1C.
Cytochrome c release and caspase-3 activation
Cytosolic cytochrome c release was evident in the cingulate cortex after a 60-minute recovery in HG, but not in NG rats. As shown in Fig. 2 (upper panel), cytochrome c was located in perikaryal and dendritic mitochondria as reflected in the punctate green dots on the NG section, whereas it was released into cytosol in the HG section, as reflected by the even distribution of the green and yellow color around the nuclei.
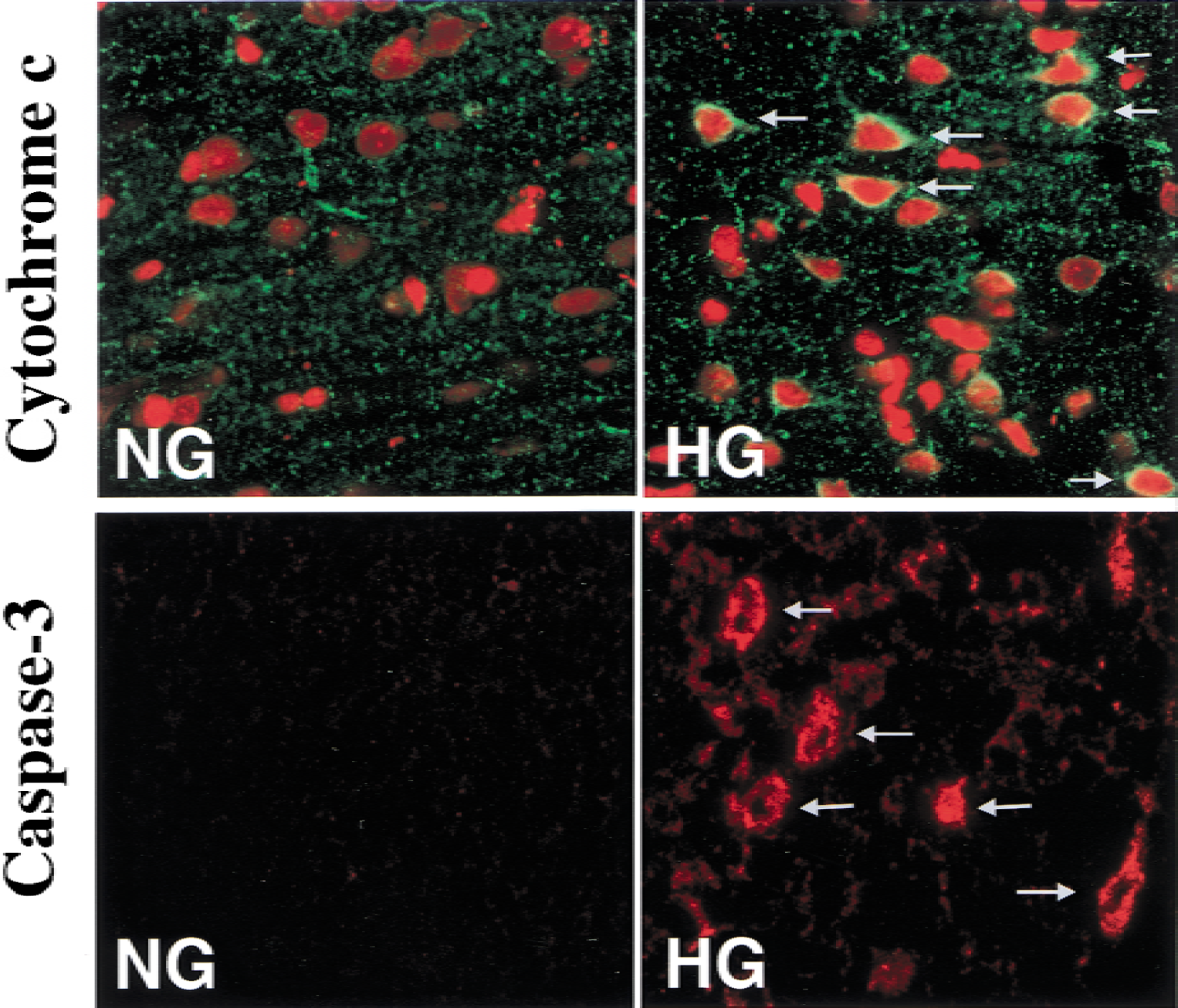
(top panel) Double immunostaining confocal images for cytochrome c in the neocortex in normoglycemic (NG) and hyperglycemic (HG) rats after 60 minutes of recovery. Red color is neuronal nuclei stained with propidium iodide. Green color is cytochrome c. Overlap of red and green generates yellow. Even distribution of the green and yellow color around the nuclei in HG animals implies release of cytochrome c into the cytoplasm. There was no observed release of cytochrome c in NG animals. (bottom panel) In situ labeling of active caspase-3 in neocortex. Caspase-3 was positively labeled after 60 minutes of reperfusion in HG, but not in NG animals.
Predictably, activated caspase-3 was detected after 60 minutes of recovery in the same brain region as cytochrome c in HG but not NG rats. As shown in Fig. 2 (lower panel), the NG section showed only light background staining, whereas the HG section demonstrated strong active caspase-3 staining.
High and low molecular weight DNA fragments
As shown in Fig. 3A, DNA fragments of 50 kbp were presented in the neocortex of HG animals, but not NG or sham-operated animals (Fig. 3, CTX). The appearance of 50-kbp fragments was seen as early as 30 minutes after ischemia and persisted for 30 and 60 minutes of recirculation in HG rats. In the hippocampus, a faint 50-kbp fragment was observed in one HG sample but not in any of the NG samples (Fig. 3, HC).
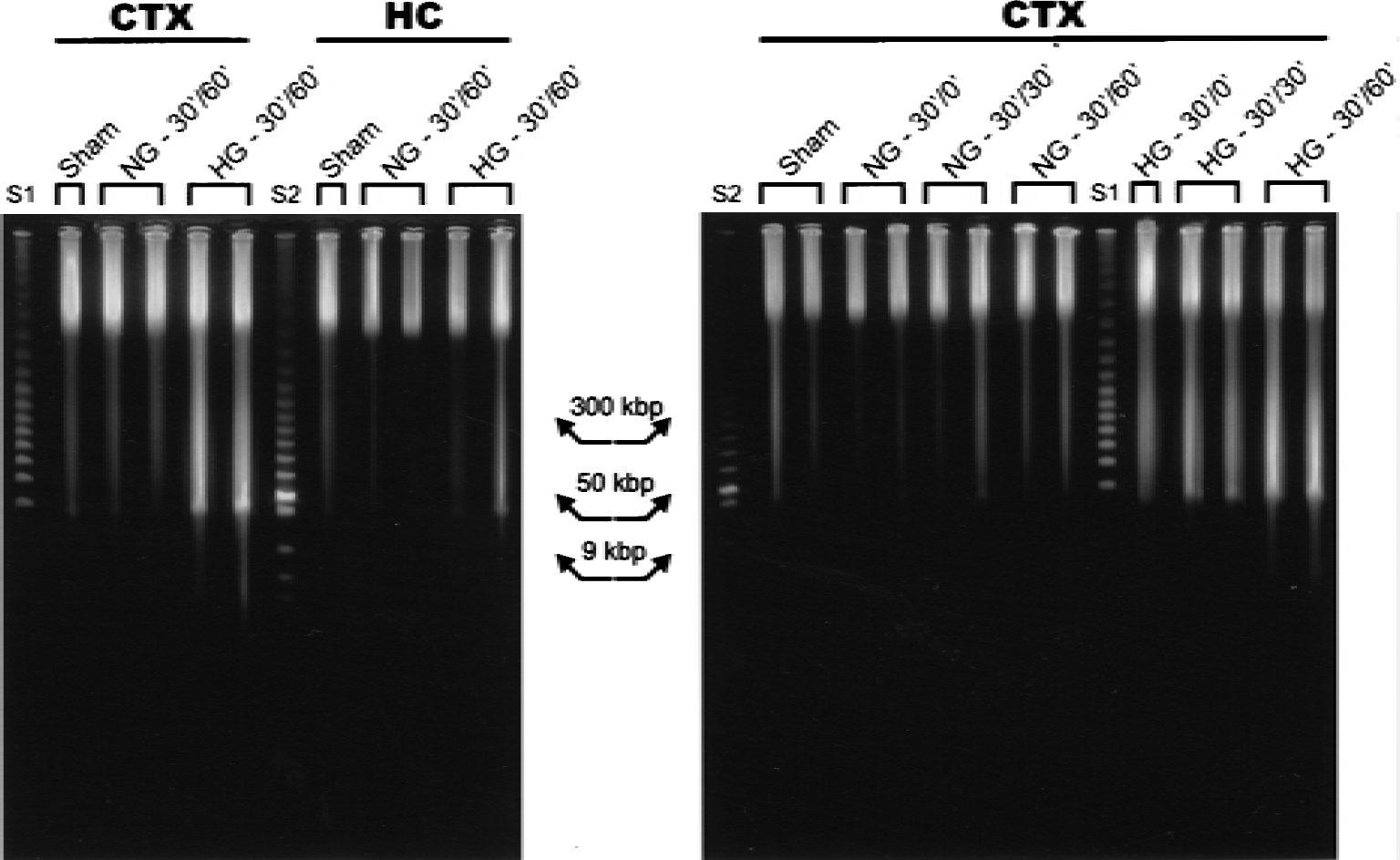
Detection of high molecular weight DNA fragments by pulse-field gel electrophoresis (PFGE) after transient forebrain ischemia in normoglycemic (NG) and hyperglycemic (HG) rats. DNA samples with sham, 0, 30, and 60 minutes of reperfusion in the neocortex (CTX) and hippocampus (HC) were electrophoresed. The appearance of 50 kbp fragments was seen after 30 and 60 minutes of reperfusion in HG, whereas faint fragments were seen in the NG condition. Lanes S1 and S2 are molecular weight markers.
Low molecular weight DNA fragmentation is illustrated in Fig. 4. Blunt-ended DNA fragments were easily detected in HG, but not in NG or sham-operated animals after 60 minutes of recovery (Fig. 4, right panel). After Klenow polymerase treatment, the laddered fragments in HG animals were clearly evident (Fig. 4, left panel); however, the pattern of the fragments was somewhat smeared. In NG animals, one of four samples showed a faint laddered DNA fragmentation after Klenow polymerase treatment.
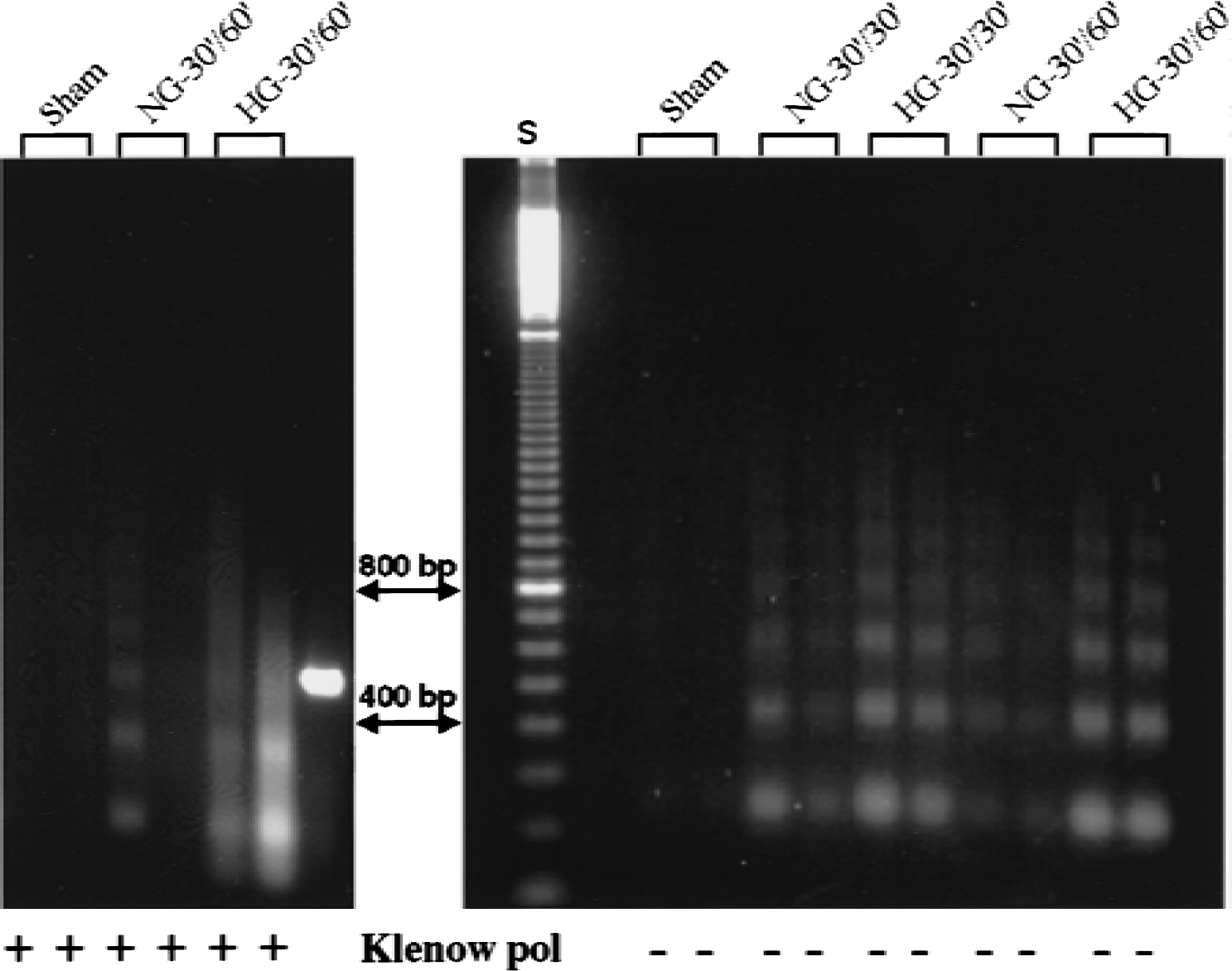
Low molecular weight DNA fragments detected by ligation-mediated polymerase chain reaction (LM-PCR) after 60 minutes of reperfusion in normoglycemic (NG) and hyperglycemic (HG) animals. Duplicates of 2.5 ng samples of DNA from neocortex were subjected to PCR with and without Klenow polymerase (Klenow pol) treatment. The HG exhibited laddered fragments without polymerase treatment, whereas the NG and sham samples showed no evidence of DNA fragments (right panel). After polymerase treatment, the laddered fragments in HG were evident. Faint fragments also were detected in a NG sample (left panel). Lane S is a molecular weight marker.
Ultrastructural alterations
To define whether punctate TUNEL-positive neurons represent apoptotic cell death, neurons in the cingulate cortex were examined under EM. Both NG and HG rats were subjected to 30 minutes of ischemia and perfusion fixed after 60 minutes of recirculation. Attempts were made to clarify whether neurons died of apoptosis or necrosis. The hallmark of apoptosis is chromatin condensation, compaction of organelles, and apoptotic bodies, whereas necrosis is diagnosed by swelling of organelles, mitochondrial flocculent densities, and membrane breaks. In the NG rats, the nucleus was relatively homogenous, the nucleolus was clearly visible (Fig. 5, NG), and mitochondrial morphology was normal (Fig. 6, NG). In the HG rats, chromatin margination was evident in the nucleus, as were severe vacuolar alterations (Fig. 5, HG). The mitochondria showed marked swelling, lucency, and loss of cristae (Fig. 6, HG). However, apoptotic bodies were not observed in any of the ultrathin sections examined.
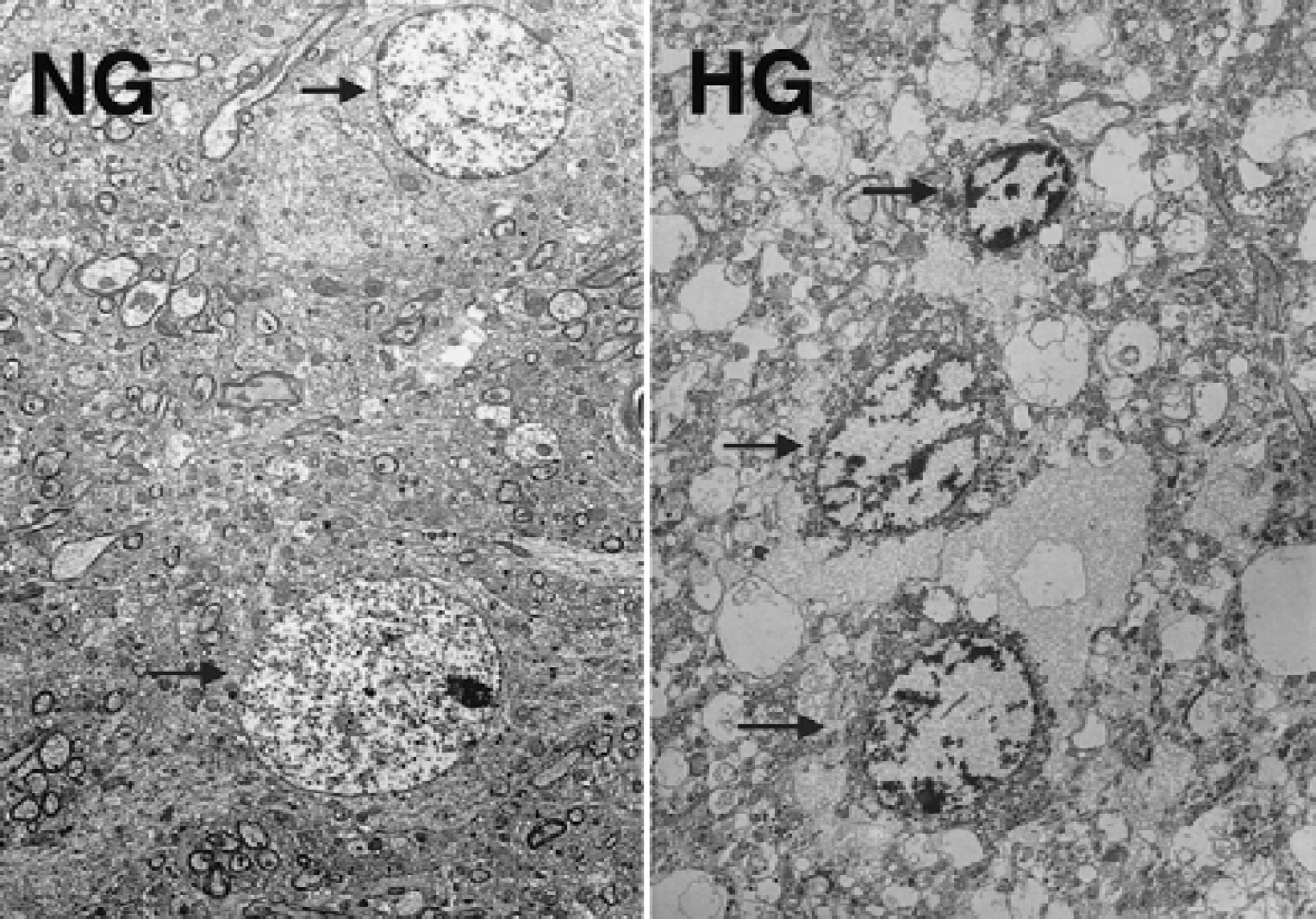
Electron micrographs of osmium-uranyl-lead stained neurons in the cingulate cortex from rats subjected to 30 minutes of ischemia, followed by 60 minutes of recovery with preischemic normoglycemic (NG) and hyperglycemic (HG) conditions. In the NG rats, nucleus is relatively homogenate and cytosolic organs are normal (arrows). In the HG rat, chromatin margination and condensation are evident (arrows), and severe vacuolar alteration is presented. Magnification ×2,000.
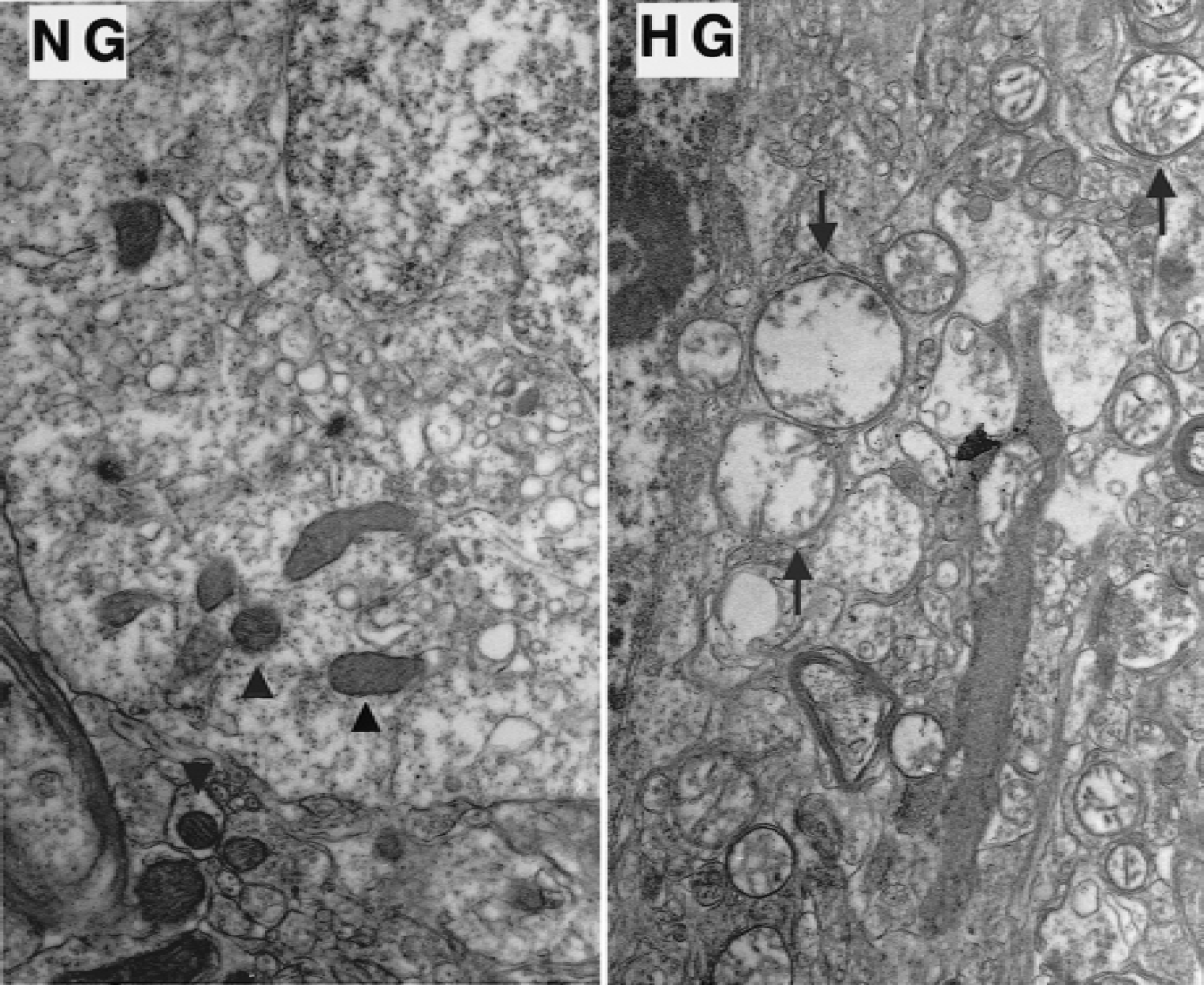
High magnification of electron micrographs of osmium-uranyl-lead stained neurons in the cingulate cortex from rats subjected to 30 minutes of ischemia, followed by 60 minutes of recovery with preischemic normoglycemic (NG) and hyperglycemic (HG) conditions. Mitochondria morphology is normal in NG rats (arrowheads). Marked swelling, lucency, and loss of cristae are the features of mitochondrial morphology in the HG rat (arrows). Magnification ×10,000.
DISCUSSION
In the current experiments, the authors have investigated whether HG exaggerates ischemia-induced DNA fragmentation in brain tissues collected from neocortex and hippocampus. For that purpose, the authors have chosen to induce 30-minute ischemia because it has previously been suggested on morphologic grounds that nuclear chromatin condensation may occur already within 1 to 2 hours after reperfusion (Kalimo et al., 1981). Results show that no, or very faint, DNA fragments were detected in NG animals in either the hippocampus or the cortex after 30 and 60 minutes of recirculation, suggesting that DNA was not fragmented at this early stage of reperfusion in NG animals. The normal appearance of nuclear and mitochondrial morphologies from the current EM studies further supported the notion. The results also show that preischemic HG causes early DNA fragmentation in the neocortex, but not in the hippocampus, as detected by TUNEL, PFGE, and LM-PCR. It is clear that hyperglycemia does not cause another type of injury but simply modifies the severity of injury at a given time point. Hyperglycemia during ischemia causes an early activation of a cell death pathway and early DNA fragmentation compared with normoglycemia. Ultrastructural studies demonstrate that HG animals show high amplitude mitochondrial swelling in the early recirculation period.
Hyperglycemia or diabetes has been known to cause exaggerated brain damage when rats or other species are subject to transient forebrain and global ischemia or short periods of focal ischemia (Siesjö et al., 1996; Li and Siesjö, 1997). The mechanisms involved have not been defined. Much effort has been made in exploring cell death pathways after ischemic insults. It is now believed that under adverse conditions, such as calcium overload or free radical generation, mitochondria form a permeability transition pore, known as MPT. Antiapoptotic genes such as bcl-2, bcl-x, or bcl-XL inhibit and proapoptotic genes such as Bax, Bad, or Bid promote the formation of MPT. MPT leads to mitochondrial membrane depolarization and the release of mitochondrial matrix toxic materials such as apoptotic inducing factor and cytochrome c into the cytoplasm. Apoptotic inducing factor, or cytochrome c, or both, further activate a family of caspases that finally results in DNA fragmentation and eventually cell death (Hara et al., 1997; Chen et al., 1998; Endres et al., 1998; Green and Reed, 1998; Hirsch et al., 1998; Namura et al., 1998; Thornberry and Lazebnik, 1998; Ouyang et al., 1999). Apoptotic inducing factor can also cause DNA fragmentation through caspase-independent mechanisms. The current results demonstrated that cytochrome c release was dramatically increased in HG animals after 60 minutes of recirculation and, correspondingly, activated caspase-3 was detected in the same brains, suggesting that DNA fragmentation may be secondary to mitochondrial damage.
Hyperglycemia induces both high and low molecular weight DNA fragmentation, with both blunt-ended and staggered fragments. These suggest that more than one endonuclease was involved. Acidosis has been shown to activate DNase II (Barry and Eastman, 1993). Recently it has been reported that acid conditions also can modulate caspase activation (Matsuyama et al., 2000) and that a caspase-activated endonuclease CAD/DFF40 is responsible for both high and low molecular weight DNA scission (Nagata, 2000; MacManus and Buchan, 2000).
It has not been clear whether brain damage that is exaggerated by ischemia in HG animals is of the apoptotic or necrotic types. The current study attempts to identify whether HG-mediated ischemic brain damage is of the apoptotic or necrotic types using well-established biochemical techniques such as PFGE and LM-PCR. The current results clearly show that both high molecular weight and low molecular weight DNA fragments and both blunt-ended and stagger-ended fragments are detected in the neocortex in HG animals. TUNEL stain reviewed that the distribution of TUNEL-positive cells is located in structures recruited by HG in the damage process, such as cingulate cortex, neocortex, and thalamus. Electron microscopy studies revealed that the positive TUNEL-stained neurons did not show the typical features of apoptosis. Thus, although nuclear chromatin margination was evident, severe edema and mitochondrial swelling coexisted in the same neuron. These findings are consistent with previous reports showing that although DNA fragments of an electrophoretic laddered pattern, which is a biochemical hallmark of apoptotic cell death (MacManus et al., 1993, 1994, 1995; Choi, 1996; Walker and Sikorska, 1997), have been detected after ischemia, ultrastructural microscopic studies have failed to prove that cell damage after ischemia is apoptosis in rats or gerbils (Desphande et al., 1992; Petito and Pulsinelli, 1984; Colbourne et al., 1999). Ischemic brain damage may have both components of apoptosis and necrosis (MacManus and Buchan, 2000).
Although EM analysis was performed to provide information on whether classical apoptosis or necrosis was responsible for the cell death, the analysis gave additional information that preischemic HG leads to massive mitochondrial swelling in the early recirculation periods in HG, but not in NG animals. The mechanisms responsible are not known, but it is tempting to speculate that the influence of a sustained decrease intracellular pH is responsible (Kristián et al., unpublished data). If the major effect of HG and enhanced acidosis is on mitochondrial integrity, it is possible, at least theoretically, to suggest that an “apoptotic” cascade triggered by apoptosis-inducing factors is responsible for the activation of endonuclease. However, it remains to be explored whether an increased glucose concentration or low pH, or both, exert a direct effect on endonucleases triggering DNA fragmentation.
In summary, preischemic HG clearly alters the pattern of cell death after a long ischemic period because TUNEL-positive cells were abundant, because both high molecular and low molecular weight DNA fragments were detected, and because both blunt-ended and stagger-ended fragments were present. Hyperglycemia accelerated the processes that are normally observed after days of recovery in NG animals. Although biochemical data suggested that HG activated an apoptotic cell death pathway, the final outcome by EM criteria may not necessarily be classical apoptosis. The results reported in the current study suggest that mitochondrial damage with release of cytochrome c is a conspicuous feature of the damage exaggerated by HG. It remains to be shown if glucose and acidosis exert a direct effect on the endonucleases involved.