
Abstract
Cyclooxygenase-2-derived prostaglandin E2 (PGE2) contributes to excitotoxic and ischemic neuronal cell death by engaging neuronal PGE2 type 1 receptors (EP1R). Our previous studies have shown that EP1R signaling resulted in disturbances of intracellular Ca2 + homeostasis and suppression of the pro-survival protein kinase AKT. The aim of this study was to investigate whether these pathophysiological mechanism have a role in the neuronal cell death after transient forebrain ischemia. Mice were subjected to ischemia/reperfusion by bilateral common carotid artery occlusion. Hippocampal cornu ammonis area 1 (CA1) neuronal cell death was determined 5 days after reperfusion. Animals treated with the EP1R antagonist SC51089 or EP1R-deficient mice (EP1 –/–) showed significantly less neuronal injury as compared to vehicle-treated wild-type controls. Benefits of EP1R blockage were still evident 14 days after injury. Better neuronal survival was correlated with reduced neuronal caspase-3 activity and decreased nuclear translocation of the apoptosis-inducing factor. Neuroprotection could be reverted by intracerebroventricular administration of the phosphoinositide 3-kinase inhibitor LY294002 and was not further increased by the calcineurin inhibitor FK506. These data implicate EP1R in postischemic neuronal apoptosis possibly by facilitating AKT inhibition.
INTRODUCTION
The PGE2 receptor subtype 1 (EP1) expressed on neurons is the downstream effector of cyclooxygenase 2 (COX-2)-mediated neurotoxicity 1 and has emerged as an attractive therapeutic target in different brain injury modalities. 2 While EP1 receptor (EP1R) null mice are protected from ischemic brain injury, inhibition of EP1R reduces focal cerebral ischemic injury with a wide therapeutic window, both in male and female mice. 3 However, the mechanisms by which EP1R contribute to brain damage have not been elucidated in full. Ischemic neuronal death is, in part, mediated by increased Ca2+ flux through the N-7V-methyl-D-aspartate (NMDA) receptor and consequent disruption of intracellular Ca2+ homeostasis. 4 There is evidence that EP1Rs are involved in the regulation of intracellular Ca2+ homeostasis. 1 For example, activation of EP1R, a G-protein-coupled receptor, leads to an increase in intracellular Ca2+, an effect that does not involve endoplasmic Ca2+ stores or phospholipase C activity, but is related to extracellular Ca2+ levels.5–7 In the setting of excitotoxicity, although EP1Rs do not contribute to the Ca2+ influx through NMDA receptors and voltage-gated Ca2+ channels, their inhibition markedly improved neuronal Ca2+ homeostasis, which was due to improved Na + /Ca2+ exchanger function. 1 These observations, collectively, indicate that EP1R could contribute to ischemic brain injury by aggravating the neuronal Ca2+ dysregulation induced by cerebral ischemia. Re-establishing Ca2+ homeostasis has obvious benefits for neuronal survival, but the downstream effectors of this beneficial effect have not been elucidated. In organotypic hippocampal slice cultures, EP1R inhibition increases AKT phosphorylation under basal conditions and after oxygen-glucose deprivation, suggesting that EP1R may worsen the injury by negatively regulating the activity of the prosurvival kinase AKT. 8 Recent evidence indicates that calcineurin, a Ca2+/calmodulin-dependent serine/threonine phosphatase highly expressed in neurons, inhibits the AKT pathway by de-phosphorylating AKT. 9 This observation raises the possibility that the reduced intracellular Ca2+ associated with EP1R inhibition suppressed calcineurin activity resulting in AKT activation and neuroprotection. However, a link between EP1R, AKT, and calciuneurin has not been established in the setting of cerebral ischemia. Furthermore, it remains unclear whether these mechanisms established in hippocampal slice cultures are also active in vivo. Therefore, in the present study, we used a mouse model of forebrain cerebral ischemia, which induces extensive damage to the hippocampal cornu ammonis area 1 (CA1) area, to investigate the role of AKT and calcineurin as downstream effectors of EP1R-mediated hippocampal damage.
MATERIALS AND METHODS
Animal Preparation
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Animal procedures followed institutional guidelines, which meet or exceed NIH standards. EP1 and COX2 null mice were obtained from in-house colonies and were congenic with the C57BL/6 strain. Therefore, C57BL/6 mice were used as wild-type controls. Experiments were performed in 10- to 12-week old male mice, housed under SPF conditions, and maintained on a 12-hour light/dark cycle with free access to food and water.
Mice were anesthetized with a mixture of isoflurane (1.8% to 2%), oxygen (30%), and nitrogen (70%). A fiber optic probe was glued to the parietal bone (3 mm lateral, 2 mm caudal to bregma on both sides) and connected to a laser-Doppler flowmeter (Periflux System 5000; Perimed, Jarfalla, Sweden) for continuous monitoring of cerebral blood flow in the neocortex. In some mice, the femoral vessels were cannulated for recording of arterial pressure and collection of blood samples. Body temperature was monitored and maintained between 36.5°C and 37.5°C using a thermostatically controlled heating pad during surgery. Through a midline incision of the neck, 4–0 surgical threads were loosely placed around both common carotid arteries (CCAs). Two minutes before bilateral CCA occlusion (BCCAo), isoflurane was adjusted to 1.1%. Both CCAs were transiently occluded by tying the threads for 20 minutes. Isoflurane levels were lowered to 0.5% after untying the threads (reperfusion). Because our preliminary study showed that the posterior communicating arteries (PcomA) were absent or atretic in mice showing less than 10% of baseline cerebral blood flow within the first minute of occlusion, mice with < 90% cerebral blood flow reduction were excluded. After closure of the incision and discontinuation of anesthesia, mice were allowed to recover for 24 hours in a temperature-controlled chamber regulated to maintain rectal temperature at 36.5°C to 37.5°C to prevent hypothermia. Thereafter, mice were returned to general housing (22°C to 24°C). Sham-operated mice underwent the same procedures, except that their arteries were not occluded. Twenty-one percent of all animals used in this study were excluded because of insufficient cerebral blood flow reduction. Overall, 10% of animals died during or immediately after surgery. No further losses were observed after recovery from surgical anesthesia. The percentage of excluded animals was similar across different genotypes and treatment groups.
Vascular Anatomy and Assessment of Neuronal Cell Death
For investigation of intracranial vasculature, mice were transcardially perfused with a short flush of saline solution and then with 4% paraformaldehyde, followed by a mixture of gelatin and India ink prepared according to Berry et al. 10 The brains were carefully removed and placed in 4% paraformaldehyde. A photographic image of the PcomA was taken and the diameter of the PcomA on each side as well as the basilar artery was determined using NIH ImageJ software (NIH, Bethesda, MD, USA). When the PcomA had no anastomosis or was less than one third of the diameter of the basilar artery, the PcomA was defined as PcomA (−). PcomA (+) was assigned when the artery diameter was more than one third of the diameter of the basilar artery and had a visible anastomosis with the middle cerebral artery. 11 Vascular anatomy was studied in 107 wild type (WT) C57BL/6 and 30 EP1 – / – mice. Fifty-five percent WT C57BL/6 and 57% EP1 –/– mice had bilateral missing or atretic PComA while 41% WT and 40% EP1 –/– mice showed unilateral functional PComA.
Hippocampal cell death was assessed according to standard protocols by a study investigator masked to the treatment allocation. 12 Briefly, the brains were quickly removed, frozen, and cut in 8-mm-thick sections at −1.6, −1.7, and − 1.8 mm posterior to bregma and stained with hematoxylin and eosin. Injury to the CA1 subfield was evaluated in the medial and intermedial portion of the CA1 spanning a distance of 1,000 μm in each section under a light microscope at 100 × magnification by manually counting the viable neurons using NIH ImageJ. Viable neurons were defined as those with a blue hue, an intact plasma membrane, and a hypochromatic oval or round nucleus. 12
Drug Treatments
Mice were randomly assigned to vehicle or treatment groups. All drugs were used at concentrations previously demonstrated to be effective in vivo. The EP1R antagonist (SC51089, 10 μg/kg in physiological saline, intraperitoneal; Biomol, Plymouth Meeting, PA, USA) was administered 5 minutes and 6 hours after reperfusion and, thereafter, twice per day at days 1, 2, 3, 4, and once at day 5. This concentration has been shown to specifically inhibit EP1R-mediated excitotoxicity in mice. 1 For determination of therapeutic time window, SC51089 administration was started 2 or 6 hours after reperfusion. The COX-2 inhibitor NS398 (20 mg/kg in H2O, intraperitoneal; Cayman Chemical, Ann Arbor, MI, USA) was given 5 minutes and 6 hours after reperfusion, and, thereafter, twice per day at days 1, 2, 3, 4, and once at day 5. 13 The COX-1 inhibitor SC560 (5 mg/kg in dimethyl sulfoxide (DMSO), intrapertioneal; Cayman Chemical) was given 5 minutes and 6 hours after reperfusion, and, thereafter, twice per day at days 1, 2, 3, 4, and once at day 5. 14 The calcineurin inhibitor FK506 (3 mg/kg in 7.5% DMSO/H2O, intraperitoneal; Sigma) or vehicle (7.5% DMSO) were given 30 minutes before ischemia. This concentration was previously shown to be effective in vivo.15,16 The PI3K inhibitor LY294002 (2 μl, 25 μM in 50% DMSO/PBS; Sigma) or vehicle (50% DMSO/PBS) were injected in both cerebral ventricles (anteroposterior 1.7 mm, lateral 1.2 mm from bregma, depth 1.7 mm) using a pulled glass micropipette 30 minutes before ischemia. The effectiveness of this concentration and administration modality have previously been reported. 17
Caspase-3 Activity Assay
Caspase-3 activity was assayed essentially as described. 18 Briefly, hippocampi were homogenized in 25 mM Hepes, pH 7.4, 0.1% Triton X-100, 5mM MgCl2, 2mM DTT, 1.5 mM EDTA, 1 mM EGTA, 2 μM antipain, 0.15 μM aprotinin, 15 μM pepstin, 20 μM leupeptin. Lysates were cleared by centrifugation at 13,000g at 4°C for 5 minutes and incubated with the fluorescent substrate ac-DEVD-afc in freshly made caspase assay buffer (20 mM Hepes, pH 7.4, 100mM NaCl, 1 mM EDTA, 0.1% Chaps, 10% sucrose, 10 mM DTT) for 60 minutes at 37°C. Protein concentration in each sample was measured using a detergent compatible protein assay kit (Bio-Rad, Hercules, CA, USA). The fluorescence intensity generated by the caspase-3 activity was measured using a fluorescence plate reader (Perkin-Elmer, Waltham, MA, USA) using a 400 nm excitation filter and a 505 nm emission filter. The fluorescence reading of each sample was normalized with its protein content and expressed as normalized caspase-3 activity in fluorescence intensity (FI/mg protein).
Immunohistochemistry
Mice were anesthetized with sodium pentobarbital (120 mg/kg, intraperitoneal) and transcardially perfused with saline followed by 4% buffered paraformaldehyde. Brains were removed and postfixed with 4% paraformaldehyde and cryoprotected in sucrose. Serial coronal sections were cut at a thickness of 14 μm starting at − 1.7 mm from the bregma. Sections were blocked and incubated overnight with cleaved caspase 3 (1:100, rabbit polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or AIF (1:50, rabbit polyclonal, Santa Cruz) specific antibodies, followed by goat anti-rabbit-F488 antibody (1:200, Invitrogen, Carlsbad, CA, USA). Nuclei were visualized using TO-PRO 3 (Invitrogen). Images were acquired on a confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA) and image stacks were collapsed as an average projection using NIH imageJ.
Western Blotting
Detection of AKT in hippocampal lysates was carried out as previously described. 8 Antibodies specific for phospho-Threonine 308-AKT (p-AKT) and total-AKT (t-AKT) were from Cell Signaling (Danvers, MA, USA).
PGE2 Measurement
Tissue PGE2 levels were measured at different reperfusion times (1, 2, 4, 6, 24, 48, and 72 hours) after transient forebrain ischemia (n = 4 per time point). At indicated time points, animals were deeply anesthetized with isoflurane, decapitated, and brains removed. Care was taken to limit the time for tissue harvesting to less than 30 seconds. After immersion in liquid nitrogen for 18 seconds, brains were equilibrated on dry ice for 30 minutes. Brains were mounted on a freezing microtome and tissue was sectioned until the appearance of the hippocampal formation. The CA1 region was collected using a brain punch (diameter 0.5 mm, Stoelting, Wood Dale, IL, USA). Tissue extracts were prepared, purified over C18 cartridge columns, and ELISA was performed according to manufacturer's instructions (Cayman Chemicals).
Statistical Analysis
Data are expressed as mean ± s.e.m. Sample numbers were determined by power analysis based on the results of a previous study. 19 We used a power of 0.8 and a significant level of 0.05. Multiple comparisons were evaluated by the analysis of variance and Tukey's multiple comparison test. Differences were considered significant at P ≤ 0.05.
RESULTS
EP1R Deletion or Pharmacological Inhibition Reduces CA1 Neuronal Death after Transient Forebrain Ischemia
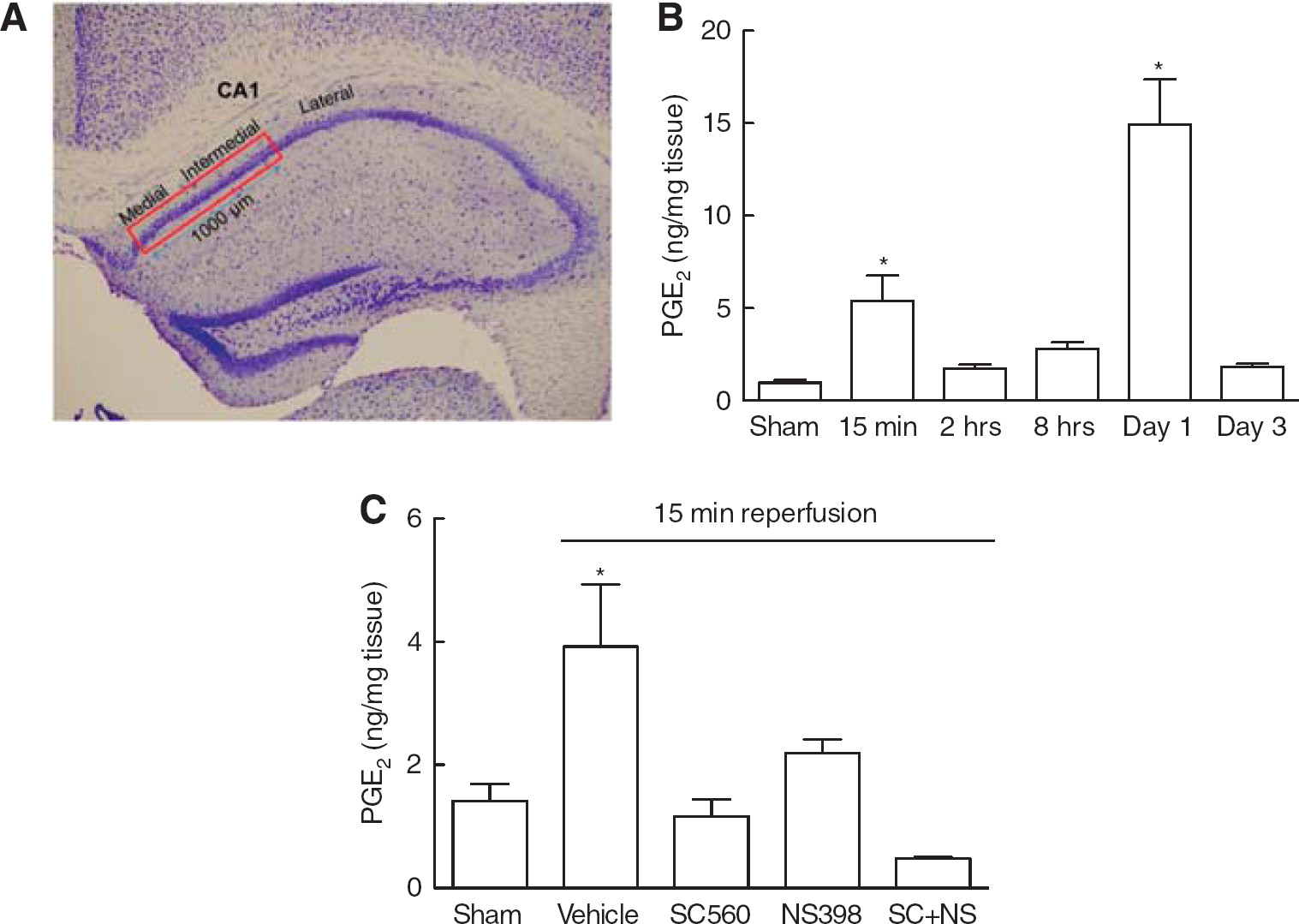
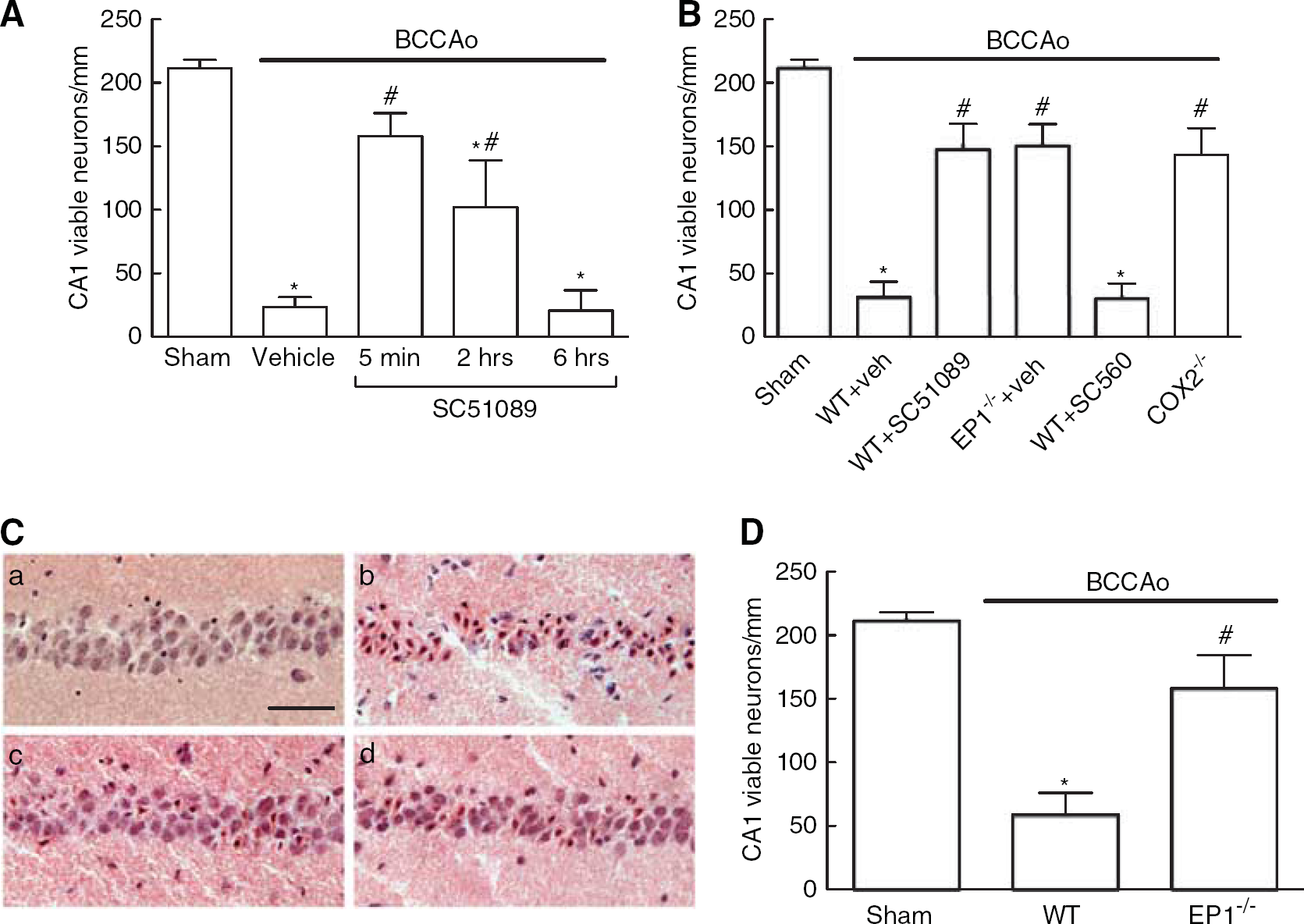
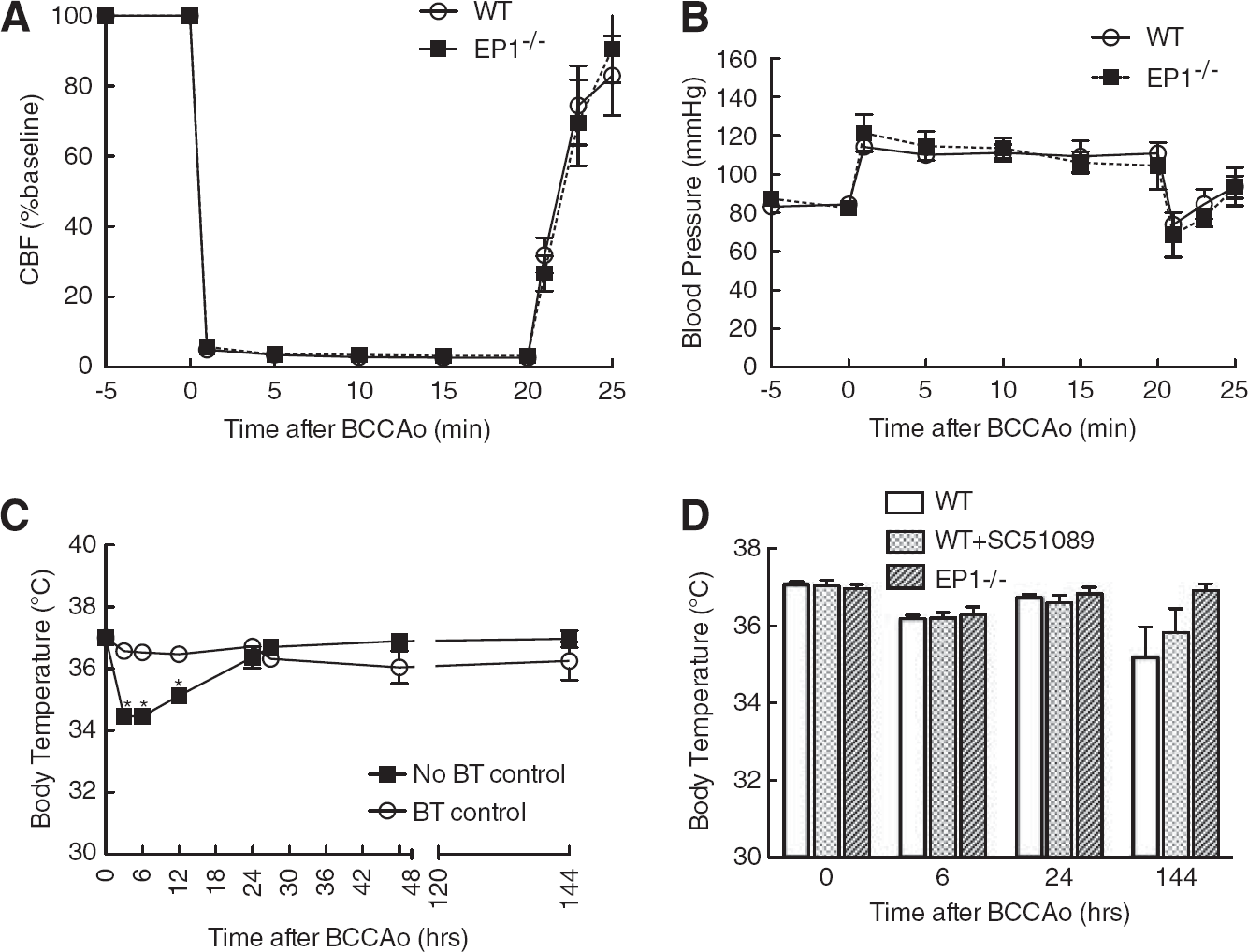
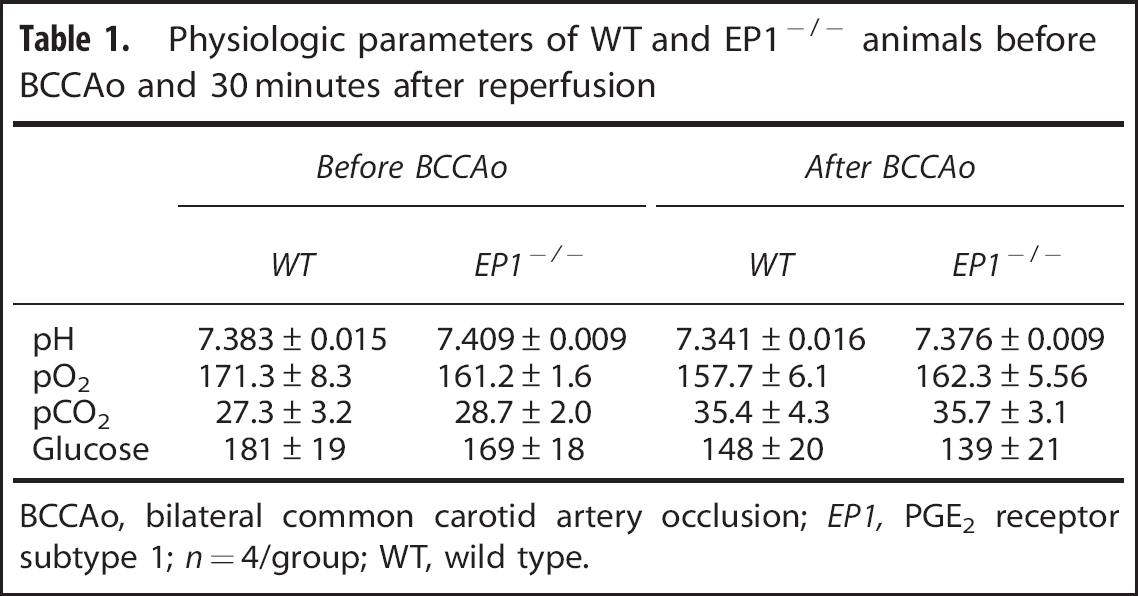
Cellular analysis of the medial and intermediate portion of the CA1 region (Figure 1A) showed maximal neuronal loss 5 days after BCCAo (data not shown). In agreement with previous studies in the gerbil, 20 post ischemic hippocampal PGE2 production in the mouse showed a biphasic profile with peaks at 15 minutes and 1 day after reperfusion (Figure 1B). Next, we analyzed the relative contribution of COX-1 and COX-2 to the PGE2 production during the early phase after ischemia by treating the animals with the selective inhibitors SC560 or NS398, respectively. PGE2 levels were suppressed in animals treated with either SC560 or NS398 indicating that both COX isoforms were contributing to the PGE2 production at 15 minutes after reperfusion (Figure 1C). Administration of the EP1R antagonist SC51089 5 minutes and 2 hours, but not 6 hours after reperfusion resulted in significant reversal of CA1 neuronal cell death after BCCAo (Figure 2A). Because the effect was strongest at 5 minutes, this time point was chosen for subsequent experiments involving pharmacological receptor inhibition. Additionally, EP1–/– mice undergoing BCCAo showed similar neuroprotection as SC51089-treated wild-type animals (Figure 2B). Wild-type animals treated with the COX-1 specific inhibitor SC560 showed no reduction in ischemic cell death, while mice deficient in COX-2 showed less damage compared to WT mice (Figure 2B). This suggests that similar to NMDA-induced excitotoxicity and cortical ischemic neuronal injury, 1 COX-2 but not COX-1-derived PGE2 contributes to neuronal cell death in this model. Reduced CA1 neuronal cell death was evident at least 14 days after BCCAo in EP1 –/– mice indicating that sustained neuroprotection has been achieved (Figure 2D). There was no difference in cerebral blood flow, body temperature, blood pressure, serum glucose, and arterial blood gas between WT mice and EP1 –/– mice undergoing BCCAo (Figures 3A, 3B, and 3D and Table 1). As previously reported, 21 stabilizing body temperature after surgery by controlling the environmental temperature for the first 24 hours was crucial to prevent post ischemic hypothermia, a potential confounder of stroke outcome (Figure 3C).
(
(
(
Physiologic parameters of WT and EP1 −/− animals before BCCAo and 30 minutes after reperfusion
BCCAo, bilateral common carotid artery occlusion; EP1, PGE2 receptor subtype 1; n = 4/group; WT, wild type.
Reduced Post-Ischemic Caspase-3 Activation and Apoptosis-Inducing Factor Nuclear Translocation in EP1 –/– Mice
Delayed ischemic cell death after global cerebral ischemia shows features of programmed cell death encompassing caspase-dependent and -independent pathways.22,23 Activation of terminal caspases has a pivotal role in neuronal cell death. 24 Consequently, caspase-3 activity was assessed in the post-ischemic hippocampus using an antibody that specifically binds to proteolytically processed (i.e. activated) caspase-3. Activated caspase-3 was observed in the cytoplasm of CA1 neurons in WT mice 3 days after ischemia and was associated with chromatin condensation and nuclear shrinkage (Figure 4A). Activated caspase-3 immunoreactivity was strongly reduced in neurons of EP1 –/– mice, while nuclear integrity and chromatin structure were largely preserved (Figure 4A). Increased caspase-3 activity was also observed using a fluorimetric activity assay at 8 and 72 hours after ischemia in WT mice, an effect significantly reduced in EP1 –/– mice at both time points (Figure 4B).
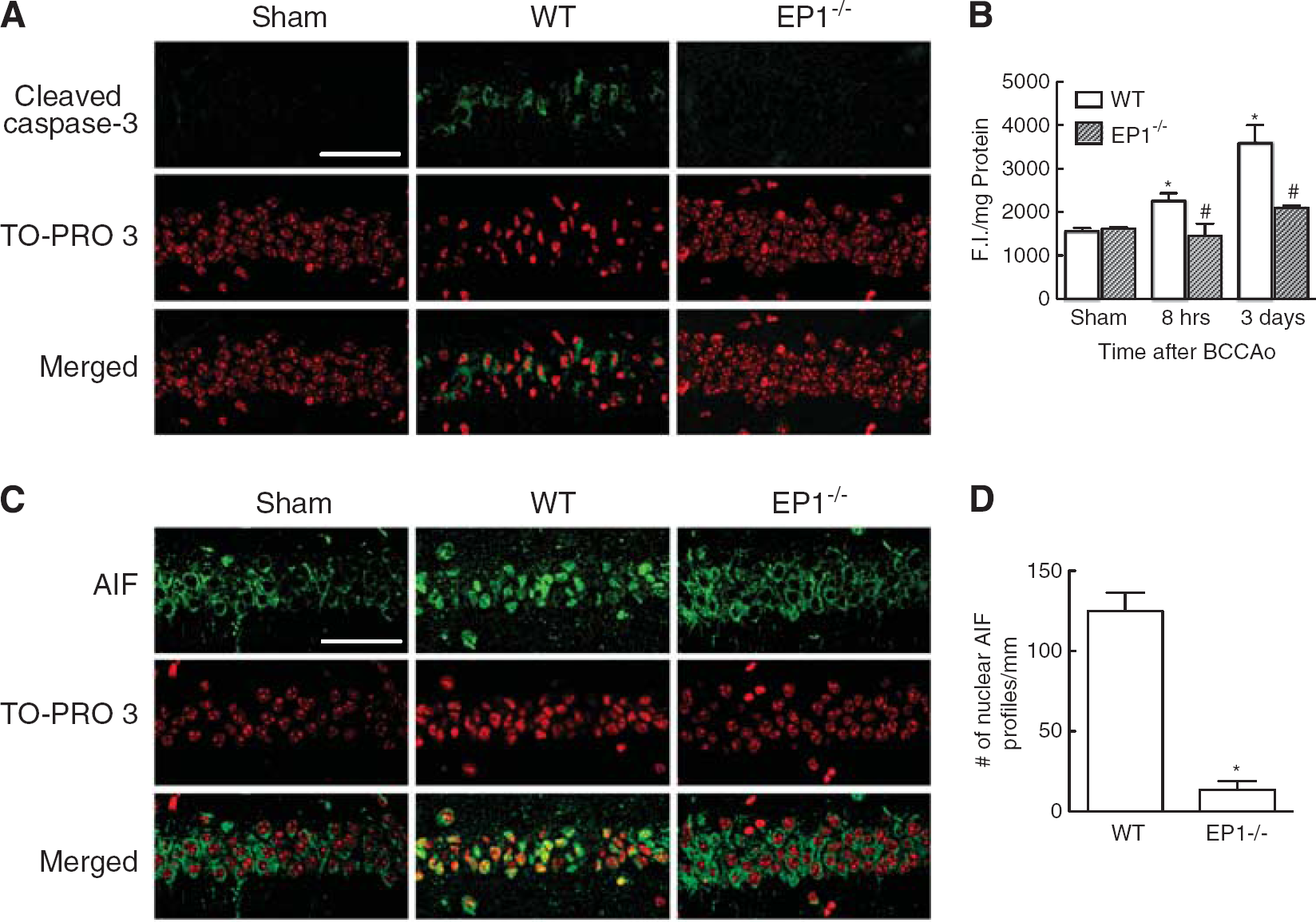
(
Caspase-independent apoptotic pathways include the activation of AIF, a mitochondrial-associated protein that translocates to the nucleus after initiation of the pro-apoptotic signaling cascade where it promotes chromatin condensation and large-scale DNA fragmentation.25,26 In cerebral ischemic injury, nuclear translocation of AIF is associated with neuronal cell death. 27 In sham-operated mice AIF immunoreactivity was localized in the cytoplasm of CA1 neurons, whereas most of AIF immunoreactivity co-localized with the nuclear marker TO-PRO 3 one day after ischemia (Figure 4C). In contrast, most of the AIF signal remained cytoplasmic in EP1 –/– mice (Figures 4C and 4D). Thus, both, caspase-dependent and -independent cell death pathways are inhibited in animals lacking EP1R.
Possible Roles for AKT and Calcineurin in EP1R Mediated Neurotoxicity
Previous in vitro studies have identified the AKT pathway as being involved in neuroprotection granted by EP1R inhibition. 8 AKT regulates various apoptotic pathways including caspase activation and AIF nuclear translocation.28,29 Therefore we hypothesized that AKT constitutes an important signaling module upstream of caspase-3 and AIF activation. Activatory AKT phosphorylation was severely diminished in WT mice after BCCAo, but was significantly less compromised in EP1 –/– mice (Figure 5A). We examined whether the protective phenotype observed in EP1 –/– mice could be counteracted by AKT inhibition. AKT activity is dependent on phosphatidylinositol-(3,4,5)-triphosphate (PIP3) generated by the phosphoinositide 3-kinase (PI3K), the activity of which can be inhibited by LY29 4 0 02.30 WT and EP1 –/– mice were administered i.c.v. with LY294002 before BCCAo and neuronal survival was determined after 5 days. LY294002 did not reduce the number of viable CA1 neurons in sham animals, but did so in WT animals undergoing ischemia and particularly it reverted the protective phenotype of EP1 –/– mice (Figure 5B). Thus, neurotoxic PGE2 signaling through the EP1R involves inactivation of the AKT pathway leading to enhancement of pro-apoptotic signaling after cerebral ischemia.
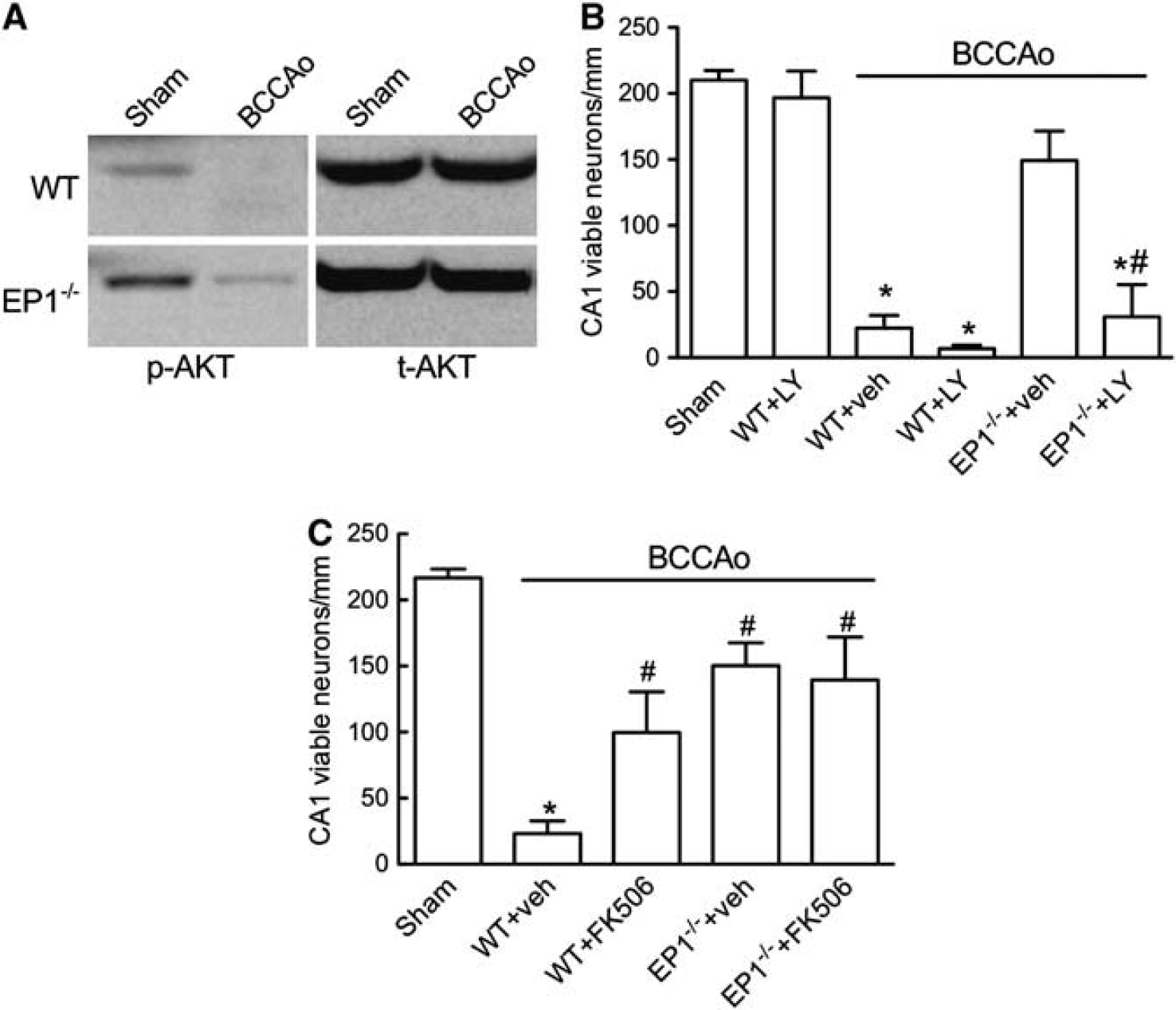
(
AKT activity is controlled by the inducible phosphorylation of serine and threonine residues. 31 Calcineurin, a Ca2+/calmodulin regulated protein phosphatase, has been implicated in suppressing AKT activity by de-phosphorylating Ser 473 and Thr 308. 9 Calcineurin is highly expressed in neurons and inhibiting calcineurin confers neuroprotection in several ischemic models. 32 Given the role of EP1R in prolonging postischemic intracellular Ca2+ dysregulation, 1 we hypothesized that EP1R signaling could contribute to AKT inactivation by enhancing calcineurin activity. To address the role of calcineurin in our injury model, we treated mice undergoing BCCAo with the calcineurin inhibitor FK506. FK506 significantly increased CA1 neuronal viability in WT mice, but failed to further improve neuronal survival in EP1 –/– mice suggesting that calcineurin activity might be already minimal in EP1 –/– animals (Figure 5C).
DISCUSSION
Transient forebrain ischemia, a model of cardiac arrest-associated neuronal injury, induces delayed neuronal cell death in the CA1 region of the hippocampus. 12 Cyclooxygenases and prostaglandins have been associated with neuronal injury in this model. In the gerbil, ischemic cell death after transient forebrain ischemia is reversed by the COX-2 inhibitor rofecoxib and the COX-1 inhibitor valeroyl salicylate. 20 Furthermore, pharmacological inhibition or genetic deletion of COX-2 is protective in global cerebral ischemia in rats and mice.33,34 Transient forebrain ischemia induces PGE2 production with COX-1 contributing to the early phase of PGE2 release minutes to hours after ischemia while COX-2 derived PGE2 peaks days after the insult.20,35 In agreement with these studies in gerbils and cats, we observed biphasic PGE2 production after BCCAo in the mouse, resulting in PGE2 peaks immediately following reperfusion and at 24 hours (Figure 1B). Because we observed that EP1R inhibition was most effective when SC51089 was administered immediately after reperfusion but not when applied after 6 hours (Figure 2A), we hypothesized that early PGE2 production can be at least partially attributed to increased COX-2 activity shown to be upstream of EP1R in other ischemic models in the mouse. 1 Indeed, we found that PGE2 levels after 15 minutes reperfusion were reduced by COX-2 and COX-1 inhibition, indicating that the activity of both isoforms was increased (Figure 1C). However, in contrast to COX-2 deletion, COX-1 inhibition could not revert the ischemic neuronal cell death after BCCAo (Figure 2B). While in the present study we did not observe an involvement of COX-1-derived prostaglandins in ischemic neuronal injury, previous studies in the gerbil have shown protection by COX-1 and COX-2 inhibition. 20 Possibly this discrepancy could result from the fact that different inhibitors were used in both studies. SC560, the COX-1 inhibitor used in this study, has an IC50 for COX-2 that is nearly 1,000-fold higher than that for COX-1. 36 In contrast, valeroyl salicylate, used as a COX-1 inhibitor in the aforementioned study, 20 shows only a 20-fold selectivity over COX-2. 37 This raises the possibility that cross-inhibition could be a factor in the protection afforded by valeroyl salicylate. The failure of COX-1 inhibition to improve neuronal survival is in agreement with the role of COX-1 in excitotoxic brain injury, where COX-1 contributes substantially to PGE2 production but does not promote neurotoxicity.14,38 The inability of COX-1-derived PGE2 to induce neurotoxicity could be due to spatial constraints that limit the interaction of COX-1-derived PGE2 with neuronal EP1R. This notion is supported by the fact that COX-1 expression in the mouse cerebral cortex is mainly associated with microglia, whereas COX-2 is associated with neurons. 14 Therefore, unlike COX-2-derived PGE2, which could act as an autocrine agent, COX-1-derived PGE2 may not have easy access to neuronal EP1R.
The EP1R has been identified as the transducer of PGE2 mediated neurotoxicity in the setting of NMDA-mediated neuronal injury and focal cerebral ischemia.1,39 Neuronal cell death in these models is partially mediated by excitotoxicity with concomitant disturbance of intracellular Ca2+ homeostasis. We have previously shown that signaling through the EP1R contributes to the disruption of intracellular ion homeostasis after NMDA receptor activation. 1 In the present study, we set out to determine whether EP1R contribute to delayed neuronal cell death after transient forebrain ischemia. Using pharmacological inhibition and EP1R deletion, our data suggest that EP1Rs are also involved in delayed neuronal cell death. Our data are in agreement with a recent study showing that EP1 –/– mice were protected after global forebrain ischemia. 40 In our study, we further characterize the role of EP1R in this model by showing that the protective effect of EP1R deletion was coupled to the inhibition of caspase-dependent and -independent pathways resulting in diminished caspase-3 activation and reduced nuclear AIF accumulation (Figure 4).
The prosurvival AKT protein kinase is a critical molecule in the control of neuronal survival in this model, and AKT activity is suppressed by EP1R activation in an in vitro model of hippocampal neuronal cell death. 8 Furthermore, AKT has been implicated in the suppression of caspase-3 and AIF-dependent cell death pathways.29,41 Here we show that baseline and post ischemic AKT phosphorylation was increased in EP1 –/– mice (Figure 5A), a finding consistent with the view that EP1R signaling suppresses AKT activity. In addition, we investigated the role of AKT in EP1R-mediated neurotoxicity after BCCAo by pharmacological inhibition of the AKT pathway by the PI3K inhibitor LY294002. We found that LY294002 abolished the protection conferred by EP1R deletion, hinting that AKT contributes to the neuroprotection observed in EP1 –/– mice (Figure 5B).
In an in vivo model of neuronal cell death the Ca2+ /calmodulin-dependent protein phosphatase calcineurin has been shown to interact with and dephosphorylate AKT thereby diminishing its activity. 9 Calcineurin is highly expressed in hippocampal neurons and has been implicated in neuronal injury in the past.32,42 Given the role of EP1R in regulating intracellular Ca2+ levels, in the present study we hypothesized that lower post ischemic intracellular Ca2+ levels in hippocampal neurons of EP1 –/– mice might decrease calcineurin activity resulting in sustained AKT activation. Consistent with this hypothesis, we found that the calcineurin inhibitor FK506 failed to increase protection in EP1 –/– mice, suggesting that calcineurin activity might already be reduced in EP1 –/– animals (Figure 5C). Although, the results are consistent with our hypothesis, we cannot exclude that inhibiting calcineurin activity might also preserve other neuro-protective pathways such as the transcription factor CREB or block proapoptotic signals such as bad activation.43,44 However, the fact that the calcineurin inhibitor FK506 was able to rescue CA1 neurons in WT but not EP1 –/– mice identifies lowered calcineurin activity as a possible mechanism for the neuroprotection afforded by EP1R deletion.
In summary, we have identified EP1R as an activator of apoptotic neuronal cell death after transient forebrain ischemia. We show that pharmacological inhibition or genetic deletion of EP1R reduces post ischemic neuronal cell death possibly in an AKT dependent manner, and we provide suggestive evidence that reduced calcineurin activity might mediate this effect. These findings add further support to the hypothesis that EP1R inhibition is a valuable strategy to counteract the deleterious effects of cerebral ischemia and, possibly, other brain pathologies as well.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
We thank Jasmine Tan for excellent technical assistance.