
Abstract
The mechanisms underlying secondary cell death after traumatic brain injury (TBI) are poorly understood. Animal models of TBI recapitulate many clinical and pathologic aspects of human head injury, and the development of genetically engineered animals has offered the opportunity to investigate the specific molecular and cellular mechanisms associated with cell dysfunction and death after TBI, allowing for the evaluation of specific cause-effect relations and mechanistic hypotheses. This article represents a compendium of the current literature using genetically engineered mice in studies designed to better understand the posttraumatic inflammatory response, the mechanisms underlying DNA damage, repair, and cell death, and the link between TBI and neurodegenerative diseases.
Keywords
Traumatic brain injury (TBI) is a major cause of mortality and disability in Europe and the United States, with more than 2.5 million individuals in the U.S. alone living with the devastating emotional and economic costs (NIH Consensus Development Panel on Rehabilitation of Persons With Traumatic Brain Injury, 1999). During the past decade, the elucidation of “primary” (mechanical) injury and “secondary” insults after head injury, improvement in pre-hospital care, classification of injury severity, refinement of radiologic imaging, prompt evacuation of surgical masses, and advances in critical care and rehabilitation have led to a proficient level of care that has been summarized in guidelines for the advanced treatment of head-injured patients (The Brain Trauma Foundation, 2000). Despite these advances, many head-injured patients die or survive with significant brain damage and behavioral impairment, even after mild or moderate head injury (Jennett, 1997; Graham et al., 2000a). Currently, no treatment is available to reverse the pathogenic cellular cascade underlying progression of cell death. A better understanding of the molecular and cellular mechanisms leading to posttraumatic cell death and their relation to behavioral impairment remains an important goal of experimental TBI research.
The pathobiologic processes that occur after brain injury are known to be associated with primary injury (caused by physical or biomechanical effects of trauma) and secondary or delayed events initiated minutes after trauma and lasting for weeks, months, or possibly years. These pathologic injury cascades are believed to be associated with alterations in gene expression and the up-regulation and release of a number of potentially damaging or restorative neurochemical factors that interact in a complex network leading to delayed cellular dysfunction, death, or both. The extended nature of these cascades offers the possibility for therapeutic interventions (alone or in combination) aimed at blocking degeneration or improving repair and regeneration (McIntosh et al., 1998; Graham et al., 2000a).
Animal models of TBI have been developed to recapitulate many clinical and pathologic aspects of head injury and have provided the basis for the dramatic increase in the understanding of the pathophysiology of brain damage after trauma (Povlishock et al., 1994; Laurer and McIntosh, 1999). Experimental models of TBI typically use rodents because of the many advantages—including small size, low cost, and the extensive amount of normative data available (Povlishock et al., 1994). Clinically relevant rodent models of TBI represent a reliable means to permit the use of sophisticated neurochemical, histopathologic, and molecular techniques, and genomic manipulation to help clarify the pathogenic mechanisms underlying trauma-associated cell death.
Properties of genetically engineered animals and rationale for their use
The use of genetically engineered mice typically involves the artificial overexpression or targeted deletion (knockout) of a specific gene. The development of this technology has revolutionized the investigation of the specific molecular and cellular mechanisms associated with cell dysfunction and death after TBI. Currently, mice are the preferred species for these studies because the murine genome has been extensively mapped and is suitable for embryonic cell technology. Different approaches with genetically engineered animals include studies of naturally occurring genetic variability or mutations and the creation of animals with targeted gene deletions or with newly introduced transgenes (Steward et al., 1999). This article summarizes the significant posttraumatic pathways investigated to date using genetically engineered mice, including inflammation and cytokines, nitric oxide and oxidative damage, and DNA damage/repair and apoptosis. In addition, we will overview the studies that have been performed using transgenic mouse technology to understand the epidemiologic link between TBI and neurodegenerative diseases.
INFLAMMATION AND CYTOKINES
Traumatic brain injury results in an acute blood–brain barrier (BBB) opening that allows the entry of leukocytes into the injured brain (Holmin et al., 1995; Soares et al., 1995). These cells (neutrophils and activated macrophages) may release oxygen free radicals causing cellular damage and inflammatory cytokines that have been implicated in posttraumatic neuropathologic damage. Posttraumatic inflammation also is associated with an increase in expression of intercellular adhesion molecules (ICAM) such as ICAM-1, which is involved in vascular adhesion, transendothelial migration of leukocytes, and the release of cytokines such as tumor necrosis factor-α (TNF-α), various interleukins, and various trophic factors (Morganti-Kossman et al., 1997). Initially, the mediators and cytokines involved in this cascade were considered to be pathogenic factors. However, it is now clear that posttraumatic inflammation also may contribute to reparative and regenerative processes after TBI (McIntosh et al., 1998; Shohami et al., 1999; Lenzlinger et al., 2001). Macrophage activation results in removal of dead tissue and debris by phagocytosis, lipid recycling, and secretion of the above-mentioned spectrum of cytokines possessing trophic, mitogenic, and chemotactic properties that may contribute to healing, plasticity, and regeneration (Lotan and Schwartz, 1994).
Intercellular adhesion molecules
Intercellular adhesion molecule-1 is a member of the immunoglobulin family that is responsible for the recruitment of leukocytes to inflammatory foci. The increase in expression of ICAM-1 reported after clinical and experimental brain trauma has been hypothesized to be involved in secondary inflammatory damage (Carlos et al., 1997; Whalen et al., 1997, 1998). Intercellular adhesion molecule-1 knockout mice have been generated by disruption of the fifth exon of the ICAM-1 locus in 129/Sv embryonic stem (ES) cells (Sligh et al., 1993). These mice do not demonstrate any susceptibility to infection, but do exhibit prominent abnormalities of the peripheral inflammatory response, including impaired neutrophil migration in response to chemical peritonitis (Sligh et al., 1993).
Platelet (P)-selectin, a member of the selectin family, mediates platelet and leukocyte rolling on endothelium and platelet–neutrophil adhesion. Generation of P-selectin knockout mice is obtained through the deletion of exons 3–5. Although dual ICAM-1 and P-selectin (−/−) mice completely lack neutrophil migration during peritoneal inflammation, they develop normally, are fertile, and appear to be healthy (Bullard et al., 1995). To clarify the role of ICAM-1 and P-selectin after TBI, ICAM-1 (−/−) and dual ICAM-1 and P-selectin (−/−) mice were subjected to controlled cortical impact (CCI) brain injury and were compared with wild type (WT) mice. Surprisingly, these studies failed to reveal any differences in brain neutrophil accumulation at 24 hours postinjury, memory and motor function, or contusion volume at 3 weeks after injury (Whalen et al., 1999a, 2000). These data suggest that after TBI, brain neutrophil accumulation may be stimulus-specific and not dependent on ICAM-1, or that these knockout mice may develop a compensatory adhesion pathway. Further studies are warranted to understand the difference between results obtained with genetically engineered mice and pharmacologic agents such as antibodies directed to ICAM-1 that reduce by almost 40% the invasion of neutrophils into the injured brain during the first 24 hours after injury (Carlos et al., 1997). However, brain edema was observed to be decreased at 24 hours after TBI in dual P-selectin and ICAM-1 (−/−) mice, suggesting a possible role for adhesion molecules (particularly P-selectin) in the pathogenesis of acute brain edema independent of leukocyte accumulation (Whalen et al., 2000). Because posttraumatic brain edema may be responsible for a considerable increase in brain volume and intracranial pressure, this pathway may be an important therapeutic target in clinical head injury. Additional studies are needed to further define the role of ICAM-1 and P-selectin in posttraumatic brain edema.
Tumor necrosis factor-α
Brain injury induces a dramatic increase in the expression of TNF-α but the exact role of this cytokine in mediating posttraumatic damage is unclear (Shohami et al., 1994; Fan et al., 1996). Tumor necrosis factor-α binds two different receptors (TNFR): p75 and p55. Activation of the p55 receptor results in activation of the nuclear factor κB, which induces genes for manganese superoxide dismutase (MnSOD) and calbindin (Das et al., 1995; Mattson et al., 1995).
To understand the role of TNF in the postinjury cascade, TNF (−/−) mice were generated using a replacement-type targeting vector containing a phosphoglycerate kinase (PGK)-neomycin (neo) expression cassette to replace the TNF gene. Scherbel et al. (1999) subjected TNF (−/−) mice and their WT littermates to CCI brain injury of mild severity and observed that the brain-injured TNF (−/−) mice exhibited attenuated cognitive deficits and motor dysfunction during the first week after injury when compared with WT littermates. However, WT mice subjected to mild TBI showed a marked recovery with time, whereas TNF (−/−) mice displayed persistent motor deficits up to 4 weeks after injury (Fig. 1). A significantly larger injury cavity also was observed in TNF (−/−) mice at 2 and 4 weeks after injury, suggesting that although this cytokine may play a deleterious role during the acute postinjury period, it may have a beneficial role in long-term behavioral recovery and tissue repair in the more chronic period after brain injury. These data are partially in agreement with the results obtained using pharmacologic agents aimed to reduce TNF production, such as pentoxifylline and the synthetic cannabinoid Dexanabinol (Jackson Laboratory, Bar Harbor, ME, U.S.A.), or its activity, such as TNF-binding protein, suggesting that TNF antagonism in the acute postinjury phase may be therapeutic (Shohami et al., 1996, 1997b).
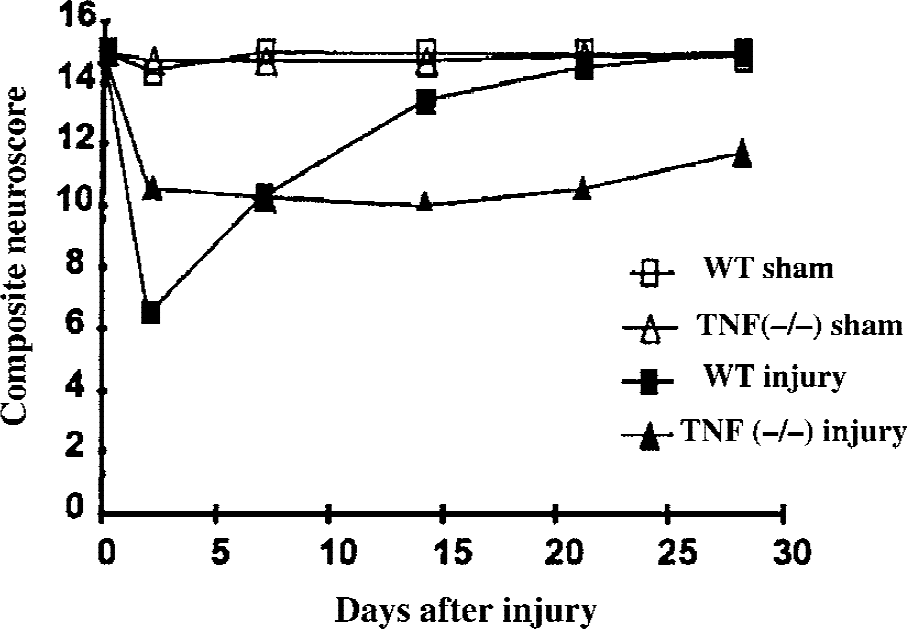
Comparison of posttraumatic recovery of neurologic motor function evaluated by composite neuroscore in tumor necrosis factor (TNF) (−/−) mice and their wild type (WT) littermates after controlled cortical impact brain injury. Reprinted with permission from Scherbel et al., 1999. Copyright ©1999 National Academy of Sciences, U.S.A.
Because the lymphotoxin (LT) may compensate for the absence of TNF by acting on the same receptors, TNF/LT double-deficient mice have been produced by inactivating both genes. These animals show splenic microarchitecture disorganization, immunoglobulin reduction, and impaired B and T cell function (Eugster et al., 1996). When TNF/LT (−/−) mice were subjected to a weight drop brain injury and were compared with brain-injured WT mice, no differences were observed in neurologic motor function (during the first 7 days), BBB permeability at 4 hours after injury, neutrophil infiltration at 24 hours after injury, or cell death. However, TNF/LT (−/−) mice have been reported to show an increased mortality at 1 week after injury, suggesting a protective role for these cytokines (Stahel et al., 2000). Although these data differ from those obtained from Scherbel et al. (1999), in which the deletion of TNF gene was suggestive of an acute detrimental effect of TNF, these differences may be caused by the effects of deletion of different genes and the use of different strains of mice and models of central nervous system (CNS) injury.
In addition to the deletion of the TNF gene, mice lacking either p75 or p55 receptors have been generated by the disruption of the TNFR genes and replacement with a neomycin (neo) resistance gene. Homozygous p75 and p55 knockout mice were subsequently mated to obtain mice that are deficient in both receptors. These knockout mice show altered response of lymphocytes to infectious agents (Zheng et al., 1995; Bruce et al., 1996), but reproduce normally, appear to be healthy, and fail to show any behavioral deficits or overt brain structural alterations compared with naïve WT littermates. After CCI brain injury, TNFR knockout mice, lacking p75 and p55 receptors, showed a greater lesion volume (7 days after injury) and alteration in BBB permeability, delayed NF-κB activation, and reduced MnSOD expression in the acute posttraumatic period compared with WT littermates (Sullivan et al., 1999). Moreover, because MnSOD overexpressing mice showed a reduced posttraumatic lesion volume at 7 days compared with WT controls, it was suggested that TNFR-mediated NF-κB activation may initiate neuroprotective pathways early in the postinjury cascade through MnSOD induction, which exerts antioxidative and antiapoptotic activity (Sullivan et al., 1999). The results obtained using TNFR (−/−) mice suggest that TNF may be neuroprotective even in the acute posttraumatic period, creating a more complex picture of the role of this cytokine in TBI. Taken together, these studies prompt the reevaluation of therapeutic strategies aimed at suppressing TNF-α production or blocking its activity and serve as a reminder that the timing of manipulation of specific cellular and molecular pathways through pharmacologic intervention is critical.
Interleukins and metallothioneins
Interleukin-1β converting enzyme (ICE or caspase-1) was the first identified member of the caspase family responsible for cleavage of pro–IL-1β into biologically active IL-1β that may play a role during cell death. Transgenic mice expressing a dominant negative inhibitor of caspase-1 have been generated by inserting cysteine in substitution for glycine at the active site (ICEC285G), under the control of a neuron-specific enolase promoter (NSE-M17Z). The embryonic development of these TG mice is normal, and although they appear neurologically normal postnatally, dorsal root ganglial neurons isolated from this strain are resistant to trophic factor withdrawal-induced apoptosis (Friedlander et al., 1997). Moreover, the neurons isolated from newborn caspase-1 knockout mice are similarly resistant to trophic factor withdrawal-induced apoptosis (Friedlander et al., 1997). Transgenic mice expressing the mutant gene of caspase-1 (ICEC285G) when subjected to CCI brain injury showed lower levels of caspase-1, reduced motor deficits, and smaller lesion volume compared with brain-injured WT mice. This neuroprotection was associated with a decrease in oxygen free radical release and was replicated by pharmacologic inhibition of caspase-1 using the selective peptide inhibitor AcYVAD-cmk and the nonselective pan-caspase inhibitor zVAD-fmk (Fink et al., 1999).
Interleukin-6 is a proinflammatory cytokine, known to protect cultured mesencephalic, catecholaminergic, and septal neurons from the toxic effect of glutamate (Hama et al., 1991; Toulmond et al., 1992). Interleukin-6 mRNA expression and protein levels are increased in the rat brain after a weight drop TBI and in cerebrospinal fluid after human head injury (Kossmann et al., 1996; Hans et al., 1999). Moreover, IL-6 induces expression of acute-phase proteins called metallothioneins (MT), which may have antioxidant properties and may regulate zinc and copper metabolism. These proteins also modulate catecholaminergic, glutamatergic, and GABAergic transmission, and their expression is altered in different pathophysiologic conditions, including neurodegenerative diseases and cortical freeze injury (Penkowa and Moos, 1995; Aschner et al., 1997).
Generation of IL-6–deficient mice has been achieved by the disruption of the IL-6 gene in 129 Sv ES cells by the insertion of neomycin into the first coding exon of IL-6. Homozygous IL-6 knockout mice develop normally, but show a compromised inflammatory acute-phase response after tissue damage (Kopf et al., 1994). After cortical freeze injury in IL-6 (−/−) knockout mice, the number of activated glial cells and brain macrophages was reduced by 3 days after injury. A reduction in MT isoforms I and II and an increase of isoform III expression with a greater neuronal cell loss in the fronto-parietal cortex was observed compared with WT littermates (Penkowa et al., 1999b). These authors suggested that IL-6 is essential for activation of microglia and recruitment of monocytes and astrocytes after brain injury (Penkowa et al., 1999b).
Metallothionein production is induced by IL-6, and to understand the role of these proteins in the response to CNS injury, MT I and II knockout mice have been generated, inactivating the genes by homologous recombination using clones lacking in MT I and II alleles. Naïve metallothionein-deficient mice do not show any anatomic or histologic difference when compared with their WT littermates (Penkowa et al., 1999 a). In a model of cortical freeze injury, MT I–II knockout mice, compared with WT-injured mice, showed a more pronounced infiltration of microglia and macrophages, a prolonged, abnormal inflammatory response up to 90 days, an increase in neuronal cell loss and apoptosis, and a reduction in wound healing and tissue regeneration due to altered angiogenesis (Penkowa et al., 1999 a, 2000). These studies suggest that these proteins may be important for the proper control of the inflammatory response, oxidative homeostasis, and apoptosis (caused by greater oxidative stress and alterations in zinc concentration) after brain injury.
Astrocytes and tissue repair
Astrocytes play an important role in maintaining metabolic homeostasis in the CNS. The biologic significance of reactive astrocytosis after TBI is of great interest because this can lead to a glial scar formation, which may putatively impede CNS regeneration, or to the release of trophic factors that support regeneration. Steward and Trimmer (1997), using a substrain of C57BL/6 mice that contains the Ola mutation and is characterized by markedly delayed Wallerian degeneration (Wlds), studied astrocyte activation in a model of denervation and showed that reactive changes in astrocytes are triggered by factors released by activated microglia. The delayed glial response to injury is believed to have detrimental consequences, including the inhibition of regeneration of the injured neurons. Fox and Faden (1998) compared the temporal course of the behavioral alterations caused by CCI brain injury in C57BL/6 and C57BL/6/Wlds mice and showed that behavioral impairment is delayed in Wlds mice. To further clarify the role of astrocytes after CNS injury, mice genetically engineered to be deficient in glial fibrillary acidic protein (GFAP)-positive astrocytes have been generated by expressing herpes simplex virus thymidine kinase from a mouse GFAP promoter. Subsequent treatment for 1 week with Ganciclovir then ablates all transgene-expressing, GFAP-positive astrocytes from the injured CNS (Bush et al., 1998). When subjected to forebrain penetrating stab injury and subsequent ablation of GFAP positive astrocytes, these mice show prolonged leukocyte infiltration, failure of BBB repair with associated brain edema, neuronal degeneration, and increased outgrowth of nerves fibers compared with injured WT mice (Bush et al., 1999). These data implicate a protective role for reactive or activated astrocytes in the postinjury period.
Insulinlike growth factors (IGF) I and II are peptides related to the hormone insulin. They are present in serum and tissue and are usually bound to IGF binding proteins (IGFBP), which can increase or inhibit IGF action. In the CNS, IGF-1 receptors are present in neurons, astrocytes, and oligodendrocytes during development (Garcia-Segura et al., 1991). In adult life, IGF-1 expression in astrocytes is reactivated in response to injury (Garcia-Estrada et al., 1992). To generate transgenic mice overexpressing IGFBP-1, the entire coding region of the rat IGFBP-1 gene was inserted downstream of the mouse phosphoglycerate kinase (PGK-I) promoter. The IGFBP-1/PGK-I fragment then was inserted into fertilized C57BL/6JXCBA/JF1 zygotes. These IGFBP-1 overexpressing mice are characterized by reduced body weight, hyperglycemia, and reduced brain size (Rajkumar et al., 1995). To clarify the potential role of IGF-1 in activating astrocytes during the postinjury process, a knife lesion was stereotaxically placed in the right cerebral hemisphere in both WT and transgenic mice overexpressing IGFBP-1. The response of astrocytes to the injury was reduced in the TG mice, suggesting an IGF-1–mediated astrocyte response to this type of injury (Ni et al., 1997).
Matrix metalloproteinases
Gelatinase (MMP 9), a member of the metalloproteinase family, is not constitutively expressed in the brain but is induced after cerebral ischemia and trauma (Wang et al., 2000; Sharp et al., 2000). After CCI brain injury in C57BL/6 mice, MMP 9 levels increase by 3 hours after injury and remain elevated for at least 1 week (Wang et al., 2000). To evaluate the role of MMP 9 in brain injury, MMP 9 knockout mice were generated by replacing part of the gene with a cassette containing the neomycin phosphotransferase cDNA (neor) driven by the PGK promoter. Matrix metalloproteinase 9 (−/−) mice exhibit an abnormal pattern of skeletal growth plate vascularization and ossification in the long bones and show a lower motor endurance compared with WT mice (Vu et al., 1998). Matrix metalloproteinases 9 (−/−) mice subjected to CCI brain injury show decreased motor deficits and smaller lesion volumes compared with brain-injured WT mice, suggesting that MMP 9 may play a deleterious role during the secondary cascade after trauma (Fig. 2) (Wang et al., 2000). Additional work is necessary to confirm whether MMP 9 may be a future target for pharmacologic intervention after TBI.
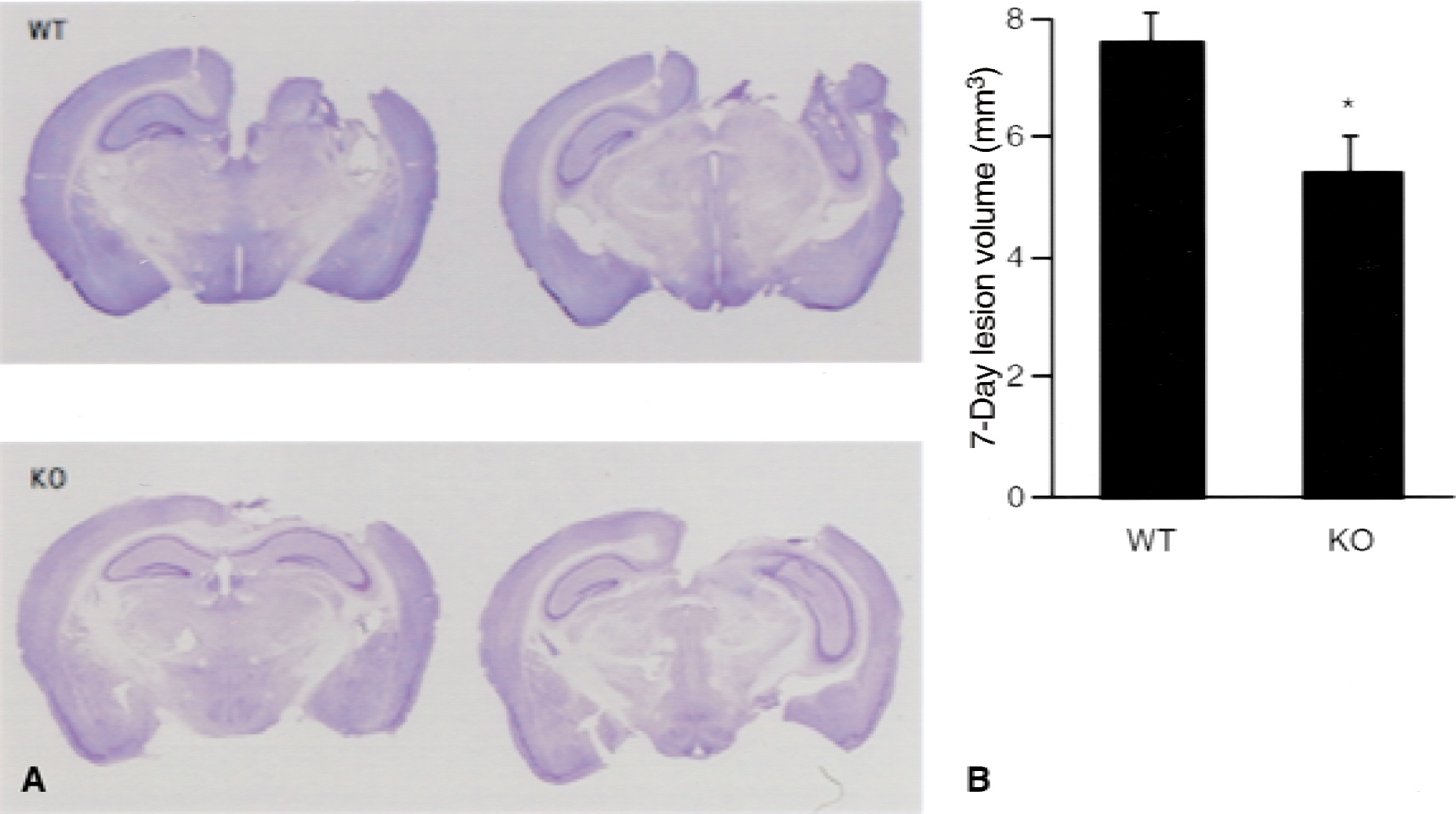
Nitric oxide
Nitric oxide (NO) has been suggested to play a role in damage, recovery, or both after brain injury (Huang et al., 1994; Mesenge et al., 1996; Holscher, 1997). Three different systems are reportedly involved in NO synthesis. Nitric oxide production by endothelial NO synthase (eNOS) appears to provide beneficial effects in models of TBI by preventing blood flow reduction (DeWitt et al., 1997), whereas neuronal NOS (nNOS)-related NO production has been suggested to exacerbate neuronal damage in models of cerebral ischemia (Huang et al., 1994). The final pathway involves inducible NOS (iNOS) which is activated in response to inflammation after experimental TBI with marked increases in expression 24 and 48 hours after injury (Clark et al., 1996).
To clarify the beneficial or detrimental effects of iNOS-related NO production, mice deficient in iNOS have been produced by targeted disruption of both the iNOS promoter and the iNOS gene (MacMicking et al., 1995). These homozygous knockout mice are healthy and perform normally on motor and spatial memory acquisition tasks when compared with WT mice. However, mice deficient in iNOS showed greater latencies, compared with WT littermates, to find a submerged platform 15 to 18 days after CCI brain injury, indicating impaired learning performance in this strain and suggesting a protective role of iNOS, although the exact mechanism remains unclear (Sinz et al., 1999). In the same study, the protective role of iNOS was confirmed in a rat model of CCI brain injury; administration of iNOS inhibitors aminoguanidine and L-N-iminoethyl-lysine resulted in reduced learning capacity and greater hippocampal cells loss at 3 weeks after injury. Because these data are contrary to those obtained in the acute posttraumatic period using the NO inhibitor aminoguanidine after lateral fluid percussion (FP) brain injury in rats (Wada et al., 1998), iNOS activity may exert detrimental effects in the acute postinjury period but show beneficial effects in the chronic phase. Further studies with knockout mice at different time points are warranted to clarify the role of NO after TBI. Figure 3 summarizes the main pathways related to the posttraumatic inflammatory response investigated using genetically engineered mice. Table 1 summarizes the results of the studies previously described in the text with the main findings in relation to the genotype used and the model of injury.
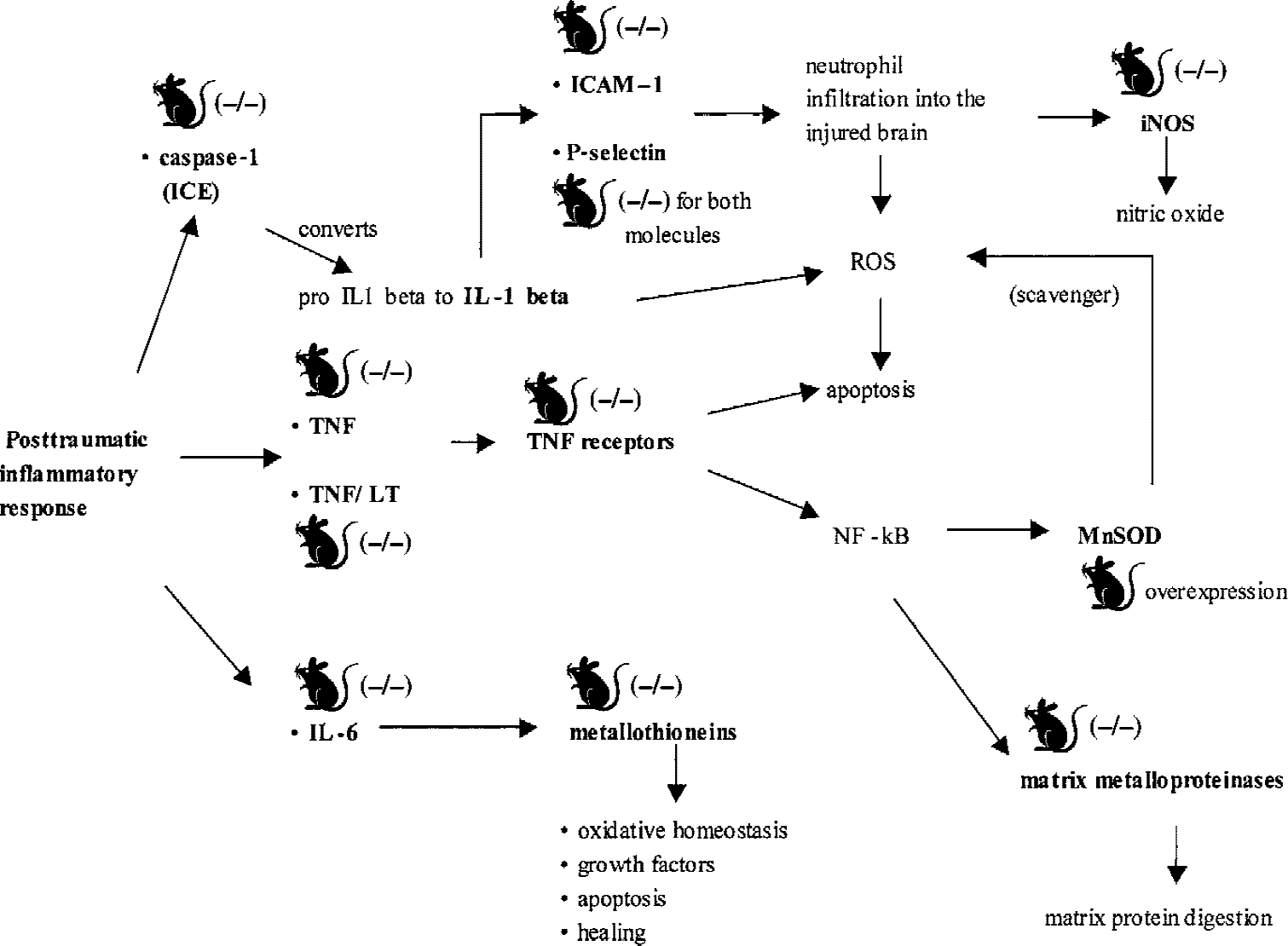
Overview of the main pathways involved in the posttraumatic inflammatory cascade investigated using genetically engineered mice. ICAM, intercellular adhesion molecules; ICE, Interleukin-1β converting enzyme; iNOS, inducible nitric oxide synthase; LT, lymphotoxin; MnSOD, manganese superoxide dismutase; ROS, reactive oxygen species; TNF, tumor necrosis factor.
Summary of studies performed with genetically engineered mice to investigate specific molecules involved in the inflammatory posttraumatic response
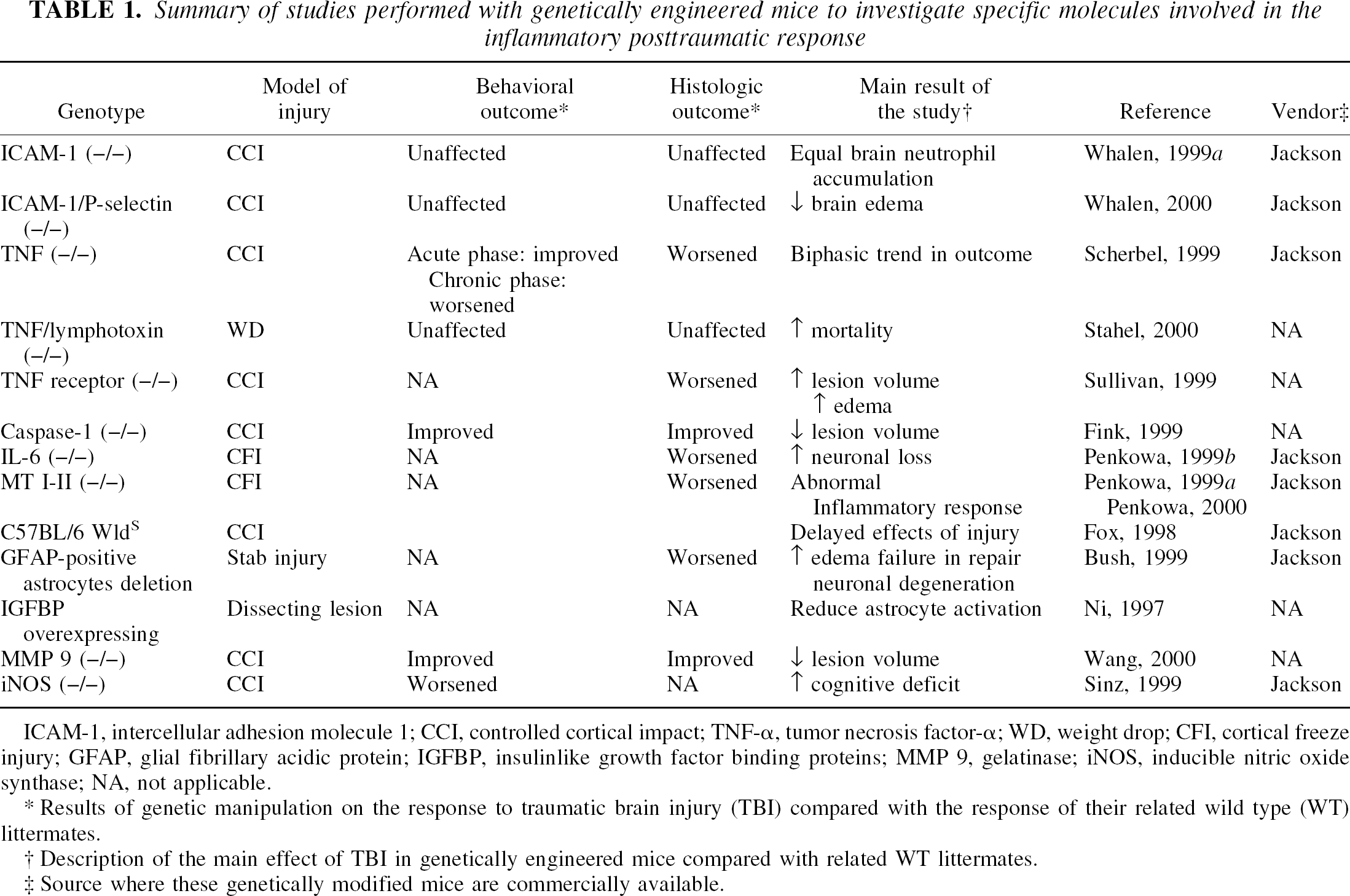
ICAM-1, intercellular adhesion molecule 1; CCI, controlled cortical impact; TNF-α, tumor necrosis factor-α; WD, weight drop; CFI, cortical freeze injury; GFAP, glial fibrillary acidic protein; IGFBP, insulinlike growth factor binding proteins; MMP 9, gelatinase; iNOS, inducible nitric oxide synthase; NA, not applicable.
Results of genetic manipulation on the response to traumatic brain injury (TBI) compared with the response of their related wild type (WT) littermates.
Description of the main effect of TBI in genetically engineered mice compared with related WT littermates.
Source where these genetically modified mice are commercially available.
OXIDATIVE DAMAGE
The formation of reactive oxygen species (ROS) is one of several major events in a biochemical cascade leading to delayed neuronal death after TBI. Oxygen free radicals are responsible for peroxidative damage to the cell membrane, oxidation of proteins, and DNA leading to cell damage and death through apoptotic and necrotic pathways (Shohami et al., 1997a; Love, 1999). Under physiologic conditions, ROS are continunously produced during cellular respiration and are subsequently removed by endogenous scavengers including superoxide dismutases (SOD), glutathione peroxidase, and catalase. Superoxide dismutase includes three isoforms: copper zinc (CuZn)-SOD in the cytosol, MnSOD in mitochondria, and high molecular weight extracellular (E)-SOD (Chan et al., 1995; Love, 1999).
Transgenic CuZn-SOD overexpressing mice have been generated through insertion of the human CuZn-SOD 14.5 Kb gene, which includes the promoter sequence. These mice express 3- to 10-fold greater than normal values of CuZn-SOD in the CNS and other tissues (Shi et al., 1994). Typically, no phenotypic differences are observed between these mice and their WT littermates, although some investigators have reported certain morphologic abnormalities of the neuromuscular junction in the tongue muscle (Avraham et al., 1988). Studies involving cold (freeze lesion) injury or weight drop brain injury have demonstrated that BBB permeability, brain edema, and lesion volume are significantly reduced and neurologic deficits are attenuated in CuZn-SOD TG mice (Chan et al., 1991; Mikawa et al., 1996; Murakami et al., 1999).
Manganese superoxide dismutase overexpressing mice have been generated by inserting the human MnSOD gene through microinjection into the pronuclei of mouse fertilized eggs. Overexpression of MnSOD in transgenic mice is associated with smaller cortical contusion volume 7 days after CCI brain injury compared with brain-injured WT controls (Sullivan et al., 1999). Although the results of these laboratory studies using TG mice have been replicated using pharmacologic treatment to inhibit ROS in the experimental setting, clinical trials with the free radical scavenger polyethylene glycol–conjugated SOD or tirilazad mesylate, a 21-aminosteroid inhibitor of free radical–mediated lipid peroxidation, have not yet produced significant benefits in patients with head injuries (Young et al., 1996; Marshall et al., 1998).
DNA DAMAGE AND REPAIR
Clinical and experimental TBI are characterized by progressive cell death. Two morphologic phenotypes of cell death have been described: necrotic and apoptotic. Both share DNA fragmentation, detectable by the (TdT)-mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) technique (Gavrieli et al., 1992). Because morphologic features of necrosis and apoptosis have been documented in the same neural cells, it has been hypothesized that a continuum exists between apoptosis and necrosis (Portera-Cailliau et al., 1997). The intracellular pathways leading to these two forms of cell death may coexist and/or overlap sequentially, and the prevalence of apoptotic or necrotic features at any single time may be related to the intensity of the insult, energy production by the mitochondria, intracellular level of calcium, and reparative mechanisms (Raghupathi et al., 2000).
As previously described, ROS that are released after TBI can induce DNA strand breaks (Love, 1999). The tumor suppressor gene p53 is up-regulated after DNA damage and can lead to DNA repair, cell growth arrest, or apoptosis (Evan and Littlewood, 1998). Napieralski et al. (1999) reported that p53 mRNA is up-regulated after lateral FP brain injury in rats, suggesting that p53 may play a role in the molecular response to TBI leading to cell death. To further evaluate the role of p53 after TBI, p53 knockout mice were generated through homologous recombination using a target vector containing 3.7 kilobases of the genomic p53 gene, interrupted in exon 5 by a PolII-neo expression cassette. Mice deficient in p53 appear healthy at birth but are prone to the spontaneous development of neoplasms by 6 months of age (Donehower et al., 1992). Because mice deficient in p53 show less histologic damage in models of excitotoxicity (subcutaneous kainic acid injection) and focal ischemia (middle cerebral artery occlusion) (Crumrine et al., 1994; Morrison et al., 1996), Tomasevic et al. (1999) compared the effects of CCI brain injury in p53 knockout mice and their WT littermates. At 7 days after injury, p53 (−/−) mice exhibited attenuated motor function deficits compared with their WT littermates, but no differences were found in cortical lesion volume or cell loss in the hippocampus and thalamus between WT and p53-deficient mice. These results may be explained, in part, by the fact that DNA damage-induced up-regulation of p53 gene may activate different pathways, such as induction of the proapoptotic bax gene family, leading to cell death (Miyashita and Reed, 1995), or induction of the wild type p53-activated fragment (WAF/p21) and the growth arrest and DNA damage-inducible gene GADD45, leading to cell survival (Artuso et al., 1995).
Poly (ADP-ribose) polymerase (PARP) is a nuclear enzyme that has been proposed to play a role in the repair of DNA damage. Normally, DNA strand breaks activate PARP, which then binds to the nicked DNA and uses NAD+ as a substrate to form Poly (ADP-ribose) (PAR). This process continues until a critical PAR chain length is reached and then PARP is released from DNA and other repair enzymes complete the repair process. Although lateral FP brain injury in the rat initially activates PARP by 30 minutes after injury, evidence suggests that the PARP molecule is subsequently degraded by caspase-3–mediated cleavage, leading to significant DNA fragmentation (LaPlaca et al., 1999). However, it has been hypothesized that excessive activation of PARP also may lead to consumption of NAD, ATP reduction, cell dysfunction, and death associated with energy failure (Berger, 1985). Poly (ADP-ribose) polymerase (−/−) mice, produced by deleting part of exon 2, develop normally and are fertile. Analysis of tissues isolated from these mice confirmed absence of PARP activity, but revealed that these mice are able to repair DNA damage induced by UV and alkylating agents (Wang et al., 1995). Genetically engineered mice deficient in PARP show attenuated functional deficits but equivalent histologic outcome after CCI brain injury, suggestive of a protective mechanism through the inhibition of posttraumatic energy failure (Whalen et al., 1999b). Further studies are warranted to elucidate the mechanisms underlying these results and to evaluate whether pharmacologic inhibition of PARP might represent a therapeutic strategy for reducing neurologic injury after TBI, as recently shown by LaPlaca et al. (2001) in a model of lateral FP brain injury in rats. Figure 4 summarizes the pathways related to oxidative brain damage and DNA damage investigated, to date, using genetically engineered mice.
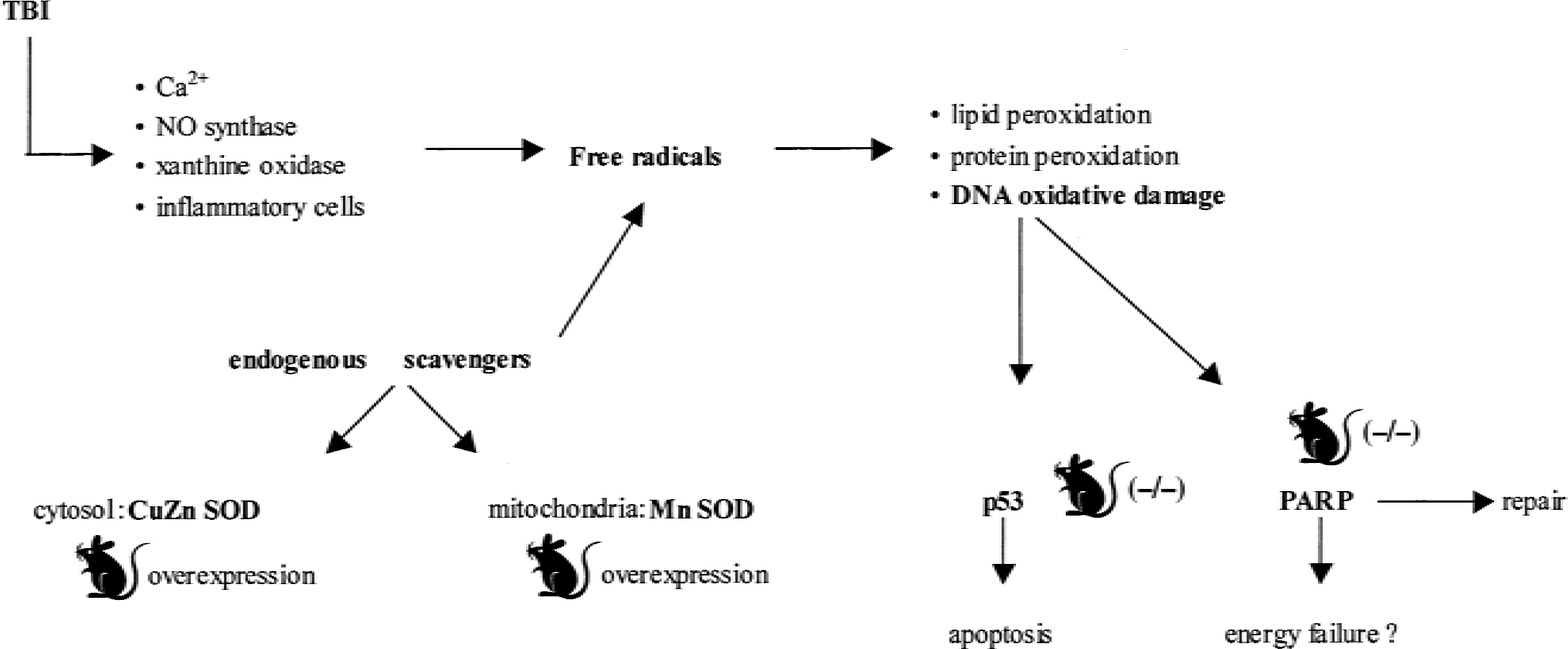
Overview of the pathways involved in the oxidative brain damage, physiologic endogenous scavenger, and DNA damage- and repair-related mechanisms evaluated using genetically engineered mice. NO, nitric oxide; CuZn SOD, copper zinc superoxide dismutase; MnSOD, manganese superoxide dismutase; PARP, poly (ADP-ribose) polymerase; TBI, traumatic brain injury.
APOPTOSIS
Apoptotic cell death has been reported to occur in the acute and the chronic posttraumatic period after lateral FP and CCI brain injury in rats and more recently in tissue from patients with head injuries (Rink et al., 1995; Colicos and Dash, 1996; Conti et al., 1998; Newcomb et al., 1999; Smith et al., 2000). Apoptosis is triggered by intracellular or extracellular signals that converge to activate a specific group of cysteine proteases called caspases. Caspase activation occurs through 2 different pathways. The first (extrinsic) pathway involves TNF-α and Fas receptors at the cell surface leading to activation of caspase-8, which then activates downstream caspases such as caspase-3, −6, and −7. The second (intrinsic) pathway involves the release of cytochrome c from the mitochondria. It is generally believed that cytochrome c binds to Apaf-1 and this complex recruits and activates procaspase-9, which finally activates the downstream caspases-3, −6, and −7 (Hengartner, 2000; Yuan and Yankner, 2000).
Apoptosis is controlled by the bcl-2 family of genes including proapoptotic factors such as bak, bim, bad, and bax and antiapoptotic factors bcl-2 and bcl-xl, whose activity is related to cytochrome c and caspase activation. An imbalance in the expression of one of these groups of genes may exacerbate or reduce the extent of cell death after neuronal injury (Graham et al., 2000b). The bcl-2 gene family may play an important role in controlling cell survival after CNS injury, and an increase in bcl-2 mRNA and bcl-2 protein has been observed after experimental and clinical TBI (Clark et al., 1997, 1999, 2000). In contrast, Raghupathi et al. (unpublished data, 2000) recently evaluated the ratio between proapoptotic and antiapoptotic members of the bcl-2 family and observed that bcl-2 immunoreactivity was decreased in the injured cortex and injured hippocampus by 2 hours after lateral FP injury. At 24 hours after injury, the authors also observed an increase in bax mRNA and proteins in the perilesion area. These data suggest that the resulting changes in the bax/bcl-2 ratio could participate in the posttraumatic pattern of apoptosis in the injured cortex and hippocampus.
Mice overexpressing human bcl-2 have been generated by transfecting the human bcl-2 gene under the control of synapsin I promoter fragment. Raghupathi et al. (1998) subjected these bcl-2 overexpressing mice to CCI brain injury, then compared them with WT littermates. Transgenic mice overexpressing bcl-2 demonstrated increased human bcl-2 expression in the cortex, hippocampus, and midbrain and reduced cortical lesion volume by 1 week after injury when compared with WT littermates. Surprisingly, no differences in neurologic motor function were observed between transgenic and WT mice. Nakamura et al. (1999 a) studied mice overexpressing human bcl-2 controlled by the neurofilament light-chain promoter. These mice (overexpressing human bcl-2 protein in the cortex, hippocampus, and thalamus) were subjected to CCI brain injury and then were compared with their WT littermates. Human bcl-2 overexpression was associated with smaller cortical lesion volume and hippocampal cell loss, confirming a protective role of the bcl-2 protein. Despite histologic protection, TG mice were as impaired as their WT littermates with respect to posttraumatic cognitive function.
Several mechanisms can explain a protective role for bcl-2, including the capacity to reduce ROS generation, an increased capacity to buffer posttraumatic intracellular calcium increases, and the blockade of cytochrome c release from the mitochondria preventing subsequent activation of caspases. Recently, the protective effect of bcl-2 was confirmed by the manipulation of its expression in a rat model of middle cerebral artery occlusion with the use of antisense oligodeoxynucleotides, which suppressed endogenous expression and exacerbated ischemic neuronal death (Chen et al., 2000).
Among the proapoptotic factors of the bcl-2 family of genes, bax has been shown to induce apoptosis by activating caspase-3 (Cregan et al., 1999). After CCI brain injury in rats, a translocation of bax into the nuclei of neurons destined to die of apoptosis was observed at 48 hours postinjury (Kaya et al., 1999). Knockout of the bax gene has been produced in bax (−/−) mice by homologous recombination substituting exons 2, 4, and part of 5 with a target vector (PGK-Neo). These mice are viable and show thymocyte hyperplasia and infertility in the males. In addition, the knockout of bax leads to a reduction in the magnitude of naturally occurring programmed cell death in sympathetic and facial motor neurons (Knudson et al., 1995). Cultures of neurons from bax (−/−) mice are resistant to excitoxicity produced by administration of glutamate or kainate and DNA damage induced by camptothecin, suggesting that bax is involved in different pathways of cell death (Xiang et al., 1998). Carbonell et al. (1999) tested the hypothesis that this proapoptotic factor is involved in the acute cortical and hippocampal damage after lateral FP brain injury by comparing the response of bax (−/−) mice and their WT littermates to TBI. As early as 10 minutes after lateral FP brain injury, bax (−/−) mice showed fewer damaged neurons than the WT-injured mice in the hippocampus but not in the cortex, suggesting that bax is involved in mediating acute posttraumatic hippocampal neuronal damage. Because the hippocampus contains a high concentration of excitatory amino acid receptors, these results might be explained by an increased resistance to excitotoxicity caused by the suppression of bax.
Downstream from caspase activation, DNA fragmentation is mediated by a heterodimeric protein composed of 40- and 45-kDa subunits. These subunits have been named DNA fragmentation factor (DFF)-40 (or caspase activated DNAase (CAD)) and DFF-45 (or inhibitor of CAD, ICAD). During apoptosis, caspase-3 is believed to cleave DFF-45, dissociating it from DFF-40 and inducing a transformation of DFF-40 into a large protein complex with DNAase activity. The proteolytic activity of DFF-40 requires DFF-45 to be reactivated, suggesting that DFF-45 is a chaperone molecule that is essential for DFF-40 synthesis and activation (Liu et al., 1997, 1998; Enari et al., 1998). Regional changes of DFF-40 and 45 have been documented in lateral FP brain injury, where a decrease in DFF-45–like proteins in rat cortex was observed in the cytosolic and nuclear fraction during the first 24 hours after injury (Zhang et al., 1999). In the hippocampus, DFF-40 was found to be reduced in the cytosolic fraction and increased in the nuclear fraction at 2 and 24 hours after brain injury, suggesting that a translocation from cytoplasm to nucleus occurs during posttraumatic apoptosis (Zhang et al., 1999).
The generation of DFF-45 mutant mice has been achieved through the deletion of 1–3 exons through homologous recombination (Zhang et al., 1998). DFF-45 (−/−) mice contain normal levels of DFF-40 (which is not functional) and caspase-3, are healthy and fertile, and grow without obvious abnormalities. No spontaneous DNA fragmentation has been observed in neurons isolated from these mice. Moreover, cells isolated from the spleen and thymus are resistant to DNA fragmentation and chromatin condensation in response to apoptotic stimuli, suggesting that DFF-45 plays a critical role during apoptosis (Zhang et al., 1998). These DFF 45 (−/−) mice were found to have a smaller lesion volume than WT mice at 1 and 4 days after CCI brain injury, suggesting a neuroprotective role of DFF 45 deletion in the acute posttraumatic period (Zhang et al., 2000).
Despite the progress that has been made during the past few years in understanding the molecular mechanisms associated with posttraumatic cell death, the complexity of these pathways and whether apoptosis also may have a beneficial or protective role—by which the brain removes injured cells with minimal damage to surrounding tissue—in the setting of CNS injury remains unclear. Further investigations with genetically engineered mice are warranted to clarify the potential targets for a rational approach to the design of new pharmacologic therapies to prevent this type of cell death. Figure 5 summarizes the apoptotic pathways investigated with genetically engineered mice. Table 2 summarizes the results of the studies related to oxidative damage, DNA damage, repair, and apoptosis described in the text with the main findings in relation to the genotype used and the model of injury.
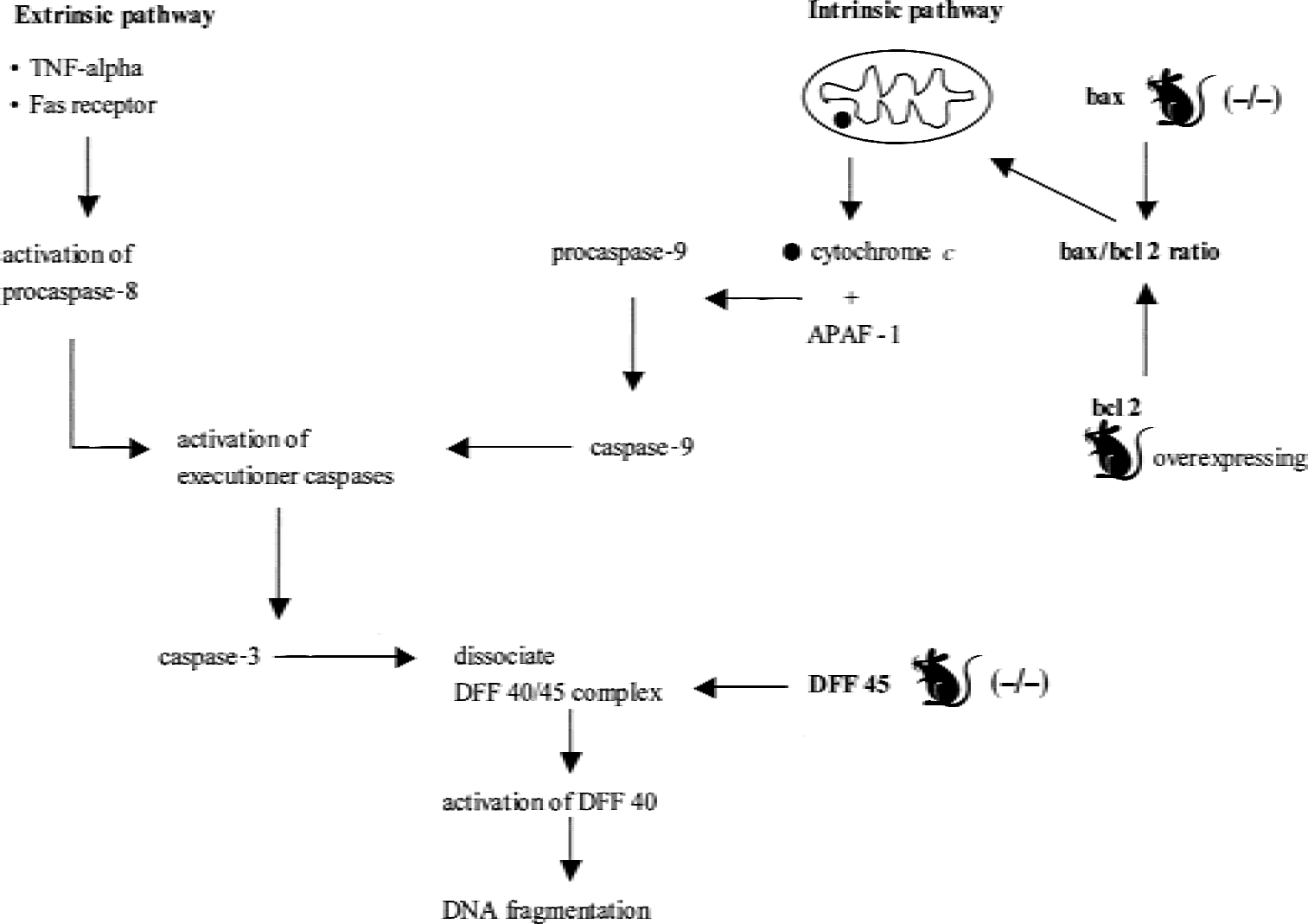
Extrinsic and intrinsic pathways of apoptosis and molecules investigated using genetically engineered mice after traumatic brain injury. DFF, DNA fragmentation factor; TNF, tumor necrosis factor.
Summary of studies performed with genetically engineered mice to investigate the pathways related to posttraumatic oxidative damage, DNA damage/repair, and apoptosis

CuZn SOD, copper zinc superoxide dismutase; CCI, controlled cortical impact; WD, weight drop; CFI, cortical freeze injury; PARP, Poly (ADP-ribose) polymerase; LFP, lateral fluid percussion; DFF, DNA fragmentation factor; NA, not applicable.
Results of genetic manipulation on the response to traumatic brain injury (TBI) compared with the response of their related wild type (WT) littermates.
Description of the main effect of TBI in genetically engineered mice, compared with related WT littermates.
Source where these genetically modified mice are commercially available.
TRAUMATIC BRAIN INJURY AND NEURODEGENERATION
Alzheimer disease
Although TBI is a known epidemiologic risk factor for Alzheimer disease (AD), the mechanistic link is not yet fully understood (Plassman et al., 2000). After a single severe head injury or a series of repetitive mild head injuries (in boxers), a cohort of TBI patients share many pathologic features with patients suffering from AD—including deposition of plaques containing β-amyloid (Aβ) derived from the proteolysis of amyloid precursor protein (APP) and composed of fibrils formed by 39–43 amino acid peptides, and the presence of neurofibrillary tangles in which paired helical filaments are composed of abnormally phosphorylated tau (Tokuda et al., 1991; Roberts et al., 1994). Alzheimer disease is believed to be profoundly influenced by genotype. In kindreds of familial AD patients, mutations are known to occur in genes encoding for APP and presenilin 1 and 2, which result in an increase in the Aβ peptides levels (Borchelt et al., 1998). Moreover, genetic studies have found a correlation between the common form of AD and the allele ε 4 of the apolipoprotein E (APOE) gene on chromosome 19 (Strittmatter et al., 1993). Recently, patients expressing the ε 4 allele of apoE have been reported to have an increased vulnerability and worse outcome after brain trauma (Teasdale et al., 1997; Lichtman et al., 2000). Because head injury is epidemiologically linked to AD, this relation recently has been studied in experimental transgenic mouse models to reveal novel and relevant pathogenetic mechanisms.
Amyloid precursor protein
Amyloid precursor protein is subjected to proteolytic degradation leading to generation and secretion of the Aβ peptide. Approximately 90% of secreted β-amyloid peptide is in the form of Aβ1–40 and 10% are Aβ1-(42–43). The Aβ1-(42–43) contains amino acids that are highly fibrillogenic, readily aggregated, and deposited early and selectively in amyloid plaques (Borchelt et al., 1998). Traumatic brain injury in humans and rodents induces accumulation of APP in perikarya of neurons and in damaged axons (Pierce et al., 1996; Bramlett et al., 1997; Sheriff et al., 1994). Despite this increase in APP, experimental models of TBI in rodents currently have not been associated with Aβ-plaque development, probably because of differences in amino acid composition between different species (Pierce et al., 1996).
Transgenic yeast artificial chromosome (YAC) mice have been generated by introducing the human APP gene into embryonic stem cells (lipofection of a 650-Kb YAC containing the APP gene). The CNS of these APP-YAC mice overexpresses APP twofold when compared with normal mice (Pearson and Choi, 1993). Murai et al. (1998) subjected APP-YAC mice to CCI brain injury and failed to observe any differences in posttraumatic cognitive function, neurologic motor deficits, APP expression, reactive astrocytosis, or cell loss between brain-injured TG mice and injured WT littermates. Moreover, no AD-like plaque deposition was observed in either brain-injured WT or APP overexpressing mice.
A second strain of mice that has been used in TBI research overexpresses a mutant APP minigene (containing the familial AD mutation Phe for Val in position 717) driven by a platelet-derived growth factor promoter (PDAPP). These mice overexpress mutant APP 10-fold when compared with normal mouse APP and develop plaques at 6 months of age (Games et al., 1995). Naïve (uninjured) PDAPP mice exhibit impaired memory function when compared with their WT littermates. However, CCI brain injury exacerbated these cognitive impairments and produced a concomitant increase in hippocampal Aβ1–40 and 1–42 that was associated with a near complete cell loss (>80%) in the CA3 region in the injured hemisphere (Smith et al., 1998). This study led the authors to develop a “two-hit hypothesis,” whereby a first “hit” or insult is represented by genetic vulnerability (that is, high concentration of amyloidogenic species of Aβ) that becomes manifest only after a second epigenetic insult or “hit” such as TBI. The resulting increase in cell loss may depend on the combination of direct neurotoxic activity of β-amyloid protein, abnormalities of the endosomal-lysosomal system, excitoxicity, activation of proteases, and/or apoptotic pathways (Borchelt et al., 1998). Using the identical strain of PDAPP mice, Nakagawa et al. (1999) observed that CCI brain injury at the same age (4 months) actually reduced plaque deposition with time (follow up at 2, 5, and 8 months after TBI). These authors hypothesized that this reduction in Aβ plaques may depend on decreased secretion of Aβ as a consequence of cell death observed in hippocampus and cingulate cortex, resulting from the combination of the previously described two insults. Subsequently, Nakagawa et al. (2000) subjected 2-year-old PDAPP mice to CCI brain injury. By 16 weeks after injury, brain-injured aged transgenic mice showed a remarkable decrease in Aβ deposits in the ipsilateral atrophic hippocampus, reflecting the massive hippocampal neuronal loss in these mice. The widespread neuronal cell loss associated with the moderate severity CCI brain injury may have offset the Aβ production preventing plaques formation.
Laurer et al. (personal communication) have recently investigated the effect of repetitive mild brain injury in a different strain of mutant APP TG mice. These mice express a human APP gene containing the double mutation Lys670 → Asn and Met671 → Leu, found in a large Swedish family with the early onset of AD (Hsiao et al., 1996). The hallmark of these TG mice is the association of elevated concentrations of Aβ, plaque formation, and neurobehavioral impairment beginning at 9 to 10 months of age (Hsiao et al., 1996). These TG mice had preinjury learning deficits by 7 months of age compared with WT littermates. A single episode of concussive brain injury, induced before plaque development, led to an acute cognitive impairment but failed to exacerbate histologic damage in this strain. However, repetitive, mild head injury induced both delayed cognitive alteration and bilateral increases in plaque deposition, suggesting that mild repetitive head injury may be a risk factor associated with an accelerated neurodegenerative process in a genetically vulnerable population (Laurer, personal communication). The results of this study encourage further evaluation of selectively vulnerable genotypes exposed to repetitive mild head injury.
Apolipoprotein E
In humans, apoE is a polymorphic protein (E2, E3, E4) in which isoforms differ from each other by 1 amino acid (Mahley, 1988). Recent epidemiologic studies have identified the ε4 allele of apoE as a major risk factor for sporadic and late-onset familial AD (Strittmatter et al., 1993). Apolipoprotein E is an important apolipoprotein constituent of cerebrospinal fluid and brain, where it is produced by glial cells. This specific apolipoprotein is involved in nervous system growth and repair through the coordination of the mobilization and redistribution of cholesterol during neuronal development. However, patients carrying apoE4 genotype have been reported to show a poorer outcome after TBI (Teasdale et al., 1997; Lichtman et al., 2000).
Genetically engineered mice have been produced and used to clarify the mechanisms by which this genotype influences posttraumatic brain damage. It has been hypothesized that apoE4 induces a loss of function of the E3 isoform; therefore, the biologic abnormalities during aging or after injury associated with the apoE4 genotype should be evident in apoE knockout mice. The deletion of the APOE gene has been achieved by replacing exon 2 of the APOE gene with a neomycin resistance gene. Apolipoprotein E (−/−) mice appear healthy at 4 to 6 weeks of age and do not have abnormalities in the septo-hippocampal system (Piedrahita et al., 1992). Homozygous apoE-deficient mice subjected to enthorinal cortex lesions exhibited persistent degeneration products (cholesterol-derived) in the deafferented hippocampus, compared with WT mice, suggesting that apoE may play a role in the clearance of cholesterol-laden neurodegeneration products after injury (Fagan et al., 1998). Moreover, apoE knockout mice subjected to weight drop brain injury showed significantly exacerbated memory impairment and motor deficits when compared with brain-injured WT controls for at least 40 days (Chen et al., 1997). Histopathologic examination revealed overt neuronal cell death bilaterally in the hippocampus of the injured apoE (−/−) mice. One possible explanation for the enhanced vulnerability of apoE (−/−) mice to TBI is the partial loss of oxidative homeostasis causing an increase in lipid peroxidation products, chronic oxidative stress, and reduction in low molecular weight antioxidants (Lomnitski et al., 1999).
One of the additional pathologic features of AD is the presence of neurofibrillary tangles composed of hyperphosphorylated tau, leading to cytoskeletal abnormalities that contribute to neuronal degeneration. Apolipoprotein E (−/−) mice typically show increased tau phosphorylation compared with WT mice. After weight drop brain injury, a transient increase in tau hyperphosphorylation has been observed in WT and apoE (−/−) mice, although the extent and time course in the two mouse groups varied markedly (Genis et al., 2000). In brain-injured WT mice, hyperphosphorylation was maximal by 4 hours after injury and reverted to basal levels by 24 hours, whereas brain-injured apoE (−/−) mice showed hyperphosphorylation of “hot spot” epitopes of the tau protein for a longer duration after TBI, even if it was reduced in its extent (Genis et al., 2000). The authors suggest that the greater hyperphosphorylation of these “hot spot” epitopes of tau protein might reflect repair mechanisms associated with an acute insult such as TBI. Although the exact pathogenic link between apoE, TBI, and AD is unclear, these studies reveal that apoE genotype may be related to the maintenance of oxidative homeostasis and repair mechanisms after injury to the CNS. Further studies with genetically engineered animals are warranted to help clarify the link between TBI, apoE genotype, and Alzheimer disease.
OTHER NEURODEGENERATIVE DISEASES LINKED TO TBI
Several neurodegenerative diseases including Parkinson disease, diffuse Lewy-body dementia, and frontotemporal dementia with Parkinsonism are characterized by intracellular neurofilament (NF)-rich filamentous inclusions in neurons. Tau protein and α-synuclein are the major building block proteins of neurofibrillary tangles and Lewy bodies, respectively, which are observed in several neurodegenerative diseases (tauopathies and synucleinopathies). These disorders share many pathogenic mechanisms and may cause neuronal toxicity through common pathways (Lee et al., 2001; Trojanowski and Lee, 2001). Because TBI can cause or accelerate latent and progressive neurodegenerative events, it appears reasonable to investigate this relation in transgenic animals showing NF inclusions.
Transgenic mice expressing a construct composed of the mouse polypeptide NF-heavy (NFH) subunit linked to the LacZ gene (NFH/LacZ) develop NF-rich inclusions in virtually all neurons of the CNS. The phenotypic features of these mice—such as tremor, ataxia, and muscular weakness—become evident by 1 year (Tu et al., 1997). Nakamura et al. (1999 b) subjected NFH/LacZ mice to CCI brain injury before the onset of behavioral signs and observed a significant and prolonged deficit in motor function up to 3 weeks after injury that was associated with increased lesion volume when compared with brain-injured WT mice. A subsequent study comparing the histologic damage between NFH/LacZ and WT brain-injured mice revealed a greater lesion cavity, increased apoptotic cell death, more pronounced hippocampal cell loss in the CA3 region and dentate gyrus, increased reactive gliosis, and greater cytoskeletal damage, as assessed by changes in immunoreactivity for MAP2, beta tubulin, and synaptophysin in NFH/LacZ mice (Galvin et al., 2000). Intracellular NF-rich inclusions may lead to increased vulnerability caused by alterations in axonal transport or impaired function of intracellular organelles entrapped in the inclusions.
Taken together, these studies suggest that TBI may be a strong risk factor for neurodegenerative disorders. It appears likely that genetic factors—such as mutations in genes for APP, presenilins, APOE4 genotype, or in genes encoding for cytoskeletal proteins—may confer a vulnerability that becomes evident after exposure to epigenetic insults. Figure 6 summarizes the relevant pathogenetic mechanisms related to the epidemiologic link between TBI and neurodegenerative diseases investigated with genetically engineered mice. Table 3 summarizes the results of the related studies described in the text with the main findings in relation to the genotype used and the model of injury.
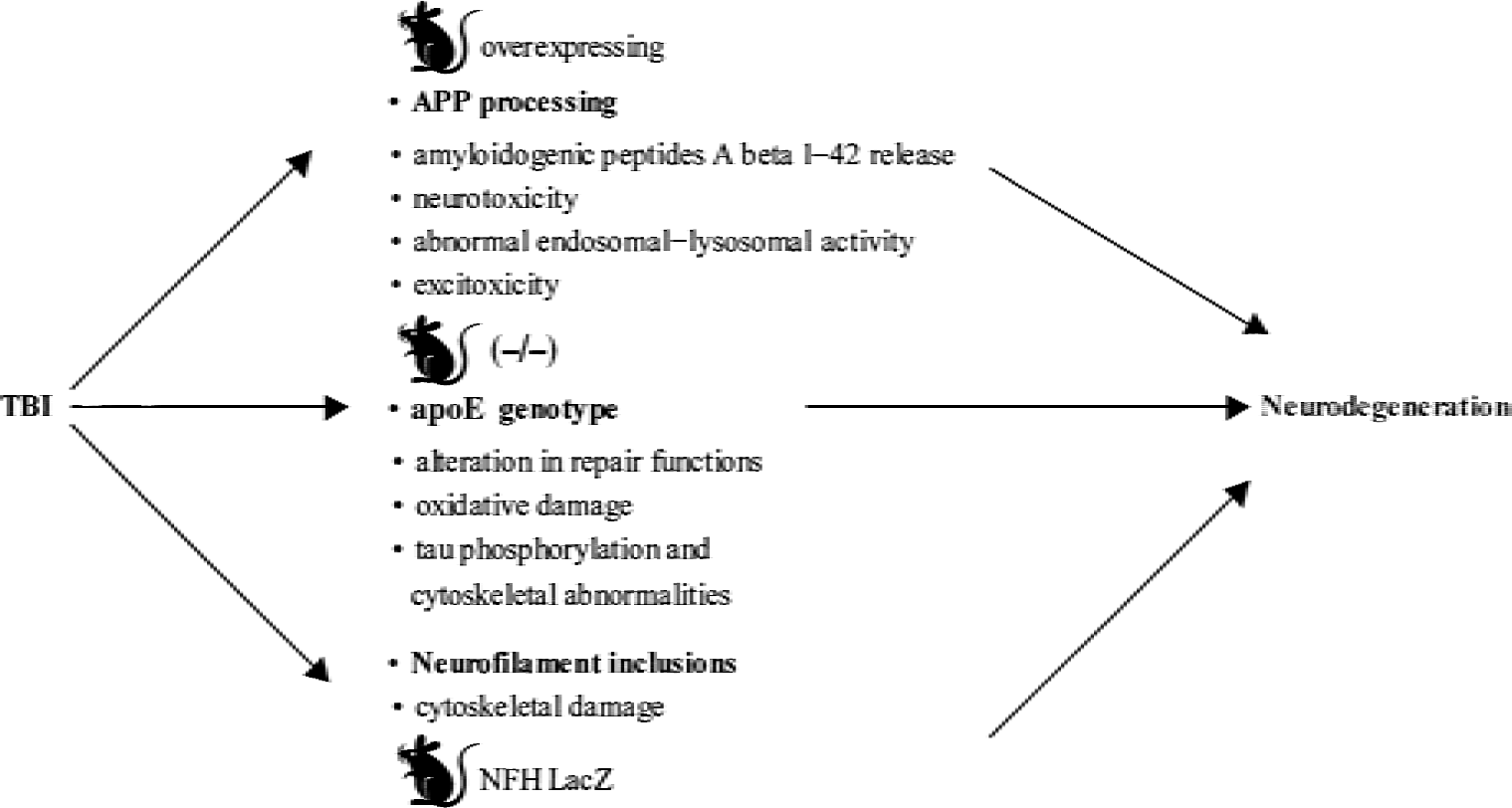
Relation between traumatic brain injury (TBI) and neurodegenerative diseases. Relevant pathogenetic mechanisms evaluated with genetically engineered mice. apoE, apolipoprotein E; APP, amyloid precursor protein; NFH, neurofilament-heavy.
Summary of studies performed to clarify the epidemiological relation between TBI and neurodegenerative diseases
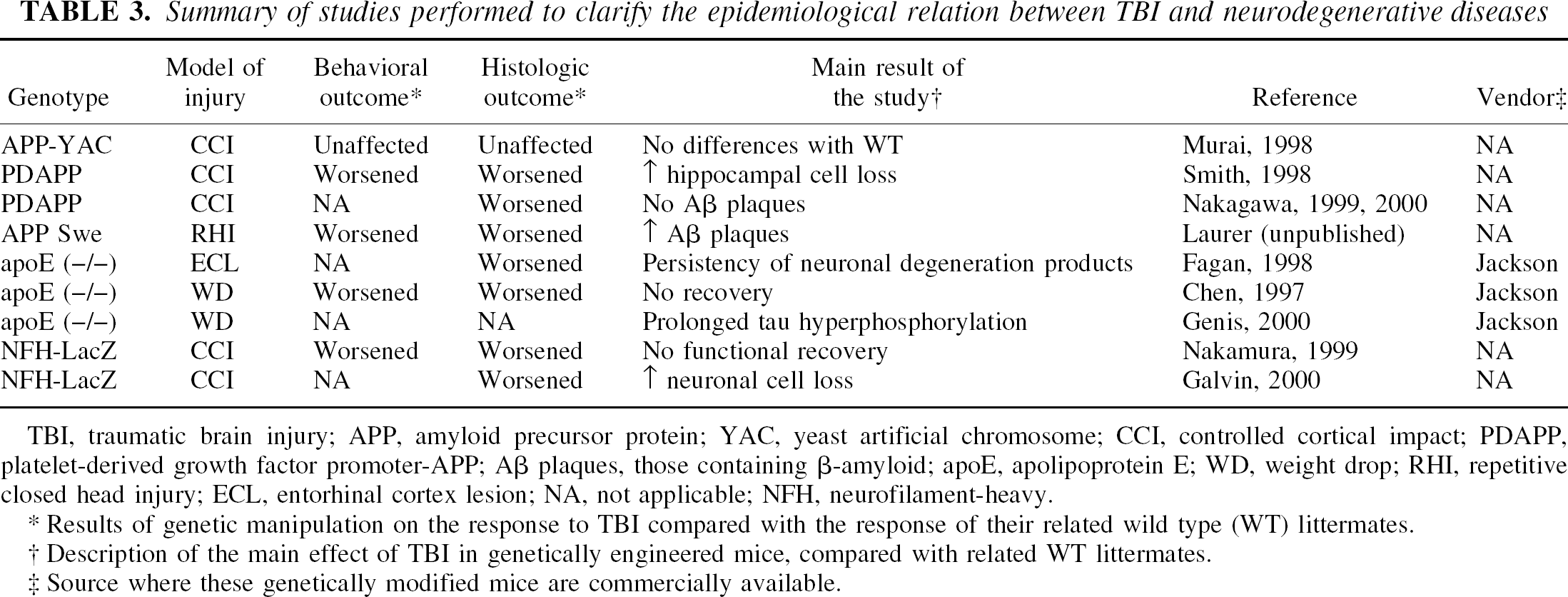
TBI, traumatic brain injury; APP, amyloid precursor protein; YAC, yeast artificial chromosome; CCI, controlled cortical impact; PDAPP, platelet-derived growth factor promoter-APP; Aβ plaques, those containing β-amyloid; apoE, apolipoprotein E; WD, weight drop; RHI, repetitive closed head injury; ECL, entorhinal cortex lesion; NA, not applicable; NFH, neurofilament-heavy.
Results of genetic manipulation on the response to TBI compared with the response of their related wild type (WT) littermates.
Description of the main effect of TBI in genetically engineered mice, compared with related WT littermates.
Source where these genetically modified mice are commercially available.
FUTURE DIRECTIONS
Traumatic brain injury activates a prolonged pathophysiologic cascade directly influencing the complex network associated with neurodegeneration and repair/regeneration pathways in the CNS. The need to explore the molecular basis of cellular dysfunction and death after injury to the CNS has led researchers to transgenic technology. Genetically engineered animals, obtained by artificial overexpression or deletion of a specific gene, offer the unique opportunity to test mechanistic hypotheses concerning cause and effect relations and to provide a mechanistic basis for the evaluation of potentially effective therapeutic agents targeted at selective secondary pathways activated after TBI. When compared with the use of pharmacologic agents to antagonize or enhance specific pathways, this technology offers several advantages, such as the possibility of overcoming obstacles related to optimal time window for treatment, cerebral tissue penetration, sex differences in metabolism, and toxicity. However, developmental compensatory mechanisms caused by a chronic alteration of the target gene may cause responses to TBI that are different from those in the WT littermates in which a molecule-mediated effect is blocked by a pharmacologic agent. Additional caveats must be acknowledged with respect to the use of genetically modified animals in TBI research. Various strains of mice (C57BL/6, C57BL/10, C57SJL, 129/SvEMS, FVB/N) have shown different and highly specific cognitive, behavioral, and histologic outcomes after excitotoxic cell death or the induction of stroke or trauma (Steward et al., 1999). For example, three background strains of mice (C57BL/6, FVB/N, and 129/SvEMS) have been reported to exhibit significantly different behavioral responses when subjected to CCI brain injury (Fox et al., 1999). These results suggest that the background strain should be carefully considered when experiments involve genetically altered mice, especially when the studies use behavioral outcome measures after CNS injury (Steward et al., 1999).
The polymorphism inherent in the genetic background of genetically engineered mouse strains can potentially make the result of gene-targeting studies difficult to interpret. A phenotypic change observed in a mutant mouse, therefore, may be related to the genetic background rather than the mutation itself. In fact, most gene targeting is performed in embryonic stem cells derived from strain 129/Sv. These cells are introduced into a blastocyst, and the derived chimeric adult mice then are mated to WT mice. If the WT mice are C57BL/6, the first derived generation will be heterozygous for the mutant allele but also will have a set of chromosomes derived from 129/Sv and C57BL/6. The mating of these heterozygous mice will produce a second generation composed of homozygous mutant, heterozygous mutant, and WT, all of whom have different loci for the targeted mutated gene. Consequently, one associated risk of these studies is the presence of false positive or negative results depending on the genetic background itself and not on the targeted mutation (Gerlai, 1996). Solutions for these potential pitfalls include increasing the number of animals, thereby increasing the power of statistical comparison and reducing the possibility of sampling error associated with differences among background strains, and carefully analyzing both genotype and phenotype of the animals to confirm that the phenotype maps consistently to the genotype at the targeted locus (Steward et al., 1999).
In spite of these potential pitfalls, the successful production of genetically modified mice lacking or overexpressing specific mediators and receptors has made it possible to investigate the cellular and molecular role of inflammation, ROS, DNA damage and repair, and cell death mediators in TBI. Several inflammatory factors—such as intercellular adhesion molecules, cytokines, and their receptors—have been investigated using this new technology, although many questions remain unresolved. Degenerative and regenerative processes associated with posttraumatic inflammation may represent a duality of inflammatory processes, which interact in a complex way during a prolonged posttraumatic time course. The goal of antagonizing (or enhancing) a specific pathway to obtain a therapeutic benefit therefore may be too simplistic, because the same molecule may exert beneficial (neuroprotective) or deleterious (neurotoxic) effects at different time points in the postinjury cascade. More work needs to be performed to clarify the role of specific pathogenic cascades such as inflammation and apoptosis in mediating cell death, to design targeted therapies that can be sequentially combined in the acute and chronic postinjury periods to reduce damage and stimulate reparative processes. Genetically engineered mice also have stimulated the exploration of the molecular basis underlying the relation between head injury and neurodegenerative diseases, including such diverse pathways as abnormalities in APP, β-amyloid, apoE genotype, and cytoskeletal proteins. Further studies are warranted to help clarify how specific genotypes and the pathways mentioned above may interact at different time points in the postinjury period and to promote the evaluation of new therapeutic strategies in well-characterized models of TBI.